

Abstract
The NHBoc group affords ortho selective C–H borylations in arenes and alkenes. Experimental and computational studies support an outer sphere mechanism where the N–H proton hydrogen bonds to a boryl ligand oxygen. The regioselectivities are unique and complement those of directed ortho metalations.
Over the past decade, advances in the transition metal-catalyzed functionalization of C–H bonds have transformed synthetic chemistry.1 In this context, the borylation of C-H bonds has shown promise because it bestows the C–H functional group with the synthetic versatility for which B–C bonds are renowned. A central challenge in these reactions is controlling their selectivity. Steric effects often dominate the regioselectivity of C-H borylations of aromatics.2 This makes C-H borylations complementary to widely applied directed ortho metalations (DoMs),3 but the intrinsic functional group and practical limitations of DoMs have intensified efforts to develop selective ortho C–H borylations.
Ortho C–H borylation has been accomplished by catalyst or substrate modification (Scheme 1).4 Catalyst control can be achieved with ligands and/or metals that make 14-electron intermediates accessible.4b,4c,4e,4f Substrates that contain a directed metalation group (DMG) likely coordinate to the metal to form a 16-electron intermediate, 1, which has a vacant site to facilitate the cleavage of an ortho C–H bond. Usually, chelation-directed selectivity is not observed for 16-electron catalytic intermediates, such as Ir(Bpin)3(dtbpy) (2, dtbpy = 4,4′-di-tert-butyl-2,2′-bipyridine, pin = pinacolate). For these catalysts ortho borylation can be accomplished by a relay-directed mechanism in which the substrate can reversibly attach to the metal by a σ-bond metathesis process.4a,4d To date, the substrates in relay-directed borylation have all contained pendant Si–H bonds, and the reactions presumably proceed through a 16-electron intermediate 3 in which the ortho C–H bond is poised for borylation. Both the chelation-directed and relay-directed mechanisms are inner sphere processes, where direction is achieved by coordination to iridium.
Scheme 1.
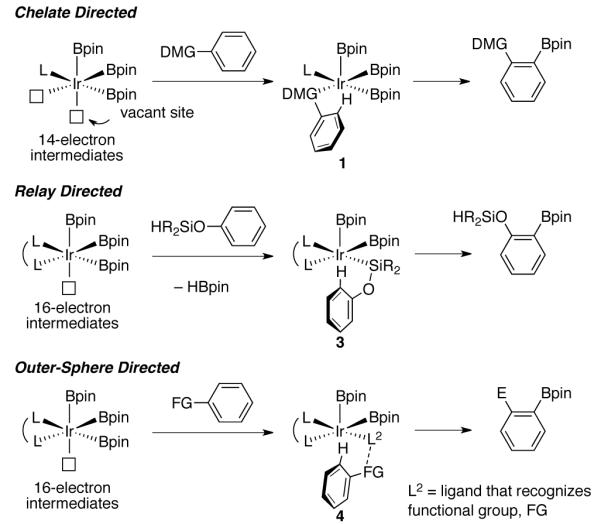
Strategies for ortho-Directed C–H Borylation
In outer-sphere direction,5 a ligand on the catalyst recognizes functionality in the substrate. This distinct paradigm for selectivity, based on ideas from molecular recognition, can provide selectivities that complement those from other directing mechanisms.6 Our efforts to understand the electronic effects in C–H borylation suggested that an outer-sphere mechanism can direct borylation.7 Herein we outline a proof-of-concept where NHBoc groups direct C–H functionalizations with selectivities that are unprecedented in the C–H borylation and DoM literature.
The potential for outer-sphere direction in Ir-catalyzed C-H borylations was suggested by an anomaly predicted for pyrrole in a recent combined computational and experimental study.7b An analysis of 21 combinations of substrates and metal complexes supported the importance of proton-transfer character in the C–H activation transition state. There was a strong linear correlation between the ΔG° and ΔG‡ for C-H activation and the NPA charge on the aryl group after activation, as reflected by the subset of five-membered heterocyclic intermediates (5) shown in Figure 1.8 This charge/reactivity relationship was remarkably predictive, but a large deviation was observed for pyrrole. Borylation at the 2-position of pyrrole is 2.3 kcal/mol more favorable than would be expected from a least-squares fit of other substrate/catalyst combinations.
Figure 1.
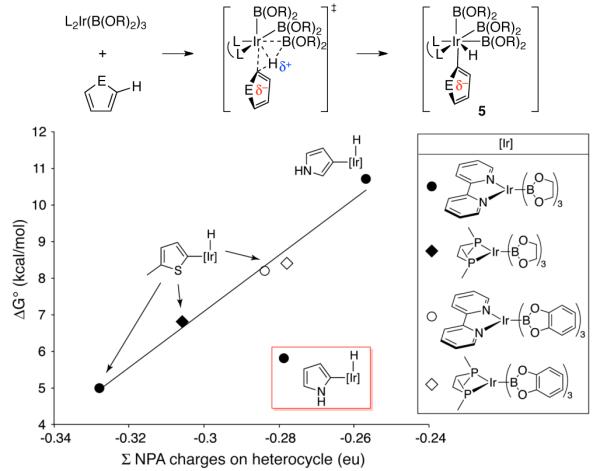
Calculated energies of intermediates (5) vs. the total Normal Population Analysis (NPA) charge on the C–H activated heterocycles in 5. Computational data are excerpted from Ref 7b.
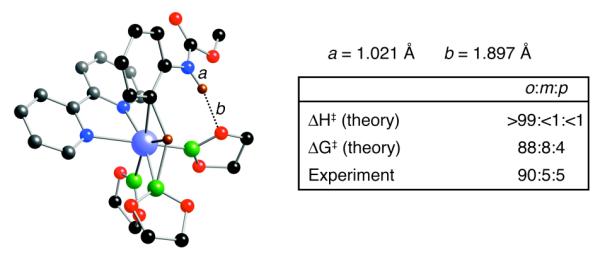
Geometries for the transition state and the resulting intermediate for pyrrole are shown in Figure 2. Two features are noteworthy. First, the NH⋯O distances between the pyrrole and one of the boryl oxygens are short, ranging from 2.05 to 2.19 Å. Second, the ∠Ir–C–N are ~ 10 ° less than the ∠Ir–C–C. Both of these indicate significant NHO hydrogen interaction in the transition state and the intermediate. This clearly accounts for the lowering of ΔG in Figure 1.
Figure 2.
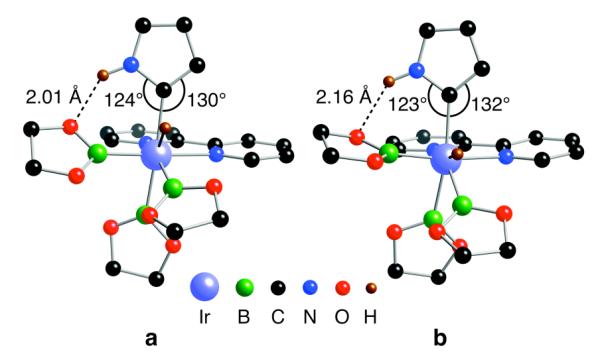
Lowest-energy M06/SDD(Ir)/6-31+G**(C H O N B) transition state (a) and intermediate (b) for C–H activation of pyrrole at the 2-position by model complex Ir(bipy)(Beg)3 (eg = ethyleneglycolate). The NH–O distances and distortions for Ir–C–C and Ir–C–N angles indicate N–H–O hydrogen bonding.
While hydrogen bonding likely accelerates C–H borylations of pyrrole, this interaction only reinforces regioselectivity that is already preferred. Because outer-sphere hydrogen bonding interactions have had significant impact in other catalytic systems,9,6b,6c we expected that similar interactions could be harnessed in C–H borylations and that the resulting regioselectivities could be complementary.
Because preliminary studies of primary anilines were hampered by poor conversion, tert-butoxycarbonyl (Boc) protected anilines were examined. N-Boc protected compounds are viable substrates for C–H borylation.10 This is illustrated in Scheme 2, where borylation of 6 gives meta functionalization typical for 1,3-disubstituted benzenes. Since one Boc group sufficiently protects primary amines,10b substrate 7 was examined next. Remarkably, replacing one Boc group with H alters the regioselectivity to favor ortho borylation product 8b.11
Scheme 2.
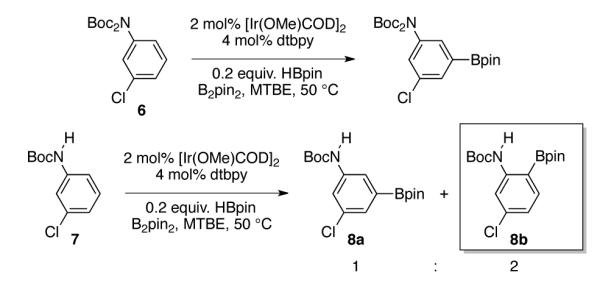
Boc Substitution and Borylation Regioselectivity
The shift in selectivity for 7 could arise from a number of mechanisms, three of which are depicted in Scheme 3. In addition to hydrogen bonding transition state 9, selectivity could arise from coordination of the carbamate O to a boryl B in transition state 10. Alternatively, inner-sphere N–H/Ir–B s-bond metathesis could account for ortho selectivity via transition state 11.
Scheme 3.

Experiments to Probe Mechanism
Substrates in 12a-c were chosen to probe for intermediates 9-11. For substrate 12a transition states 9 and 11 are impossible since H has been replaced by CH3. The only product detected is meta isomer 13a, consistent with pathways via 9 or 11. For 12b, the NH and O groups of 7 are transposed. This would affect selectivity via 9 or 11, because the ring sizes in the transition states increase. Conversely, the effects on transition state 10 should be slight. Exclusive formation of meta product 13b makes participation of 10 unlikely. The meta selectivity for amide 12c eliminates 10 because its carbonyl O is more basic than those in carbamates 12a-b,12 but it calls 9 to question because the hydrogen bonding mechanism of 9 seems equally plausible for 12c.
Transition states 9 and 11 can be distinguished by isotopic labeling. As indicated in Scheme 3, a pathway involving 11 requires N–D scission. Consequently, C–H borylation and product elimination would produce significant quantities of 8b-d0. When 7-d1 is subjected to borylation, greater than 95% of product is N–deuterated.
The experimental data in Scheme 3 excludes 10 and 11 but it provides no support for 9. If the hydrogen-bonding mechanism is correct, then it might be expected that the acidity of the N-H bond would affect the selectivity. However, attempts to improve selectivity with more acidic N–H bonds were unsuccessful. For example, N-aryl triflamides, gave N–borylation. Since solutions containing both HBpin and N-aryl triflamides are stable, an Ir complex catalyzes N–borylation. In a plausible mechanism, σ-bond metathesis of the Ir-B bond with the highly acidic triflimide N-H bond generates intermediate 14 (Scheme 3) and subsequent N–B elimination yields the N-borylated triflamide
An experimental result supporting the hydrogen-bonding mechanism of 9 was the observation that increased basicity of the dipyridyl ligands enhances ortho selectivity (Figure 3). We propose that the pinacolate oxygens in complexes with more electron rich dipyridyl ligands are more basic, and this accounts for the increased ortho selectivity. While more electron rich dipyridyl ligands have been shown to increase borylation rates,13 this is the first case where dipyridyl electronic effects significantly alter regioselectivities.
Figure 3.
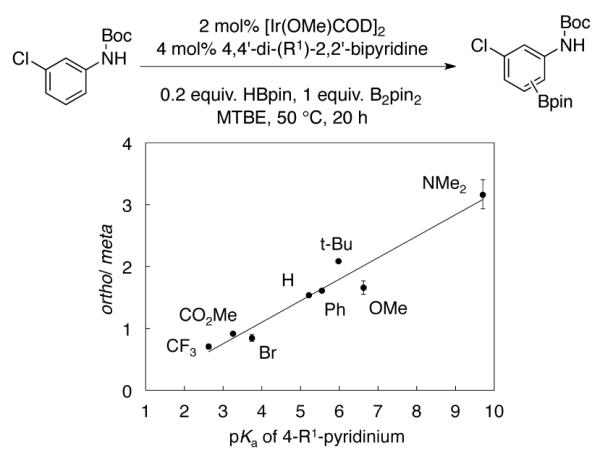
Plot of the ortho/meta product ratios using 4,4′-di(R1)-2,2′-dipyridyl catalysts vs. pKas of 4-R1-pyridium ions.
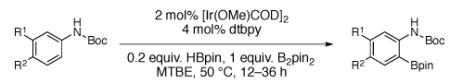
The mechanism of the ortho direction and an explanation of its failure with 12c was clarified by theoretical calculations. In M06/SDD(Ir)/6-31+G**(C H O N B) calculations, a series of transition states were located for C-H activation of PhNHCO2Me by the model complex (bpy)Ir(Beg)3 (15, eg = ethyleneglycolate). These calculations predict the ortho borylation to be strongly favored, and the transition state (Fig. 4) shows a clear NH-O hydrogen bond. The predicted o:m:p ratio (based on the lowest-energy transition states for each possibility) is 88:8:4, which is very close to the experimental value for borylation of PhNHCO2Me by 2 (o:m:p = 90:5:5 from GC-FID). The experimental ratio is a direct measurement of relative rates for directed and non-directed borylation. The agreement between theory and experiment strongly supports the NH–O hydrogen-bonding mechanism. The calculated ortho transition state also suggest a steric explanation for the failure of hydrogen-bonding direction in 12c; when the OCH3 of the carbamate is replaced by an ethyl group and the Beg groups are replaced by BPin groups, there is a tight steric interaction (with H-H distances less than 1.0 Å) that would preclude the ortho structure. An interesting observation is that the ortho transition structure is very strongly favored enthalpically, but the restricted motion associated with the hydrogen bond is disfavored entropically, so the final selectivity is limited by entropy-enthalpy compensation.
Figure 4.
Lowest energy transition state for C–H activation of PhNHCO2Me by model complex 15. The o:m:p ratios predicted by theory and those found for borylation catalyzed by 2 are given.
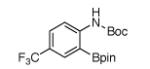
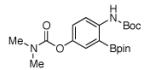
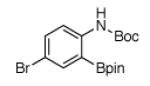
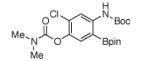
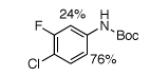
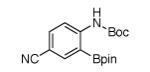
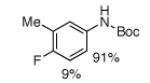
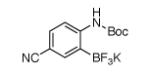
For Boc-protected anilines with a single meta substituent, there is a tradeoff between the hydrogen bond direction and the usual preference for reaction at the least hindered position (Table 1, entry 1). This is not an issue for 4-substituted substrates where the ortho selectivity is high. Except for the fluorine-substituted substrates in entries 5 and 6, these products were isolated as single regioisomers. For entries 11 and 12, 3 equivalents of arene were used to minimize diborylation. Converting Bpin products to their BF3K salts can in cases ease isolation, leading to the higher yield in entry 12 versus 11. All regiochemical assignments were based on NMR spectroscopy and confirmed by X-ray crystallography for entries 4 and 7-9.
Table 1.
ortho-Borylation of N-(Boc)-Anilinesa
| entry | product | yield | entry | product | yield |
|---|---|---|---|---|---|
| 1 |
|
92%b | 7 |
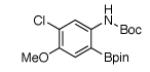
|
95% |
| 2 |
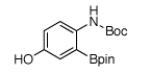
|
78% | 8 |
|
82% |
| 3 |
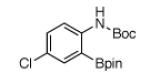
|
79% | 9 |
|
44% |
| 4 |
|
89% | 10 |
|
62% |
| 5 |
|
68% c | 11 |
|
40% |
| 6 |
|
70% d | 12 |
|
51% |
See supporting information for details on conditions. Yields refer to isolated material except as noted
Yield determined by 1H NMR
Yield is for the major isomer
Isolated as a 92:8 mixture.
Entry 2 in Table 1 shows that substrates with more acidic protons are viable through in situ protection as borates by excess pinacolborane. Entries 1, 3-7, and 10 produce boronates with halogen groups that can be further manipulated. Entries 11 and 12 are noteworthy because the selectivity diverges from that of the acetamide analog, which borylates ortho to CN exclusively.14
The comparison with DoM in Scheme 4 highlights the new selectivity offered by hydrogen-bond direction. When there is a distinguishable choice among ortho C-H bonds, DoM is dominated by acidity. Thus, for substrates 16 and 17 (entries 5 and 7 in Table 1), the more acidic C–H bonds flanked by NHBoc and halogen will metalate first.15 In contrast the Ir-catalyzed process is selective for the less hindered C–H bond ortho to NHBoc. For substrates 18 and 19, the O-aryl carbamate is a stronger director for DoM than NHBoc, but it provides no hydrogen bond. Consequently, the preferred site for C–H borylation is the least reactive position for DoM. These examples clearly show that C–H borylation via an outer-sphere hydrogen-bonding mechanism gives regioselectivities that are unprecedented for DoM. It is noteworthy that C–H borylations of substrates in Scheme 4 do not require low temperatures that must be maintained during DoM to minimize generation of benzyne intermediates.15,16
Scheme 4.
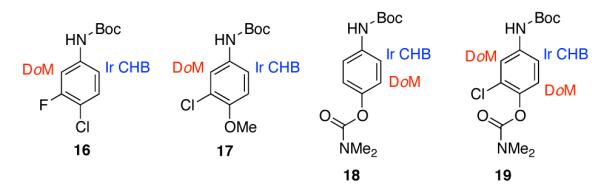
C–H borylation and DoM regioselectivities.
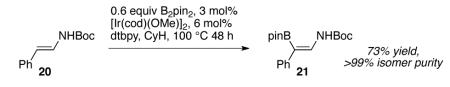
Finally, this chemistry can be extended to Boc-protected enamines such as 20 where the regioselectivity for the vinyl C–H that is beta to N exceeds 99:1. Remarkably, the Ph C–H bonds and the stereochemistry of the double bond in product 21 are unperturbed. Recent reports reflect interest in borylenamines and related compounds,17 including a route using C-H borylation,17c but the reaction here provides a cis disposition of N and B groups that is unavailable by other methods.
In summary, we have shown that the NHBoc group offers ortho selectivity in Ir-catalyzed C–H borylation that cannot be obtained with DoM, or any other methodology. Experiment and theory make a convincing case for an outer-sphere mechanism were the NHBoc proton bonds to Bpin oxygen in the transition state. We currently are applying this chemistry in synthesis and are pursuing more general strategies for outersphere directed C–H borylations.
Supplementary Material
ACKNOWLEDGMENT
We thank the NIH (GM63188) for generous financial support and BASF for gifts of HBpin and B2pin2. We thank Mr. Peter Heisler for experimental assistance, Dr. Daniel Holmes for help with NMR characterization, and Dr. Richard Staples for solving crystal structures.
Footnotes
Supporting Information Available: Full characterization, copies of all spectral data, experimental procedures and X-ray crystallographic data. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- (1) (a).Diaz-Requejo MM, Perez PJ. Chem. Rev. 2008;108:3379. doi: 10.1021/cr078364y. [DOI] [PubMed] [Google Scholar]; (b) Lyons TW, Sanford MS. Chem. Rev. 2010;110:1147. doi: 10.1021/cr900184e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Mkhalid IAI, Barnard JH, Marder TB, Murphy JM, Hartwig JF. Chem. Rev. 2010;110:890. doi: 10.1021/cr900206p. [DOI] [PubMed] [Google Scholar]
- (3) (a).Whisler MC, MacNeil S, Snieckus V, Beak P. Angew. Chem., Int. Ed. 2004;43:2206. doi: 10.1002/anie.200300590. [DOI] [PubMed] [Google Scholar]; (b) Hurst TE, Macklin TK, Becker M, Hartmann E, Kügel W, Parisienne-La Salle JC, Batsanov AS, Marder TB, Snieckus V. Chem. Eur. J. 2010;16:8155. doi: 10.1002/chem.201000401. [DOI] [PubMed] [Google Scholar]
- (4) (a).Boebel TA, Hartwig JF. J. Am. Chem. Soc. 2008;130:7534. doi: 10.1021/ja8015878. [DOI] [PubMed] [Google Scholar]; (b) Kawamorita S, Ohmiya H, Hara K, Fukuoka A, Sawamura M. J. Am. Chem. Soc. 2009;131:5058. doi: 10.1021/ja9008419. [DOI] [PubMed] [Google Scholar]; (c) Ishiyama T, Isou H, Kikuchi T, Miyaura N. Chem. Commun. 2010;46:159. doi: 10.1039/b910298a. [DOI] [PubMed] [Google Scholar]; (d) Robbins DW, Boebel TA, Hartwig JF. J. Am. Chem. Soc. 2010;132:4068. doi: 10.1021/ja1006405. [DOI] [PubMed] [Google Scholar]; (e) Ros A, Estepa B, Lopez-Rodriguez R, Alvarez E, Fernandez R, Lassaletta JM. Angew. Chem., Int. Ed. 2011;50:11724. doi: 10.1002/anie.201104544. [DOI] [PubMed] [Google Scholar]; (f) Dai H-X, Yu J-Q. J. Am. Chem. Soc. 2012;134:134. doi: 10.1021/ja2097095. [DOI] [PubMed] [Google Scholar]; (g) Ros A, Lopez-Rodriguez R, Estepa B, Alvarez E, Fernandez R, Lassaletta JM. J. Am. Chem. Soc. 2012;134:4573. doi: 10.1021/ja300308c. [DOI] [PubMed] [Google Scholar]; (h) Xiao B, Li Y-M, Liu Z-J, Yang H-Y, Fu Y. Chem. Commun. 2012;48:4854. doi: 10.1039/c2cc31737k. [DOI] [PubMed] [Google Scholar]
- (5).Although “outer-sphere” commonly refers to eletron transfer, it has been used to describe mechanisms for hydrogen transfer and nucleophilic addition to π-allyl Pd compounds: Samec JSM, Backvall JE, Andersson PG, Brandt P. Chem. Soc. Rev. 2006;35:237. doi: 10.1039/b515269k. Trost BM, Xu JY, Schmidt TJ. Am. Chem. Soc. 2009;131:18343. doi: 10.1021/ja9053948.
- (6) (a).Das S, Incarvito CD, Crabtree RH, Brudvig GW. Science. 2006;312:1941. doi: 10.1126/science.1127899. [DOI] [PubMed] [Google Scholar]; (b) Fackler P, Berthold C, Voss F, Bach TJ. Am. Chem. Soc. 2010;132:15911. doi: 10.1021/ja107601k. [DOI] [PubMed] [Google Scholar]; (c) Vetticatt MJ, Desai AA, Wulff WD. J. Am. Chem. Soc. 2010;132:13104. doi: 10.1021/ja103863j. [DOI] [PubMed] [Google Scholar]
- (7) (a).Chotana GA, Vanchura BA, II, Tse MK, Staples RJ, Maleczka RE, Jr., Smith MR., III Chem. Commun. 2009:5731. doi: 10.1039/b914736e. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Vanchura BA, II, Preshlock SM, Roosen PC, Kallepalli VA, Staples RJ, Maleczka RE, Jr., Singleton DA, Smith MR., III Chem. Commun. 2010;46:7724. doi: 10.1039/c0cc02041a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).The originally proposed IrIII/V mechanism8a has been supported both computationally8b and exprimentally8c:Cho JY, Tse MK, Holmes D, Maleczka RE, Jr., Smith MR., III Science. 2002;295:305. doi: 10.1126/science.1067074. Tamura H, Yamazaki H, Sato H, Sakaki S. J. Am. Chem. Soc. 2003;125:16114. doi: 10.1021/ja0302937. Boller TM, Murphy JM, Hapke M, Ishiyama T, Miyaura N, Hartwig JF. J. Am. Chem. Soc. 2005;127:14263. doi: 10.1021/ja053433g.
- (9) (a).Cook GR, Yu H, Sankaranarayanan S, Shanker PS. J. Am. Chem. Soc. 2003;125:5115. doi: 10.1021/ja028426w. [DOI] [PubMed] [Google Scholar]; (b) Hoveyda AH, Lombardi PJ, O’Brien RV, Zhugralin AR. J. Am. Chem. Soc. 2009;131:8378. doi: 10.1021/ja9030903. [DOI] [PubMed] [Google Scholar]
- (10) (a).Beck EM, Hatley R, Gaunt MJ. Angew. Chem., Int. Ed. 2008;47:3004. doi: 10.1002/anie.200705005. [DOI] [PubMed] [Google Scholar]; (b) Kallepalli VA, Shi F, Paul S, Onyeozili EN, Maleczka RE, Jr., Smith MR., III J. Org. Chem. 2009;74:9199. doi: 10.1021/jo901822b. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Tomita D, Yamatsugu K, Kanai M, Shibasaki M. J. Am. Chem. Soc. 2009;131:6946. doi: 10.1021/ja901995a. [DOI] [PubMed] [Google Scholar]
- (11).In Ref. 3b the following observation was noted: “For reasons currently unappreciated, N-tert-butoxycarbonyl 3-methoxyaniline led to mixtures of unidentified products.”
- (12).Lequestel JY, Laurence C, Lachkar A, Helbert M, Berthelot MJ. Chem. Soc.-Perkin Trans. 1992;2:2091. [Google Scholar]
- (13).Ishiyama T, Takagi J, Hartwig JF, Miyaura N. Angew. Chem., Int. Ed. 2002;41:3056. doi: 10.1002/1521-3773(20020816)41:16<3056::AID-ANIE3056>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- (14).Chotana GA, Rak MA, Smith MR., III J. Am. Chem. Soc. 2005;127:10539. doi: 10.1021/ja0428309. [DOI] [PubMed] [Google Scholar]
- (15).Mayes BA, Chaudhuri NC, Hencken CP, Jeannot F, Latham GM, Mathieu S, McGarry FP, Stewart AJ, Wang JY, Moussa A. Org. Process Res. Dev. 2010;14:1248. [Google Scholar]
- (16) (a).Clark RD, Caroon JM. J. Org. Chem. 1982;47:2804. [Google Scholar]; (b) Reavill DR, Richardson SK. Synth. Commun. 1990;20:1422. [Google Scholar]; (c) Yoshida Y, Hamada Y, Umezu K, Tabuchi F. Org. Process Res. Dev. 2004;8:958. [Google Scholar]
- (17) (a).He Z, Yudin AK. J. Am. Chem. Soc. 2011;133:13770. doi: 10.1021/ja205910d. [DOI] [PubMed] [Google Scholar]; (b) Hussain N, Hussain MM, Ziauddin M, Triyawatanyu P, Walsh P. J. Org. Lett. 2011;13:6464. doi: 10.1021/ol202766g. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Takaya J, Kirai N, Iwasawa N. J. Am. Chem. Soc. 2011;133:12980. doi: 10.1021/ja205186k. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.