

Abstract
The need for a rapid detection and characterization of biowarfare (BW) agents cannot be over emphasized. With diverse array of potential BW pathogen available presently, rapid identification of the pathogen is crucial, so that specific therapy and control measures can be initiated. We have developed a multiplex polymerase chain reaction based reverse line blot macroarray to simultaneously detect four pathogens of BW importance viz. Bacillus anthracis, Yersinia pestis, Brucella melitensis and Burkholderia pseudomallei. The multiplex PCR utilizes 14 pairs of primers targeting 18 specific markers. These markers include genes which are genus specific, species-specific chromosomal sequences and virulence markers of plasmid origin. The assay was evaluated on various human, environment and animal isolates. The assay w successful in simultaneous detection and characterization of isolates of the four pathogens on as a single platform with sensitivity ranging from 0.3 pg to 0.3 ng of genomic DNA. The assay was able to detect 5 × 102 cfu/ml for B. anthracis, 8 × 102 cfu/ml for Yersinia sp., 1.4 × 102 cfu/ml for B. melitensis and 4 × 102 cfu/ml for B. pseudomallei.
Keywords: Reverse line blot, Bacillus anthracis, Yersinia pestis, Brucella, Burkholderia, Biowarfare
Introduction
Incidence of anthrax spore attack in the US and the discussions that ensued has fuelled great interests in the need to develop a rapid, sensitive and robust multiplex assays for screening biological warfare (BW) agents. The advantage of multiplex molecular assays over the classical method of isolation and culturing is the ability of the former to characterize the virulence of the identified species simultaneously on a single platform. A number of reports on the usage of real-time PCR assay for simultaneous detection of multiple targets are available [1]. Presently, the drawback of the real-time PCR assay for multiple target detection is the limited number of dyes and detector available commercially. Microarray based multiplex detection assays has the advantage of screening large number of targets on a single platform [2]. However, the complication in putting up a microarray assay, the need for qualified trained person and the cost involved makes the assay unsuitable for small health centres and low-funded laboratories.
Multiplex PCR based reverse line blot hybridization assay (mPCR/RLB) is a DNA hybridization assay where labelled products of multiplex PCR are allowed to hybridize with membrane bound amine modified target probes. The assay has been used for characterization and genotyping study on a number of organisms [3, 4]. In this paper, we describe the development of a much simpler; less expensive mPCR/RLB assay to identify and characterize simultaneously four potential bioweapons classified pathogens, Bacillus anthracis, Yersinia pestis, Brucella melitensis and Burkholderia pseudomallei.
Materials and Methods
Bacterial Isolates
The pathogens included in the present study consist of both field isolates and standard strains along with their closely related species (Table 1). Commonly encountered pathogens were also included for specificity studies. All culturing and propagation of the organisms was done using brain heart infusion broth and agar (Difco, MD, USA) at 37 °C for 12–14 h.
Table 1.
Organisms used for the development of mPCR/RLB assay
| Organism | Isolate name | Source |
|---|---|---|
| B. anthracis | BA-1 to BA-18 and BA-21 to BA-29 | Human isolates |
| B. anthracis | WB and O2 | Soil isolates |
| B. anthracis | Sterne | Vaccine strain |
| Y. pestis | Yp4, Yp8, Yp9, Yp101 to Yp108, YpS1 to YpS3 | Human isolates |
| Y. pestis | Yp111 to Yp117 | Rodent isolates |
| B. melitensis | Bm1 to Bm7 | Human isolates |
| B. melitensis | 16 M | Vaccine strain |
| B. pseudomallei | Bps 2 to Bps 12 | Human isolates |
| B. pseudomallei | Bps Soil | Soil isolate |
| B. cereus | ATCC 10876, ATCC 10371, ATCC 13061, ATCC 11778 | |
| B. thuringiensis | MTCC 869 | |
| Y. enterocolitica | ATCC 23715, MTCC 3101, MTCC 3100 | |
| Y. pseudotuberculosis | Strain 1b | |
| Y. kristensenii | ATCC 33639 | |
| Y. federicksenii | ||
| B. abortus | U38, U39 | Cattle isolate |
| B. abortus | NCTC 10093 | |
| B. abortus | S19 | Vaccine strain |
| B. mallei | NCTC 10230 | |
| Salmonella typhimurium | MTCC98 | |
| Salmonella typhi | MTCC734 | |
| Shigella boydi | ATCC9207 | |
| Escherichia coli | BL-21 | |
| Leptospira interogans | ATCC23470 |
ATCC American type culture collection, MTCC microbial type culture collection, NCTC national culture type collection
Primers and Probes
The probes consist of 5′-terminal amino modified oligonucleotides with size ranging from 20 to 30 bases. They were designed to have similar melting temperature of approximately 60 °C. For mPCR, 14 pairs of primers, 20–30 bases in length with 5′-terminal biotin modified, were designed with similar melting temperature. The primers were designed in such a way that the amplicon size does not exceed 500 bp. The 16S rRNA universal probes and primers were designed from the available alignment data [5]. All the in silico analysis of the primers and probes were carried out using Lasergene, version 8.1 software (DNASTAR, Madison, USA). The specificity of the probes and primers were checked thoroughly on all available sequences in the database using NCBI BLAST programme (http://blast.ncbi.nlm.nih.gov/blast.cgi). All the primers and probes were synthesized from Sigma-Aldrich. The details of the probes and primers used are given in Table 2.
Table 2.
Oligonucleotide probes and primers used for the study
| Probe/primer | Target gene | Organism | 5′–3′ Sequence | Genbank accession no. |
|---|---|---|---|---|
| 16S1Fa | 16S rRNA | Most eubacteria | AGAGTTTGATCCTGGCTCAG | |
| 16S1Ra | CGTATTACCGCGGCTGCTGGCAC | |||
| 16S2Fa | 16S rRNA | Most eubacteria | ATTGACGGGGGCCCGCACAAG | |
| 16S2Ra | TGACGTCATCCCCACCTTCCT | |||
| BaChFa | dhp61.183 | B. anthracis | TGCAATCGATGAGCTAATGAACAAT | NC003997 |
| BaChRa | CAGGATGGGTCTCGATTTTGTG | |||
| BaPaFa | pag | B. anthracis | TTCGAAAAGGTTACAGGACGGATT | AF268967 |
| BaPaRa | GCTTTAATTGTCGCGAGTGTTTG | |||
| BaLfFa | Lethal factor | B. anthracis | TCCCGTCTTTATCCCCCTTGT | M29081 |
| BaLfRa | ACGGGTTCATATCCTTCTTTTGCAT | |||
| BaEfFa | Edema Factor | B. anthracis | TCCGTTTGCATCCCGTTTTGTAT | M24074 |
| BaEfRa | TCTCCTTTCAGCACATCAATCCTAT | |||
| BaCapFa | capA | B. anthracis | CAGGAGCTATTGCAACGAAAGAAC | NC003981 |
| BaCapRa | GGTTTTGGTGATCCCTCTTGAATATT | |||
| YpesChFa | YihN | Y. pestis | CCAAGTAATGACGCCAACCAATATC | NC003143 |
| YpesChRa | CTATGCAACAAGACTGGGGGCTAAC | |||
| YpesCafFa | caf1 | Y. pestis | CGTTATCGCCATTGCATTATTTG | AL117211 |
| YpesCafRa | ACGGTTACGGTTACAGCATCAGTG | |||
| YpesPlaFa | pla | Y. pestis | GGCAACCATTATAACTATTCTGTCCG | AL109969 |
| YpesPlaRa | GAACCACCTGTAGCTGTCCAACTG | |||
| YpesLcrFa | lcrV | Y. pestis | ATCGCCGAATACACAATGGGAAT | AL117189 |
| YpesLcrRa | CCCCGGTTCTTTTATTCTCACTTCC | |||
| BcspFa | BCSP31 | Brucella spp. | TGCGGGAAGAGGACTGGTATTATGA | M20404 |
| BcspRa | GGGTAAAGCGTCGCCAGAAGG | |||
| BmelFa | IS117 | Brucella spp. | TGCCGATCACTTAAGGGCCTTCAT | AE008917 |
| BmelRa | BMEI1162 | B. melitensis | TCGGCTCAGAATAATCCACAGAAGG | AE008917 |
| BpsFa | Orf11 | B. pseudomallei | GCCTGCACCGGATTCTCGATA | AF074878 |
| BpsRa | ACGTGCATCGGTTTCCAGTGT | |||
| 16SUniv1b | 16S rRNA | Most eubacteria | GACTCCTACGGGAGGCAGCAG | |
| 16SUniv2b | 16S rRNA | Most eubacteria | TTAAGTCCCGCAACGAGCGCAACCCT | |
| 16SBacb | 16S rRNA | Bacillus sp. | AAGTGCTAGTTGAATAAGCTGGCAC | |
| 16SYerb | 16S rRNA | Y. Sp. | AGCGGGAAGTAGTTTACTACTTTG | |
| 16SBrub | 16S rRNA | Brucella sp. | GTCGCGGTTAGTGGAGACA | |
| 16SBpsb | 16S rRNA | Burkholderia sp. | ATCATTCTGGCTAATACCCGGAGT | |
| BaChb | dhp61.183 | B. anthracis | CCCACAGCGGCTAAAGAGACCAGTA | |
| BaPAb | pag | B. anthracis | ACCCCCTTGTGGCAGCTTATCCG | |
| BaLFb | Lethal factor | B. anthracis | AGGGGAGGAAGCTGTTAAAAAAGAGGCA | |
| BaEFb | Edema factor | B. anthracis | AATTTCAGCATGCGTTTTCTTTAGCG | |
| BaCapAb | capA | B. anthracis | ACGCACTGGGGGGAAGAATACGA | |
| YpChb | YihN | Y. pestis | GGCCCACACCGATCAACCCTACG | |
| YpCaf1b | caf1 | Y. pestis | CGGGCAGCCAGGATTTCTTTGT | |
| YpPlab | pla | Y. pestis | AGGGGGTGGACGTCTCTGGCTTC | |
| YpLcrb | lcrV | Y. pestis | TCAGGTGGATGGGAGCGAGAAAAA | |
| BmBCSPb | BCSP31 | Brucella sp. | TTGCACAGGCCCCGACATTTTTCCGTATC | |
| Bmelitensisb | BMEI1162 | B. melitensis | GCAGGGCGAAGATTGGCGGGTAAGCT | |
| BpsOrf11b | Orf11 | B. pseudomallei | CGCCCAGCAGCAACCCCTCCTCCAG |
F forward, R reverse
aPrimers
bProbes
Specificity and Sensitivity Analysis
Single colonies were picked up from brain heart infusion agar plate and resuspended in 100 μl of 10 mM TE buffer (pH 8.0) for DNA extraction. The extracted DNA was tested by PCR with all the primers given in Table 2. The sensitivity analysis was performed based on both DNA dilution and colony forming units of the organisms. For determining DNA detection limit, total genomic DNA concentration was determined and ten fold serial dilution was prepared. The diluted DNA was used directly for the assay. In order to determine the sensitivity, based on colony forming units, cfu was determined from cultures of B. anthracis, Y. pestis, B. melitensis and B. pseudomallei and was spiked into human blood of healthy volunteers at concentration starting from 105 to 101 cfu/ml.
DNA Extraction and Multiplex PCR
DNA was extracted either from 100 μl of spiked blood or colonies of each organism using DNeasy Blood and Tissue kit (Qiagen, GmbH) as per the manufacturer’s instructions. Approximately, 5 μl of each DNA was used as a template for the assay. The remaining PCR reaction mixture consisted of 0.2 μM each of primers, 3 mM MgCl2, 200 μM each of dNTP, 20 mM Tris–HCl (pH 8.3), 20 mM KCl, 50 mM (NH4)2 SO4 and 1 U of True Start Taq polymerase (Fermentas, Lithuana) in a total volume of 25 μl. The mPCR was run with the following conditions: Initial denaturation at 95 °C for 3 min; 40 cycles of 95 °C for 45 s, 60 °C for 90 s and 72 °C for 45 s; final extension for 10 min at 72 °C. From this reaction, 2 μl of the product was run on 1.5 % agarose gel prepared in TBE buffer and the remaining was diluted up to 150 μl with 2 × SSPE/0.1 % SDS for reverse line blot.
Reverse Line Blot Hybridization
Reverse line blot hybridization was carried out as per the standard protocol of Kong and Gilbert [6]. Briefly, 150 μl of diluted 5′ amino modified oligonucleotide probes at a final concentration of 1–20 pmol/μl were bound to EDAC activated Biodyne C membrane (Pall Gelman Laboratory, USA). After removing the excess oligonucleotide probes, the membrane was deactivated by incubating in 100 mM NaOH for 8 min in a rolling bottle. The membrane was then washed once with 250 ml 2 × SSPE/0.1 % SDS for 5 min at 60 °C. PCR products were diluted to 150 μl volume with 2 × SSPE/0.1 % SDS and denatured at 99 °C in a thermal cycler for 10 min. The denatured were loaded on to their respective lanes and allowed to hybridize with the membrane bound probes. Hybridization was carried out at 52 °C while a more stringent washing step at 60 °C removed any nonspecific binding. Signals were obtained with the help of streptavidin–horseradish peroxidase conjugate (Roche Diagnostics GmBH, Germany) and exposing the membrane to X-Ray film (Kodak, USA) for 5–10 min. All the assembly for probe and sample loading including the hybridization steps were carried out using a 45 lane Miniblotter (Hoefer Inc, CA, USA).
Results
Specificity of the Probes and Primers
Fourteen pairs of primers and 18 probes which did not have any significant hits with other targets in the database were chosen for our assay. The specificity of the primers was first checked by PCR individually prior to hybridization. The primer pairs amplified only the corresponding gene from the specific organisms and did not show any non specific amplification other than the intended targets.
Multiplex PCR
The multiplex PCR amplicons consisted of fragments with size ranging from 280 to 540 bp while in non related organisms only the two internal control bands, corresponding to the 16S rRNA gene, were amplified (Fig. 1). Due to only slight difference in the size of the PCR amplicon, some of the bands overlapped and cannot be differentiated well enough on agarose gel.
Fig. 1.
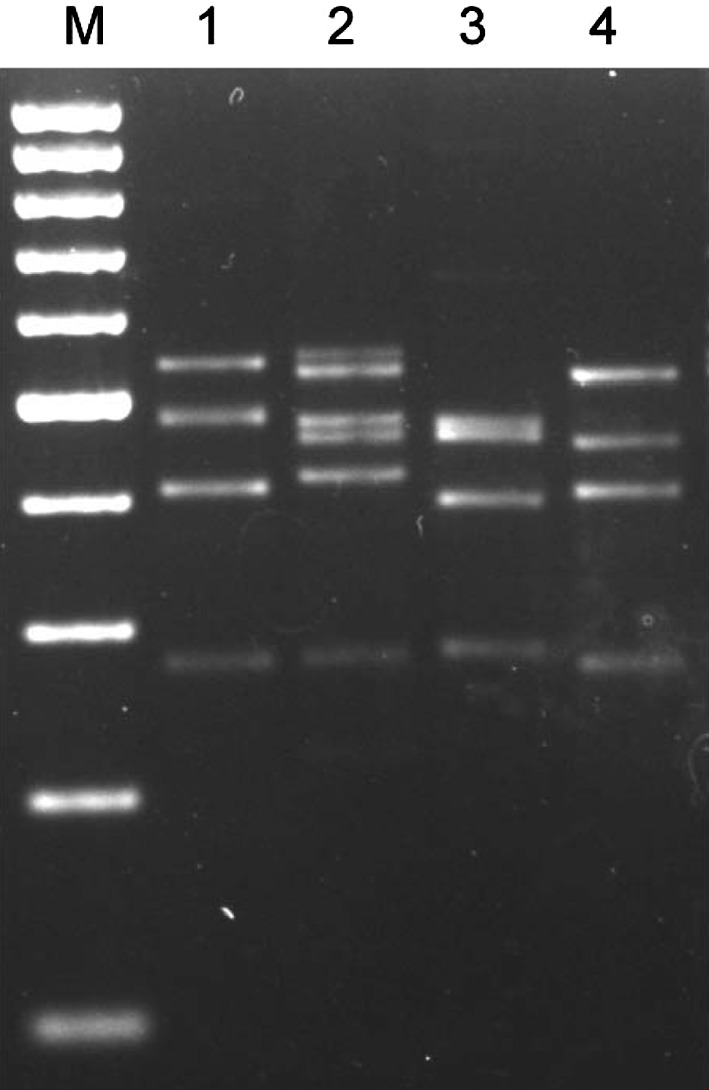
Agarose gel showing multiplex PCR products from the four organisms individually Lane M: 100 bp DNA ladder; Lane 1: Multiplex PCR with B. anthracis DNA; Lane 2: Multiplex PCR with Y. pestis DNA; Lane 3: Multiplex PCR with B. melitensis DNA; Lane 4: Multiplex PCR with B. pseudomallei DNA
Reverse Line Blot Hybridization
The hybridization temperature was kept low at 52 °C for proper hybridization of the amplified DNA to the probes. However, the washing step was carried out at 60 °C in order to eliminate any non specific hybridization. Hybridization assay with purified DNA from bacterial isolates did not show any cross reaction other than their respective target probes. The assay could differentiate the organism distinctively from their closely related species as shown in the Fig. 2. All the other non related but common pathogens which were used for this study did not react with any of the probes except with the 16S rRNA universal control probes.
Fig. 2.

Reverse line blot with representative organisms for each species Probes 16S Univ 1 and 16S Univ 2 are two universal control probes which are suppose to hybridize all Eubacter 16S rRNA. Probe 16S Bac is Bacillus genus specific while BaCh, BaPA, BaEF, BaLF, and BaCapA are B. anthracis specific probes. Sterne strain of B. anthracis does not show positive signal for BaCapA since it is plasmid pXO2 negative. For Y. pestis, 16S Yer is a genus specific probe while YpCh, YpPLA, YpCaf1 and YpLcrV are species specific probes. For B. melitensis, 16S Bru and BmBCSP are genus specific probes while B. melitensis is a species specific probe. For B. pseudomallei, 16S Bps is a genus specific probe while BpsOrf11 is a species specific probe. Salmonella, Leptospira, Shigella and E. coli samples were included as an unrelated but common pathogen negative control
Sensitivity of the Assay
The sensitivity of the assay was assessed both with purified DNA and with bacteria spiked blood samples. When purified DNA from picked colonies was used, the minimum concentration of detection limit ranged from 0.3 pg to 0.3 ng for different targets. With spiked blood, the detection level was 5 × 102 cfu/ml for B. anthracis, 8 × 102 cfu/ml for Yersinia species, 1.4 × 102 cfu/ml for B. melitensis and 4 × 102 cfu/ml for B. pseudomallei.
Discussion
The aim of the current study was to develop a simple, robust and rapid diagnostic platform capable of simultaneous detection and characterization of four BW classified pathogens. A total of 29 isolates of B. anthracis, 22 isolates of Y. pestis, seven isolates of B. melitensis and 13 isolates of B. pseudomallei along with their closely related species were included in the study. Since the assay has been designed with the intention of simultaneous detection of genetically unrelated organism, it was not possible to locate a universal gene where upon a single pair of primers can be used to differentiate the pathogen to the species level. Although, 16S rRNA subunit gene has been used for broad classification of organisms, discrimination up to the species level is not possible as the selected organisms for the present work share a very close genetic make up with other members of their respective genus. Firstly, we have used 16S rRNA probes to classify the pathogens based on genera. Our 16S rRNA probes are meant to hybridize the corresponding gene, at least from the pathogenic species within the genus, if not all. We have designed two pairs of universal primers, 16S1F and 16S1R along with 16S2F and 16S2R to amplify two different regions on 16S rRNA gene of most eubacteria. The organism specific 16S rRNA probes lie within these regions. Since the assay is based on two step method, we consider it essential to include a control hybridization probe to eliminate any false negative cases. Two universal probes, 16S Univ1 and 16S Univ2, were designed from the region encompassed by the amplified fragment and served as an internal control (IC) for the assay. For Brucella, in addition to the 16S rRNA gene, we have included a probe from BCSP 31 gene which is considered genus specific for all Brucella species [7]. These probes and primers proved very specific in identification of organisms to the genus level.
In order to specifically differentiate the pathogen from its closely related species, we have targeted chromosomal gene sequences specific for each pathogen. For B. anthracis, we have chosen a unique genomic locus, dhp61.183 [8] and for Y. pestis, a chromosomal gene target, YihN [9]. B. melitensis specific sequence was designed from BMEI1162 gene [10]. The probe and primer for this gene, targets the unique insertion of IS711 element in the B. melitensis genome. The B. pseudomallei probe has been designed from a gene coding for its type III secretion system [11]. Plasmid based virulence factors have usually been targeted for both immunological and PCR based detection assays. But recent discovery of virulence plasmids cured B. anthracis isolates [12], B. cereus harbouring anthrax virulence plasmids [13] and atypical strains of Y. pestis lacking virulence plasmids [14] has made the inclusion of a specific chromosomal sequence in the assay vital. Detection of virulence factors along with a species specific chromosomal marker will help to pinpoint the exact identity of the pathogen. This is especially important from biodefence perspective where the use of genetically altered organisms cannot be ruled out. In the case of B. melitensis and B. pseudomallei, all the targets chosen in this study are chromosomal sequences. Our probes based on these gene sequences were very specific and clearly differentiated the species of the organism within the genus.
Apart from these chromosomal targets, the virulence factor based probes includes capA (Capsular antigen), pag (Protective antigen), lef (lethal factor) and cya (Edema factor) for B. anthracis. These gene sequences originate from two mega plasmids, the pXO1 and pXO2. The pag, lef and cya genes, located in the pXO1 plasmid, are components of a tripartite anthrax toxin complex and together in binary combination produce the lethal toxin and edema toxin. The capA gene, which codes for poly-d-glutamic acid capsule is located on the plasmid pXO2 and plays a role in the pathogen evasion of host immune response. The absence of either of the two plasmids renders the organism avirulent [15]. We have included capA gene to differentiate the virulent isolates of from avirulent vaccine strains of B. anthracis. The virulence factors of Y. pestis includes caf1 (Fraction 1 antigen), pla (Plasminogen activator) and lcrV (V antigen) which are located on plasmids pMT1, pCP1 and pCD1 respectively. These virulence factors’ probes and primers also did not show any cross hybridization except for Y. pestislcrV probe which reacted lightly with Y. pseudotuberculosis. However, this cross reaction was eliminated by increasing the level of stringency of the assay. This cross reaction is due to the presence of a homologue of the pCD1 plasmid, a type III secretion system encoding plasmid, in Y. enterocolitica and Y. pseudotuberculosis [16]. The plasmids, pMT1 and pPCP1, however, exists only in Y. pestis and the detection of these targets will confirm the identity of the species involved. The reason for the inclusion of the lcrV gene in our assay, in spite of the lcrV gene cross reacting with Y.pseudotuberculosis even in our BLAST analysis (95 %), is to complement the characterization of the virulence factors of Y. pestis in the assay, and not for differentiation of the species. Given the intra-genus similarity in the genetic make up of the pathogens included in this study, none of the probes and primers cross reacted with their closely related organisms except for lcrV gene mentioned above. The assay also did not show cross reactivity with any of the commonly encountered clinical pathogens tested in the study either.
In conclusion, the mPCR/RLB assay demonstrated capable of simultaneous detection and thorough characterization of four selected BW agents. The assay has proven excellent, both in terms of specificity and sensitivity. Although, the present work includes only four BW categorized agents, it can be improved to cover more organisms and targets. It can serve as an important diagnostic platform in BW attack scenario where the threat can include a diversity of organisms and the disease-specific symptoms are not expected to be seen initially. In critical situation where rapid identification of pathogen is required, the assay will have a crucial role in preventing further spread by timely administration of specific therapy and control measures as it can easily be carried out even in small laboratories and health subcentres.
Acknowledgment
The authors would like to thank the Director, Defence Research and Development Establishment, Gwalior for his support and encouragement.
References
- 1.Skottman T, Piiparinen H, Hyytiainen H, Myllys V, Skurnik M, Nikkari S. Simultaneous real-time PCR detection of Bacillus anthracis, Francisella tularensis and Yersinia pestis. Eur J Clin Microbiol. 2007;26:207–211. doi: 10.1007/s10096-007-0262-z. [DOI] [PubMed] [Google Scholar]
- 2.Tomioka K, Peredelchuk M, Zhu X, Arena R, Volokhov D, Selvapandiyan A, Stabler K, Mellquist-Riemenschneider J, Chizhikov V, Kaplan G, Nakhasi H, Duncan R. A multiplex polymerase chain reaction microarray assay to detect bioterror pathogens in blood. J Mol Diagn. 2005;7:486–494. doi: 10.1016/S1525-1578(10)60579-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kong F, Brown M, Sabananthan A, Zeng X, Gilbert GL. Multiplex PCR-based reverse line blot hybridization assay to identify 23 Streptococcus pneumoniae polysaccharide vaccine serotypes. J Clin Microbiol. 2006;44:1887–1891. doi: 10.1128/JCM.44.5.1887-1891.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xiong L, Kong F, Yang Y, Cheng J, Gilbert G. Use of PCR and reverse line blot hybridization macroarray based on 16S–23S rRNA gene internal transcribed spacer sequences for rapid identification of 34 Mycobacterium species. J Clin Microbiol. 2006;44:3544–3550. doi: 10.1128/JCM.00633-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chakravorty S, Helb D, Burday M, Connell N, Alland D. A detailed analysis of 16S ribosomal RNA gene segments for the diagnosis of pathogenic bacteria. J Microbiol Methods. 2007;69:330–339. doi: 10.1016/j.mimet.2007.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kong F, Gilbert GL. Multiplex PCR-based reverse line blot hybridization assay (mPCR/RLB)—a practical epidemiological and diagnostic tool. Nat Protoc. 2006;1:2668–2680. doi: 10.1038/nprot.2006.404. [DOI] [PubMed] [Google Scholar]
- 7.Morata P, Queipo-Ortuno MI, Reguera JM, Garcia-Ordonez MA, Cardenas A, Colmenero JD. Development and evaluation of a PCR-enzyme-linked immunosorbent assay for diagnosis of human brucellosis. J Clin Microbiol. 2003;41:144–148. doi: 10.1128/JCM.41.1.144-148.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Antwerpen MH, Zimmermann P, Bewley K, Franqoulidis D, Meyer H. Real-time PCR system targeting a chromosomal marker specific for Bacillus anthracis. Mol Cell Probes. 2008;22:313–315. doi: 10.1016/j.mcp.2008.06.001. [DOI] [PubMed] [Google Scholar]
- 9.Stewart A, Satterfield B, Cohen M, O’Neill K, Robison R. A quadruplex real-time PCR assay for the detection of Yersinia pestis and its plasmids. J Med Microbiol. 2008;57:324–331. doi: 10.1099/jmm.0.47485-0. [DOI] [PubMed] [Google Scholar]
- 10.Probert WS, Schrader KN, Khuong NY, Bystrom SL, Graves MH. Real-time multiplex PCR assay for detection of Brucella spp., B. abortus and B. melitensis. J Clin Microbiol. 2004;42:1290–1293. doi: 10.1128/JCM.42.3.1290-1293.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thibault FM, Valade E, Vidal DR. Identification and discrimination of Burkholderia pseudomallei, B. mallei and B. thialandensis by real-time PCR targeting type III secretion system genes. J Clin Microbiol. 2004;42:5871–5874. doi: 10.1128/JCM.42.12.5871-5874.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Turnbull PC, Hutson RA, Ward MJ, Jones MN, Quinn CP, Finnie NJ, Duggleby CJ, Kramer JM, Melling J. Bacillus anthracis but not always anthrax. J Appl Bacteriol. 1992;72:21–28. doi: 10.1111/j.1365-2672.1992.tb04876.x. [DOI] [PubMed] [Google Scholar]
- 13.Hoffmaster AR, et al. Identification of anthrax toxin genes in a Bacillus cereus associated with an illness resembling inhalation anthrax. Proc Natl Acad Sci. 2004;101:8449–8454. doi: 10.1073/pnas.0402414101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Garcia E, Worsham P, Bearden S, Malfatti S, Lang D, Larimer F, Lindler L, Chain P. Pestoides F, an atypical Yersinia pestis strain from the former Soviet Union. Adv Exp Med Biol. 2007;603:17–22. doi: 10.1007/978-0-387-72124-8_2. [DOI] [PubMed] [Google Scholar]
- 15.Green BD, Battisti L, Koehler TM, Thorne CB, Ivins BE. Demonstration of a capsule plasmid in Bacillus anthracis. Infect Immun. 1985;49:291–297. doi: 10.1128/iai.49.2.291-297.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hu P, Elliot J, McCready P, Skowronski E, Garnes J, Kobayashi A, Brubaker RR, Garcia E. Structural organization of virulence-associated plasmids of Yersinia pestis. J Bacteriol. 1998;180:5192–5202. doi: 10.1128/jb.180.19.5192-5202.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]