

Abstract
AIM: To determine the effect and molecular mechanism of ezrin-radixin-moesin-binding phosphoprotein-50 (EBP50) in hepatocellular carcinoma (HCC).
METHODS: Three human HCC cell lines, i.e., SM-MC7721, HepG2 and Hep3B, were used. We transfected the Pbk-CMV-HA-EBP50 plasmid into SMMC7721 cells with Lipofectamine 2000 to overexpress EBP50. Western blotting were performed to determine the effects of the plasmid on EBP50 expression and to detect the expression of β-catenin and E-cadherin before and after the transfection of the plasmid into SMMC7721 cells. In vitro cell proliferation was assessed with a Cell Counting Kit-8 (CCK-8) assay. Cell cycle distribution was assessed with flow cytometry. Invasion and migration ability of before and after the transfection were determined with a transwell assay. Cell apoptosis was demonstrated with Annexin V-FITC. The effect of EBP50 overexpressing on tumor growth in vivo was performed with a xenograft tumor model in nude mice.
RESULTS: The transfection efficiency was confirmed with Western blotting (1.36 ± 0.07 vs 0.81 ± 0.09, P < 0.01). The CCK8 assay demonstrated that the growth of cells overexpressing EBP50 was significantly lower than control cells (P < 0.01). Cell cycle distribution showed there was a G0/G1 cell cycle arrest in cells overexpressing EBP50 (61.3% ± 3.1% vs 54.0% ± 2.4%, P < 0.05). The transwell assay showed that cell invasion and migration were significantly inhibited in cells overexpressing EBP50 compared with control cells (5.8 ± 0.8 vs 21.6 ± 1.3, P < 0.01). Annexin V-FITC revealed that apoptosis was significantly increased in cells overexpressing EBP50 compared with control cells (14.8% ± 2.7% vs 3.4% ± 1.3%, P < 0.05). The expression of β-catenin was downregulated and E-cadherin was upregulated in cells overexpressing EBP50 compared with control cells (0.28 ± 0.07 vs 0.56 ± 0.12, P < 0.05; 0.55 ± 0.08 vs 0.39 ± 0.07, P < 0.05). In vivo tumor growth assay confirmed that up-regulation of EBP50 could obviously slow the growth of HCC derived from SMMC7721 cells (28.9 ± 7.2 vs 70.1 ± 7.2, P < 0.01).
CONCLUSION: The overexpression of EBP50 could inhibit the growth of SMMC7721 cells and promote apoptosis by modulating β-catenin, E-cadherin. EBP50 may serve asa potential therapeutic target in HCC.
Keywords: Hepatocellular carcinoma, Ezrin-radixin-moesin-binding phosphoprotein-50, Growth, Migration, Invasion
INTRODUCTION
Hepatocellular carcinoma (HCC) is the third most common cause of mortality due to cancer in the world[1]. The low rate of early diagnosis renders the tumors unresectable and results in metastasis to the lymph nodes or distant organs. In addition, HCC cells are relatively resistant to chemotherapy; thus, the overall survival rate is poor. Recently, targeted molecular therapy has been shown to be effective in treating HCC, which encouraged us to study the cellular and molecular mechanisms of HCC and identify more effective treatments.
The etiology of hepatocarcinogenesis is still not fully understood, the activation of oncogenes, dysfunction of tumor suppressor genes, and aberrant activation of signal transduction pathways have been described. β-catenin is a key effector molecule in the Wnt signaling pathway and also plays an important role in cell-cell adhesion[2,3]. E-cadherin is a member of the cadherin family and is a classical type I cadherin. Both β-catenin and E-cadherin are membrane-associated proteins that are essential for regulating and providing cellular adhesion. Membrane-associated adhesion proteins regulate cell motility, including metastasis and invasion, and they link the extracellular matrix and actin cytoskeleton, which is critical in many biological signal transduction pathways. β-catenin and E-cadherin dysfunction has been described in several tumors, such as breast[4], prostate[5], gastric[6], colorectal[7], lung[8], oral[9] and hepatocellular carcinoma[10].
Ezrin-radixin-moesin-binding phosphoprotein-50 (EBP50, also known as NHERF1) is a 55-kDa phosphoprotein, and it belongs to the family of PDZ scaffolding proteins. Previous studies suggest that EBP50 is a potential tumor suppressor in several types of tumors[11-15]. Our previous studies also showed that EBP50 can inhibit the growth of gastric and pancreatic cancer cells[16,17]. EBP50 is mainly localized at the apical plasma membrane in human epithelial tissues[18]. The loss of normal apical membrane expression and/or distribution to the cytoplasm and nuclear overexpression was found in several human cancers[12-15,18]. Shibata et al[19] found that 45% of HCC cases expressed high levels of EBP50 mRNA, and 55% of HCC cases showed no difference. The cytoplasmic and nuclear accumulation of the EBP50 protein was present in 55% of HCC tissues compared with the surrounding noncancerous liver tissue, and EBP50 interacts with β-catenin to participate in hepatocarcinogenesis. The role of EBP50 as an oncogene or a tumor suppressor in HCC remains unclear.
In this study, we examined the effects of EBP50 up-regulation on cell growth, migration, invasion, and apoptosis in the HCC cell line SMMC7721 and determined the expression of β-catenin and E-cadherin to elucidate the role of EBP50 in HCC.
MATERIALS AND METHODS
Cell lines and plasmids
The human HCC cell lines Hep3B, SMMC7721, HepG2 were obtained from the Cell Bank of Shanghai Institute for Biological Sciences (Shanghai, China). Of the three cells lines, the SMMC7721 cell line has relatively low EBP50 protein expression levels. These cells were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum at 37 °C in a humidified atmosphere of 5% CO2. The pBK-CMV-HA-EBP50 plasmid was kindly provided by Dr. Randy Hall from Emory University (Atlanta, GA, United States).
Stable transfection
The pBK-CMV-HA-EBP50 plasmid or the pBK-CMV-HA vector was transfected into SMMC7721 cells with LipofectamineTM 2000 (Invitrogen, CA, United States) according to the manufacturer’s protocol. G418 (350 μg/mL) was used to select stably transfected cells. Single-cell clones were isolated for clonal expansion. The transfection efficiency was determined with Western blotting. Subsequently, stably transfected cell clones were cultured and amplified in culture medium supplemented with 350 μg/mL G418.
Cell proliferation assay
The cell-counting kit-8 (CCK-8, Dojindo, Japan) colorimetric assay was used to measure cell proliferation and viability with triplicate experiments for each set of conditions. We seeded 5 × 103 cells per well in a 96-well plate. Cell viability was assessed using the Cell Counting Kit after culture for 24 h. The absorbance at 450 nm wasmeasured with a plate reader.
Western blotting
Western blotting was performed as described previously[20]. The specific antibodies were rabbit anti-EBP50 antibody (1:1000, Abcam; Pierce, GA, United States), rabbit anti-β-catenin antibody (1:1000, Abcam), mouse anti-E-cadherin antibody (1:1000, Abcam), and mouse anti-β-actin antibody (1:1000, Abcam).
Transwell assay
Invasion through a Matrigel-coated filter was measured in transwell chambers (Corning, New York, NY, United States) as described previously[14]. Cells (1 × 104) were trypsinized and suspended in the upper chambers. After incubation for 24 h, the transwell chambers were fixed with 4% paraformaldehyde and stained with Giemsa (Sigma). Cells in five randomly selected fields were photographed, and the number of trans-membrane cells wascounted.
Cell cycle assay
Cells were trypsinized and fixed in 70% ethanol on ice and then stained with 50 μg/mL propidium iodide (Sigma) and 0.1 μg/mL RNase A (Sigma). The cells were then analyzed with flow cytometry using a FACStar Plus, and Cell Quest software was used to calculate the percentage of the cell population in each phase.
Cellular apoptosis assay
Apoptosis was assessed with the Annexin V-FITC kit according to the manufacturer’s protocol. A total of 1 × 106 cells/mL were washed twice with cold PBS and resuspended in binding buffer. A total of 5 mL of Annexin V-FITC and 10 mL of propidium iodide (PI) were added (BioVision, Mountain New, CA, United States), and the cells were incubated for 10 min at room temperature in the dark. At the end of the incubation, 200 μL of binding buffer was added, and the cells were immediately analyzed with flow cytometry. Cell Quest software was used to analyze flow cytometry data.
Ex vivo tumor growth assay
Male nude mice (4-6 wk old, n = 6) were (Beijing HFK Biosciencs Co., Ltd) used in the experiments. After alcohol preparation of the skin, 1 × 106 SMMC cells (pBK-CMV-HA-EBP50, pBK-CMV-HA vector and control cells), suspended in 100 μL PBS, were subcutaneously inoculated into the dorsal area of the nude mice. Six weeks after injection, At the end of the experiment, tumors were harvested and weighed. All experiments were performed according to the recommendations of the Institutional Animal Care and Use Committee, and the study protocol was approved by the Ethics Committee for Animal Research of Wuhan University, China.
Statistical analysis
The results were analyzed using SPSS 13.0 statistical software. All of the data are presented as the means ± SD. Statistically significant differences between groups in each assay were compared with a one-way analysis of variance (ANOVA), and P < 0.05 was considered to be statistically significant.
RESULTS
EBP50 expression in HCC cells
We used Western blotting to evaluate the expression of EBP50 in human HCC cell lines, with SMMC7721 expressing the lowest of the three cell lines (Figure 1). To investigate the role of EBP50 in HCC, we overexpressed EBP50 in SMMC7721 cells, which expressed relatively low levels of endogenous EBP50. SMMC7721 cells were transfected with pBK-CMV-HA-EBP50 or the pBK-CMV-HA vector to establish vector control cells. To evaluate the transfection efficiency, we determined the protein expression of EBP50 with Western blotting (Figure 1). The data showed that after transfection for 48 h, the protein levels of EBP50 were upregulated in pBK-CMV-HA-EBP50-transfected cells compared with pBK-CMV-HA vector-transfected cells and parental cells (1.36 ± 0.07 vs 0.81 ± 0.09 or 0.75 ± 0.06, P < 0.01).
Figure 1.
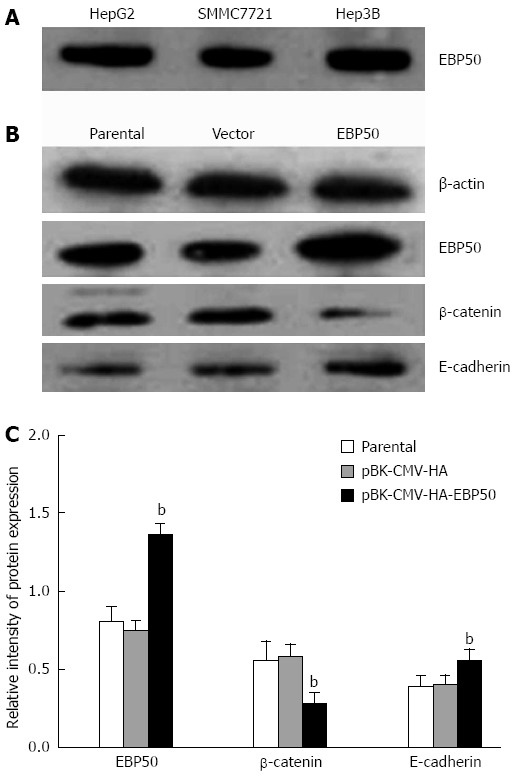
Ezrin-radixin-moesin-binding phosphoprotein-50, β-catenin, and E-cadherinprotein levels were detected with Western blotting. A: The expression of ezrin-radixin-moesin-binding phosphoprotein-50 (EBP50) protein in Hep3B, SMMC7721, HepG2; B: Western blotting analysis of EBP50, β-catenin, and E-cadherinprotein protein before and after transfection; C: Analysis of the expression of proteins. bP < 0.01 vs parental or pBK-CMV-HA.
Expression of β-catenin and E-cadherin in HCC cells
To investigate the biological effect of EBP50 in HCC and the potential signaling pathway through which it acts, we analyzed the expression of β-catenin and E-cadherin in pBK-CMV-HA-EBP50-and pBK-CMV-HA-transfected cells and untransfected cells. The protein levels of β-catenin was downregulated (0.28 ± 0.07 vs 0.56 ± 0.12 or 0.58 ± 0.08, P < 0.05) and E-cadherin (0.55 ± 0.08 vs 0.39 ± 0.07 or 0.40 ± 0.06, P < 0.05) was upregulated in pBK-CMV-HA-EBP50-transfected cells (Figure 1). The expression level was the same as previously reported by Hayashi et al[12].
Overexpression of EBP50 suppresses cancer cell growth
To assess the potential effects of EBP50 overexpression on cell proliferation and survival, we measured the number of viable cells at different times in vitro with CCK-8 assays. The pBK-CMV-HA vector had no effect on the proliferative ability of SMMC7721 cells, whereas pBK-CMV-HA-EBP50-transfected cells showed a dramatic reduction in proliferation (P < 0.01, Figure 2).
Figure 2.
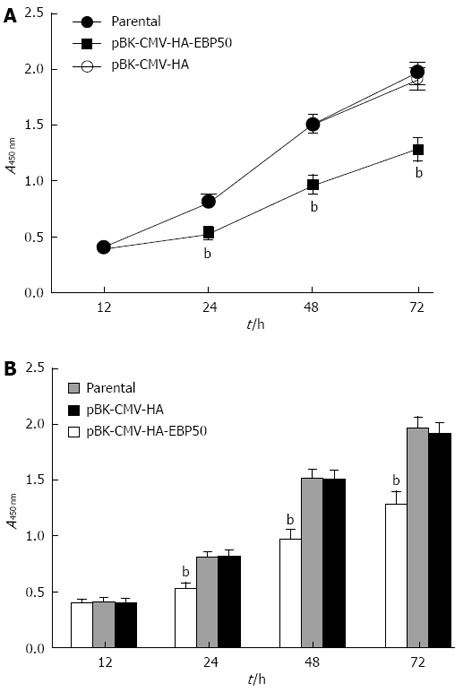
Growth rate comparison of parental, pBK-CMV-HA-EBP50, pBK-CMV-HA cells with a Cell Counting Kit-8 assay. A: Growth rate comparison of parental, pBK-CMV-HA-EBP50, pBK-CMV-HA cells; B: Analysis of the growth rate. bP < 0.01 vs parental or pBK-CMV-HA.
The cell cycle distribution was measured to determine whether the growth-inhibitory activity of EBP50 on SMMC7721 cell proliferation was due to cell cycle alterations. Compared with the control cells, the percentage of pBK-CMV-HA-EBP50 cells in G0/G1 phase was increased to 61.3% ± 3.1% from 54.0% ± 2.4% in parental cells and 54.5% ± 1.9% in mock cells (P < 0.05, Figure 3).
Figure 3.
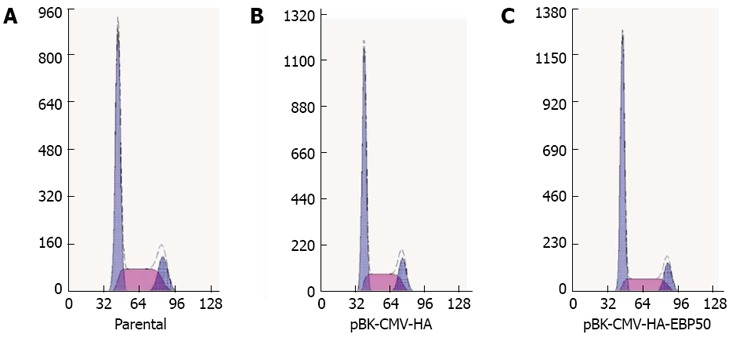
Effect of ezrin-radixin-moesin-binding phosphoprotein-50 over-expression on cell cycle distribution. A: Cell cycle analysis of parental (G0/G1, 54.2%); B: pBK-CMV-HA (G0/G1, 56.2%); C: pBK-CMV-HA-EBP50 (G0⁄G1, 61.4%) cells with flowcytometry.
Overexpression of EBP50 suppresses cancer cell migration and invasion
We explored whether the over-expression of EBP50 resulted in decreased invasion and migration in cancer cells with a transwell assay. As shown in Figure 4A-D, the number of migrated pBK-CMV-HA-EBP50 cells (5.8 ± 0.8 cells/HP) was lower than the pBK-CMV-HA vector cells (20.4 ± 1.1 cells/HP) and parental cells (21.6 ± 1.3 cells/HP) (P < 0.01), which indicates that EBP50 could significantly suppress the invasive ability of SMMC7721 cells. No significant difference was found between SMMC7721 parental cells and the pBK-CMV-HA vector cells. The result showed that upregulating EBP50 expression in SMMC7721 cells inhibited tumor cell invasion and migration.
Figure 4.
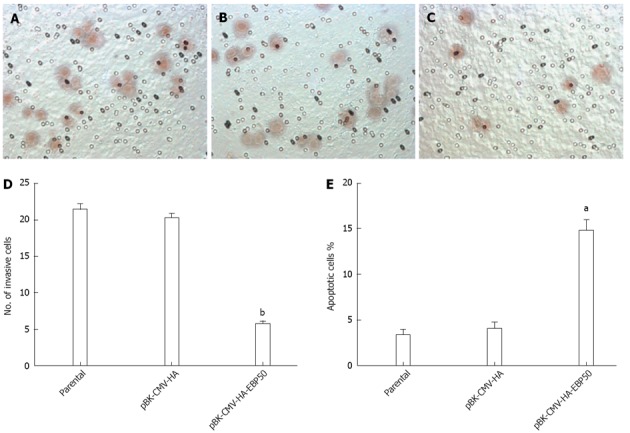
Ezrin-radixin-moesin-binding phosphoprotein-50 over-expression suppressed the migration and invasion of cancer cells. A: Migration and invasion analysis of parental (21.6 ± 1.3 cells/HP); B: pBK-CMV-HA (20.4 ± 1.1 cells/HP); C: pBK-CMV-HA-EBP50 (5.8 ± 0.8 cells/HP) cells with transwell assey; D: Analysis of the number of invasive cells; E: Apoptotic analysis of parental (3.4% ± 1.3%), pBK-CMV-HA (4.1% ± 1.5%) and pBK-CMV-HA-EBP50 (14.8% ± 2.7%) cells with flowcytometry. aP < 0.05, bP < 0.01 vs parental or pBK-CMV-HA.
Overexpression of EBP50 promotes cell apoptosis
Apoptosis was investigated with an annexin V/propidium iodide assay. The pBK-CMV-HA-EBP50 cells showed an approximately fourfold increase in apoptosis (P < 0.05) compared with the parental group and the pBK-CMV-HA vector group (Figure 4E), suggesting that the pBK-CMV-HA-EBP50 plasmid enhances SMMC7721 apoptosis.
Overexpression of EBP50 decreased the growth of the tumor ex vivo
The cell growth response as result of NHERF1 overexpression was also examined in the nude mice model. We compared the growth of SMMC7721 cells infected with pBK-CMV-HA-EBP50, pBK-CMV-HA vector and control cells ex vivo. At 6 wk after inoculation, obvious tumors were formed in the two control groups of mice inoculated with SMMC7721 cells or the pBK-CMV-HA vector, while tumor growth was obviously suppressed in mice inoculated with pBK-CMV-HA-EBP50 cells. As shown in Figure 5, tumors formed from pBK-CMV-HA-EBP50 (28.9 ± 7.2 mg) were significantly smaller than those from pBK-CMV-HA and control cells (68.9 ± 7.9 mg, 70.1 ± 7.2 mg, n = 6) (P < 0.01), suggesting that expression of EBP50 suppressed tumor growth ex vivo.
Figure 5.
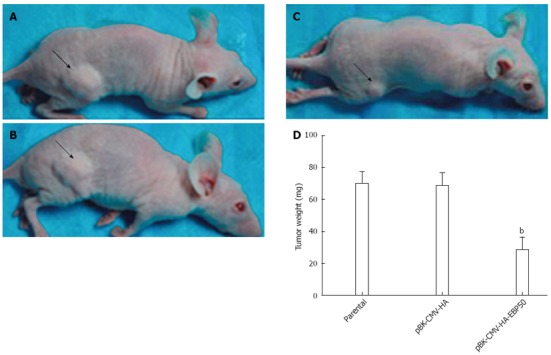
Tumorigenicity was inhibited by overexpression of ezrin-radixin-moesin-binding phosphoprotein-50 ex vivo (arrow). A: Tumor weight in nude mice of parental (70.1 ± 7.2 mg); B: pBK-CMV-HA (68.9 ± 7.9 mg); C: pBK-CMV-HA-EBP50 (28.9 ± 7.2 mg); D: Tumor weight of the nude mice in each group. bP < 0.01 vs parental or pBK-CMV-HA.
DISCUSSION
In the present study, to determine the role of EBP50 in HCC as an oncogene or a tumor suppressor, we used the pBK-CMV-HA-EBP50 plasmid to increase the expression of EBP50 in SMMC7721 cells. Western blotting analysis proved that EBP50 protein levels were significantly upregulated in pBK-CMV-HA-EBP50-transfected SMMC7721 cells. The proliferation rate of cells over-expressing EBP50 was lower than the pBK-CMV-HA vector and parental SMMC7721 cells (Figure 2). In a transwell assay, the number of migrated cells over-expressing EBP50 was significantly lower than control cells, indicating that the over-expression of EBP50 can inhibit the anchorage-independent growth of HCC cells (Figure 4). Cellular apoptosis analysis showed that EBP50 overexpression could increase apoptosis and induce G0/G1 cell cycle arrest in HCC cells (Figures 3 and 4). Ex vivo tumor growth assay confirmed that cells overexpressing EBP50 could obviously slow the growth of HCC cells (Figure 5). Zheng et al[11] reported that EBP50 overexpression could inhibit breast cancer cell proliferation and promote cell apoptosis through decreasing ERK activation. The current study also suggests EBP50 as a potential tumor suppressor in HCC.
The molecular mechanisms through which EBP50 exerts its tumor suppressor functions are diverse. EBP50 contains two PDZ domains and an ERM domain. PDZ domains have been implicated in protein-protein interactions, and the ERM domain binds to ezrin through its ERM-binding region[21]. In normal cells, EBP50 interact with cyclin dependent kinase and PP2A through its reversible phosphorylation of the protein to regulate cell division[22]. EBP50 interacts with many tumor-related proteins through these domains, including PTEN, PDGFR, EGFR, podocalyxin,and GPCRs, to participate in tumorigenesis[23-29]. EBP50 forms a complex with beta-catenin to promote Wnt signaling and may participate in the development of HCC[19].
Both β-catenin and E-cadherin play an essential role in cell-cell junctions in Wnt signal pathway. β-catenin binds directly to the cytoplasmic tail of E-cadherin to form a bridge between E-cadherin and the actin cytoskeleton and stabilizes adherens junctions[30]. The plasma membrane localization of β-catenin allows it to function as a tumor suppressor by interacting with E-cadherin, and the phosphorylation of β-catenin results in its accumulation in the cytoplasm and translocation to the nucleus, which allows it to act as an oncogenic protein by increasing the transcription of genes involved in growth control[31]. The loss of membranous E-cadherin expression was found in highly aggressive tumors including HCC and contributed to their high metastatic potential[32-34]. Aberrant activation of Wnt/β-catenin signaling has been reported in a wide range of HCC[35].
In our study, EBP50 upregulation resulted in an increase in E-cadherin levels and a decrease in β-catenin levels. EBP50 is required to maintain a fraction of the cortical β-catenin protein at the membrane; EBP50 binds to the C-terminal PDZ motif of β-catenin through its PDZ2 domain, and this association is required for β-catenin localization at cell-cell junctions[36]. In EBP50-depleted cells, there was a shift of β-catenin to the nucleus and an increased invasiveness of cells[12]. In EBP50-deficient MEFs, disorganization of E-cadherin-mediated adherens junctions and an increase in β-catenin transcriptional activity was observed[26], suggesting that the depletion of EBP50 decreased the interaction between β-catenin and E-cadherin. Because of the contributing role of β-catenin and E-cadherin in HCC hyperplasia and tumorigenesis[10,19], it is conceivable that the regulation of β-catenin/E-cadherin as a result of EBP50 overexpression contributes to HCC initiation or progression.
Our results suggest a tumor-suppressing role of EBP50 and establish that it may be a promising target for therapeutic intervention in HCC. Additionally, the interactions of EBP50 with β-catenin/E-cadherin enhance the tumor suppressor properties of EBP50 in HCC.
ACKNOWLEDGMENTS
The Pbk-CMV-HA-EBP50 plasmid was kindly provided by Dr. Randy Hall from Emory University.
COMMENTS
Background
Hepatocellular carcinoma (HCC) is the fifth most common cancer globally and relatively resistant to chemotherapy, it encouraged us to study the cellular and molecular mechanisms of HCC targeted molecular therapy. ezrin-radixin-moesin-binding phosphoprotein-50 (EBP50) is a PDZ scaffolding protein, previous studies suggest that EBP50 is a potential tumor suppressor in colorectal, prostatic, biliary, breast, gastric and pancreatic cancer, and it has an abnormal expression in HCC. But EBP50 is an oncogene or a tumor suppressor in HCC remains unclear. The authors aimed to examine the role of EBP50 and whether it interact with the β-catenin/E-cadherin pathway in HCC.
Research frontiers
Targeted molecular therapy is a new effective treatment for cancer including HCC. EBP50 is a potential tumor suppressor in several cancers, but the role of EBP50 is an oncogene or a tumor suppressor remains controversial and its exact role in HCC remains unknown.
Innovations and breakthroughs
Authors used a pBK-CMV-HA-EBP50 plasmid to overexpress EBP50 in SMMC7721 cells. The results showed that overexpression of EBP50 could inhibit the growth, migration, invasion of SMMC7721 cells and promote cell apoptosis, and modulate the expression of beta-catenin, E-cadherin. EBP50 may serve as a potential therapeutic target in HCC. It suggested that EBP50 is a tumor suppressor and interact with β-catenin/E-cadherin in HCC.
Applications
The results showed that EBP50 is a tumor suppressor and interact with β-catenin/E-cadherin in HCC. It may contribute to the future research of HCC and be a promising target for therapeutic intervention in HCC.
Terminology
Ezrin-radixin-moesin-binding phosphoprotein-50: EBP50 (also known as NHERF1) is a 55-kDa phosphoprotein; β-catenin: A adhesion protein, a key mediator in the canonical Wnt signal pathway; E-cadherin: A member of the cadherin family and is a classical type I cadherin.
Peer review
The function of EBP50 as a tumor suppressor has been studied in other cancers, but not in HCC. The expression in HCC was not significantly different between patient cases reported by Shibata et al. Thus they examined the effects of EBP50 up-regulation on cell growth, migration, invasion, and apoptosis in the HCC cell line SMMC7721 and determined the conclusion. This conclusion is meaningful in the sense characterized the function of EBP in HCC cells.
Footnotes
Supported by Fundamental Research Funds for the Central Universities of China, No. 201130202020005
P- Reviewer Yu DY S- Editor Gou SX L- Editor A E- Editor Zhang DN
References
- 1.El-Serag HB. Hepatocellular carcinoma. N Engl J Med. 2011;365:1118–1127. doi: 10.1056/NEJMra1001683. [DOI] [PubMed] [Google Scholar]
- 2.Tsanou E, Peschos D, Batistatou A, Charalabopoulos A, Charalabopoulos K. The E-cadherin adhesion molecule and colorectal cancer. A global literature approach. Anticancer Res. 2008;28:3815–3826. [PubMed] [Google Scholar]
- 3.Kikuchi A. Regulation of beta-catenin signaling in the Wnt pathway. Biochem Biophys Res Commun. 2000;268:243–248. doi: 10.1006/bbrc.1999.1860. [DOI] [PubMed] [Google Scholar]
- 4.Morrogh M, Andrade VP, Giri D, Sakr RA, Paik W, Qin LX, Arroyo CD, Brogi E, Morrow M, King TA. Cadherin-catenin complex dissociation in lobular neoplasia of the breast. Breast Cancer Res Treat. 2012;132:641–652. doi: 10.1007/s10549-011-1860-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lee J, Ju J, Park S, Hong SJ, Yoon S. Inhibition of IGF-1 signaling by genistein: modulation of E-cadherin expression and downregulation of β-catenin signaling in hormone refractory PC-3 prostate cancer cells. Nutr Cancer. 2012;64:153–162. doi: 10.1080/01635581.2012.630161. [DOI] [PubMed] [Google Scholar]
- 6.Czyzewska J, Guzińska-Ustymowicz K, Ustymowicz M, Pryczynicz A, Kemona A. The expression of E-cadherin-catenin complex in patients with advanced gastric cancer: role in formation of metastasis. Folia Histochem Cytobiol. 2010;48:37–45. doi: 10.2478/v10042-010-0017-z. [DOI] [PubMed] [Google Scholar]
- 7.Aamodt R, Bondi J, Andersen SN, Bakka A, Bukholm G, Bukholm IR. The Prognostic Impact of Protein Expression of E-Cadherin-Catenin Complexes Differs between Rectal and Colon Carcinoma. Gastroenterol Res Pract. 2010;2010:616023. doi: 10.1155/2010/616023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shapiro M, Akiri G, Chin C, Wisnivesky JP, Beasley MB, Weiser TS, Swanson SJ, Aaronson SA. Wnt pathway activation predicts increased risk of tumor recurrence in patients with stage I nonsmall cell lung cancer. Ann Surg. 2013;257:548–554. doi: 10.1097/SLA.0b013e31826d81fd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.González-Moles MA, Bravo M, Ruiz-Avila I, Gil-Montoya JA, Acebal F, Esteban F. E-cadherin in non-tumor epithelium adjacent to oral cancer as risk marker for the development of multiple tumors. Br J Oral Maxillofac Surg. 2013;51:157–163. doi: 10.1016/j.bjoms.2012.05.001. [DOI] [PubMed] [Google Scholar]
- 10.Wang XQ, Zhang W, Lui EL, Zhu Y, Lu P, Yu X, Sun J, Yang S, Poon RT, Fan ST. Notch1-Snail1-E-cadherin pathway in metastatic hepatocellular carcinoma. Int J Cancer. 2012;131:E163–E172. doi: 10.1002/ijc.27336. [DOI] [PubMed] [Google Scholar]
- 11.Zheng JF, Sun LC, Liu H, Huang Y, Li Y, He J. EBP50 exerts tumor suppressor activity by promoting cell apoptosis and retarding extracellular signal-regulated kinase activity. Amino Acids. 2010;38:1261–1268. doi: 10.1007/s00726-009-0437-2. [DOI] [PubMed] [Google Scholar]
- 12.Hayashi Y, Molina JR, Hamilton SR, Georgescu MM. NHERF1/EBP50 is a new marker in colorectal cancer. Neoplasia. 2010;12:1013–1022. doi: 10.1593/neo.10780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bartholow TL, Becich MJ, Chandran UR, Parwani AV. Immunohistochemical analysis of ezrin-radixin-moesin-binding phosphoprotein 50 in prostatic adenocarcinoma. BMC Urol. 2011;11:12. doi: 10.1186/1471-2490-11-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lin YY, Hsu YH, Huang HY, Shann YJ, Huang CY, Wei SC, Chen CL, Jou TS. Aberrant nuclear localization of EBP50 promotes colorectal carcinogenesis in xenotransplanted mice by modulating TCF-1 and β-catenin interactions. J Clin Invest. 2012;122:1881–1894. doi: 10.1172/JCI45661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Clapéron A, Guedj N, Mergey M, Vignjevic D, Desbois-Mouthon C, Boissan M, Saubaméa B, Paradis V, Housset C, Fouassier L. Loss of EBP50 stimulates EGFR activity to induce EMT phenotypic features in biliary cancer cells. Oncogene. 2012;31:1376–1388. doi: 10.1038/onc.2011.334. [DOI] [PubMed] [Google Scholar]
- 16.Lv XG, Ji MY, Dong WG, Lei XF, Liu M, Guo XF, Wang J, Fang C. EBP50 gene transfection promotes 5-fluorouracil-induced apoptosis in gastric cancer cells through Bax- and Bcl-2-triggered mitochondrial pathways. Mol Med Report. 2012;5:1220–1226. doi: 10.3892/mmr.2012.805. [DOI] [PubMed] [Google Scholar]
- 17.Ji MY, Fan DK, Lv XG, Peng XL, Lei XF, Dong WG. The detection of EBP50 expression using quantum dot immunohistochemistry in pancreatic cancer tissue and down-regulated EBP50 effect on PC-2 cells. J Mol Histol. 2012;43:517–526. doi: 10.1007/s10735-012-9424-0. [DOI] [PubMed] [Google Scholar]
- 18.Stemmer-Rachamimov AO, Wiederhold T, Nielsen GP, James M, Pinney-Michalowski D, Roy JE, Cohen WA, Ramesh V, Louis DN. NHE-RF, a merlin-interacting protein, is primarily expressed in luminal epithelia, proliferative endometrium, and estrogen receptor-positive breast carcinomas. Am J Pathol. 2001;158:57–62. doi: 10.1016/S0002-9440(10)63944-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shibata T, Chuma M, Kokubu A, Sakamoto M, Hirohashi S. EBP50, a beta-catenin-associating protein, enhances Wnt signaling and is over-expressed in hepatocellular carcinoma. Hepatology. 2003;38:178–186. doi: 10.1053/jhep.2003.50270. [DOI] [PubMed] [Google Scholar]
- 20.Liu QS, Zhang J, Liu M, Dong WG. Lentiviral-mediated miRNA against liver-intestine cadherin suppresses tumor growth and invasiveness of human gastric cancer. Cancer Sci. 2010;101:1807–1812. doi: 10.1111/j.1349-7006.2010.01600.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Weinman EJ, Steplock D, Tate K, Hall RA, Spurney RF, Shenolikar S. Structure-function of recombinant Na/H exchanger regulatory factor (NHE-RF) J Clin Invest. 1998;101:2199–2206. doi: 10.1172/JCI204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Boratkó A, Gergely P, Csortos C. Cell cycle dependent association of EBP50 with protein phosphatase 2A in endothelial cells. PLoS One. 2012;7:e35595. doi: 10.1371/journal.pone.0035595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Takahashi Y, Morales FC, Kreimann EL, Georgescu MM. PTEN tumor suppressor associates with NHERF proteins to attenuate PDGF receptor signaling. EMBO J. 2006;25:910–920. doi: 10.1038/sj.emboj.7600979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pan Y, Weinman EJ, Dai JL. Na+/H+ exchanger regulatory factor 1 inhibits platelet-derived growth factor signaling in breast cancer cells. Breast Cancer Res. 2008;10:R5. doi: 10.1186/bcr1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Curto M, McClatchey AI. Nf2/Merlin: a coordinator of receptor signalling and intercellular contact. Br J Cancer. 2008;98:256–262. doi: 10.1038/sj.bjc.6604002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lazar CS, Cresson CM, Lauffenburger DA, Gill GN. The Na+/H+ exchanger regulatory factor stabilizes epidermal growth factor receptors at the cell surface. Mol Biol Cell. 2004;15:5470–5480. doi: 10.1091/mbc.E04-03-0239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.James MF, Beauchamp RL, Manchanda N, Kazlauskas A, Ramesh V. A NHERF binding site links the betaPDGFR to the cytoskeleton and regulates cell spreading and migration. J Cell Sci. 2004;117:2951–2961. doi: 10.1242/jcs.01156. [DOI] [PubMed] [Google Scholar]
- 28.Rochdi MD, Parent JL. Galphaq-coupled receptor internalization specifically induced by Galphaq signaling. Regulation by EBP50. J Biol Chem. 2003;278:17827–17837. doi: 10.1074/jbc.M210319200. [DOI] [PubMed] [Google Scholar]
- 29.Hsu YH, Lin WL, Hou YT, Pu YS, Shun CT, Chen CL, Wu YY, Chen JY, Chen TH, Jou TS. Podocalyxin EBP50 ezrin molecular complex enhances the metastatic potential of renal cell carcinoma through recruiting Rac1 guanine nucleotide exchange factor ARHGEF7. Am J Pathol. 2010;176:3050–3061. doi: 10.2353/ajpath.2010.090539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Provost E, Rimm DL. Controversies at the cytoplasmic face of the cadherin-based adhesion complex. Curr Opin Cell Biol. 1999;11:567–572. doi: 10.1016/s0955-0674(99)00015-0. [DOI] [PubMed] [Google Scholar]
- 31.Yang SZ, Kohno N, Yokoyama A, Kondo K, Hamada H, Hiwada K. Decreased E-cadherin augments beta-catenin nuclear localization: studies in breast cancer cell lines. Int J Oncol. 2001;18:541–548. [PubMed] [Google Scholar]
- 32.Da Silva L, Parry S, Reid L, Keith P, Waddell N, Kossai M, Clarke C, Lakhani SR, Simpson PT. Aberrant expression of E-cadherin in lobular carcinomas of the breast. Am J Surg Pathol. 2008;32:773–783. doi: 10.1097/PAS.0b013e318158d6c5. [DOI] [PubMed] [Google Scholar]
- 33.Humar B, Blair V, Charlton A, More H, Martin I, Guilford P. E-cadherin deficiency initiates gastric signet-ring cell carcinoma in mice and man. Cancer Res. 2009;69:2050–2056. doi: 10.1158/0008-5472.CAN-08-2457. [DOI] [PubMed] [Google Scholar]
- 34.Vasioukhin V. Adherens junctions and cancer. Subcell Biochem. 2012;60:379–414. doi: 10.1007/978-94-007-4186-7_16. [DOI] [PubMed] [Google Scholar]
- 35.Nejak-Bowen KN, Monga SP. Beta-catenin signaling, liver regeneration and hepatocellular cancer: sorting the good from the bad. Semin Cancer Biol. 2011;21:44–58. doi: 10.1016/j.semcancer.2010.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kreimann EL, Morales FC, de Orbeta-Cruz J, Takahashi Y, Adams H, Liu TJ, McCrea PD, Georgescu MM. Cortical stabilization of beta-catenin contributes to NHERF1/EBP50 tumor suppressor function. Oncogene. 2007;26:5290–5299. doi: 10.1038/sj.onc.1210336. [DOI] [PubMed] [Google Scholar]