

Abstract
Jaspamide (jasplakinolide; NSC-613009) is a cyclodepsipeptide that has antitumor activity. A narrow margin of safety was observed between doses required for efficacy in mouse tumor models and doses that caused severe acute toxicity in rats and dogs. We explored the hypothesis that the observed toxicity was due to cardiotoxicity. Jaspamide was tested in a patch clamp assay to determine its effect on selected cardiac ion channels. Jaspamide (10 μM) inhibited Kv1.5 activity by 98.5%. Jaspamide also inhibited other channels including Cav1.2, Cav3.2, and HCN2; however, the Kv11.1 (hERG) channel was minimally affected. Using spontaneously contracting human cardiomyocytes derived from induced pluripotent stem cells, effects on cardiomyocyte contraction and viability were also examined. Jaspamide (30 nM to 30 μM) decreased cardiomyocyte cell indices and beat amplitude, putative measurements of cell viability and cardiac contractility, respectively. Concentration-dependent increases in rhythmic beating rate were noted at ≤6 h of treatment, followed by dose-dependent decreases after 6 and 72 hours exposure. The toxic effects of jaspamide were compared with that of the known cardiotoxicant mitoxantrone, and confirmed by multiparameter fluorescence imaging analysis. These results support the hypothesis that the toxicity observed in rats and dogs is due to toxic effects of jaspamide on cardiomyocytes.
Keywords: stem cell derived cardiomyocytes, jaspamide, cyclodepsipeptide, in vitro alternatives, predictive toxicology, cardiotoxicity
1. Introduction
Jaspamide (jasplakinolide, NSC-613009), a cyclodepsipeptide isolated from the marine sponge Jaspis johnstoni (Crews et al., 1986) has been extensively investigated as a potential cancer therapeutic agent. Jaspamide exhibits antitumor activity in multiple in vitro tumor models for prostate and breast carcinomas and acute myeloid leukemia (Takeuchi et al., 1998; Bubley et al., 1996; Stingl et al., 1992; Fabian et al., 1995). Jaspamide inhibits the growth of prostate carcinoma PC-3 cells by disrupting the actin cytoskeleton (Senderowicz et al., 1995) and acts as a radiosensitizer against prostate and lung carcinoma cells in vitro (Takeuchi et al., 1998).
In vivo, a 7-day continuous subcutaneous infusion of 31.5 mg/m2 jaspamide resulted in a 5-day tumor growth delay in mice bearing Lewis lung carcinoma xenografts (Takeuchi et al., 1998). However, in Fischer 344 rats given intravenous (iv) injections (0.8–4.0 mg/m2/dose) every eight hours for three doses, a total dose of 4.5 mg/m2/dose was lethal (Schindler-Horvat et al., 1998). A slightly lower dose was minimally toxic with signs of toxicity limited to hunched posture. Pulmonary edema and cardiac hemorrhage and congestion were present in rats that received lethal doses; however, jaspamide-related microscopic lesions were not noted in rats that survived to Day 15. In beagle dogs, a dose of 0.026 mg/kg/h (12.5 mg/m2) given as a 24-hour continuous iv infusion was not lethal, whereas 0.030 mg/kg/h (14.4 mg/m2) given on the same route and schedule was a lethal dose. Pulmonary edema and cardiac hemorrhage and congestion were present in dogs that received lethal doses. Dogs that survived to the end of the study were not euthanized, therefore histopathology data are not available for sub-lethal doses (Schindler-Horvat et al., 1998). Based on the narrow therapeutic index observed in these studies, jaspamide was dropped from consideration for further development as an anticancer agent at the National Cancer Institute.
Given the observation of cardiotoxicity with jaspamide, mechanistic studies were undertaken to determine the effect of jaspamide on cardiac ion channel function and on the viability and contractile function in human induced pluripotent stem cell-derived cardiomyocytes (iPSC-CMs). Human iPSC-CMs have the complement of ionic currents and channel gating properties required for synchronized cardiomyocyte contractions (Ma et al., 2011). Action potentials from these cells have atrial-, nodal-, and ventricular-like properties, indicative of a heterogeneous cell population. Furthermore, the cells maintain synchronized contraction in a 96-well dish for more than 7 days, allowing high-throughput investigations of the putative cardiotoxic compounds in a functional human cardiomyocyte system (Ma et al., 2011; Guo et al., 2011). Herein, we report the results of mechanistic in vitro investigations of jaspamide on cardiomyocyte function and propose a mechanism of jaspamide induced cardiotoxicity.
2. Materials and Methods
2.1 Compounds
Jaspamide (jasplakinolide, NSC-613009) was extracted from a sponge sample provided by the Coral Reef Research Foundation under a National Cancer Institute collection contract. Jaspamide was isolated and purified by the Natural Products Extraction Laboratory (SAIC-Frederick), and supplied as a solid powder in amber glass capsules. The capsules were capped with nitrogen and stored protected from light at −20°C.
2.2 Patch clamp assay
The in vitro effects of jaspamide on cardiac ion channel activity were evaluated (Chantest Inc., Cleveland, OH) using an automated patch clamp system (PatchXpress 7000A, Molecular Devices, Sunnyvale, CA). Single mammalian cells (CHO or HEK293), each expressing one of 12 cardiac ion channels (calcium, potassium, or sodium, Table 1), were exposed to 10 μM jaspamide for 5 minutes at room temperature. Experiments were conducted in duplicate for each ion channel. The inhibition by jaspamide of the current flow through each ion channel was calculated as the mean percent inhibition ± the standard deviation. The sensitivity of each channel was confirmed using a positive control specific for each ion channel.
Table 1.
Conditions for Cardiac Ion Channels Tested in the Patch Clamp Assay
| Channel | Function | Expressed in Cell Type | Positive Control |
|---|---|---|---|
| Cav1.2 | ICa, L, high threshold calcium current | CHO | Nifedipine |
| Cav3.2 | ICa, T, low threshold calcium current | HEK293 | Nickel |
| HCN2 | If, hyperpolarization-activated potassium current | CHO | Zatebradine |
| HCN4 | If, hyperpolarization-activated potassium current | HEK293 | Zatebradine |
| hERG | IKr | HEK293 | E-4031 |
| Kir2.1 | IK1, inwardly rectifying potassium current | HEK293 | Barium |
| Kir3.1/3.4 | IACh, inwardly rectifying potassium current | HEK293 | Barium |
| Kir6.2/SUR2A | ATP-sensitive current, IKATP | HEK293 | Glybenclamide |
| Kv1.5 | IKur, ultra-rapid delayed rectifier potassium current | CHO | 4-AP |
| Kv4.3 | Ito, transient outward potassium current | HEK293 | Flecainide |
| KvLQT/mink | IKs, slow delayed rectifier potassium current | CHO | Chromanol 293B |
2.3 Cardiomyocyte cell culture
Cryopreserved human induced pluripotent stem cell-derived cardiomyocytes (iCell Cardiomyocytes®, Cellular Dynamics International, Madison, WI) were used in these studies. These cardiomyocytes exhibit the biochemical and electrophysiological characteristics of normal human heart cells, and form electrically connected layers that beat in synchrony (Anson et al., 2011). The cells were plated on a 0.1% gelatin-coated 6-well plate (2×106 cells/well) with plating medium (iCell Cardiomyocytes Plating Medium (iCPM); Cellular Dynamics International) and cultured at 37°C with iCell Cardiomyocytes Maintenance Medium (iCMM), Cellular Dynamics International).
Cells were replated 1–3 weeks later to a tissue culture E-plate (ACEA Biosciences, San Diego, CA) at a concentration of approximately 5.0×104 cells/well. Plates were coated with 0.1% gelatin for approximately 3 h at 37°C before cell plating. Cells were maintained in iCMM, which was changed every 2–3 days.
2.4 Assessment of cardiomyocyte viability
Jaspamide was dissolved in iCMM at a concentration of 60 μM. Half of the volume of maintenance medium in each well was replaced with the vehicle or jaspamide (final concentrations of 30, 100, 300 nM; and 1, 10, 30 μM). Each concentration was tested initially in triplicate for 72 h in one E-plate, and repeated in duplicate in an additional two E-plates seeded with cells from two different lots. The following positive controls were also tested: mitoxantrane (structural cardiotoxicant), E-4031 (hERG blocker), TTX (Na+ channel blocker), nifedipine (L-type Ca2+ channel blocker) and S9947 (Kv1.5 blocker). Impedance technology (xCELLigence RTCA Cardio-96 System, ACEA/Roche) was used to monitor in real time the viability of cells treated with jaspamide or vehicle. An IC50 was calculated as cell index in treated wells vs. cell index in untreated (vehicle control) wells at selected time-points during 72 h exposure. The cell index is an impedance measurement whose increase reflects increases in the number of cells attached to the electrode of the well. The cell index is zero when there are no cells present or cells do not adhere to the electrodes. In addition, changes in cell morphology, cell adhesion, cell-cell interaction and/or cell viability can be reflected by a resultant change in cell index (Xi et al., 2008). The cell viability in each well of the E-plate was also assessed after 72 h exposure to jaspamide by ATP quantification using a CellTiter-Glo® Luminescent kit (Promega, Madison, WI).
The cardiotoxicity of jaspamide and mitoxantrone was further examined using a Cellomics® ArrayScan VTI HCS Reader with the Multiparameter Cytotoxicity 2 Kit (Cat. #8400202, Thermo Fisher Scientific, Pittsburgh, PA). In this experiment, cells from three different lots were plated on 6-well plates and cultured for one week, then replated into three 0.1% gelatin-coated 96-well back/clear plates as described above. Three days after replating the cells were treated with jaspamide or mitoxantrone for 72 h. Cells were then live-stained with nuclear (blue), membrane permeability (green) and mitochondrial membrane potential (red) dyes according to the kit instructions, and the fluorescent images were analyzed using the multiparameter cytotoxicity BioApplication module.
2.5 Assessment of cardiomyocyte function
As part of the cardiomyocyte studies described above, impedance was measured using the xCELLigence RTCA Cardio-96 System (Roche Applied Sciences, Indianapolis, IN). The change in beat amplitude (a measure of cardiac contractility) and beat rate (a measure of rate of rhythmic contraction of cardiomyocytes) was calculated as a percentage of the time-matched vehicle control group at four selected time points between one and 72 h drug exposure.
2.6 Data analysis
Data are expressed as plus or minus the standard error of the mean. Dose-dependent reductions in cell index (at 1, 12, and 72 h) and in ATP content, cell density and mitochondrial transmembrane potential (at 72 h) were fitted with the Hill equation using Add-in Solver in Microsoft Excel 2010 to generate an IC50. Jaspamide and mitoxantrone-induced, dose-dependent changes in cell index, ATP content, cell density (nuclei count), membrane permeability and mitochondrial membrane potential fluorescence intensity, and cytotoxicity index were compared with the time-matched vehicle group at selected time points using the standard Student t-test (paired). Statistical significance was defined at a P < 0.05.
3. Results
3.1 Jaspamide Inhibits the Activity of Several Cardiac Ion Channels
Jaspamide at 10 μM inhibited the activity of the Kv1.5 ion channel at a level greater than the positive control (98.5% inhibition, vs. 87.0% with the positive control 4-AP at 2 mM) (Table 2). Other channels, primarily Cav1.2, Cav3.2, and HCN2, were also inhibited by jaspamide, although the percent inhibition was lower than that caused by the respective positive controls (64.3% vs. 87.7% by 1 μM nifedipine, 48.8% vs. 88.3% by 100 μM nickel, and 41.3% vs. 81.2% by 100 μM zatebradine, respectively). The hERG channel was minimally affected (4.2% inhibition) by jaspamide.
Table 2.
In Vitro Inhibition of Cardiac Ion Channels by Jaspamide
| Channel | Mean % inhibition by Jaspamidea | S.D. | S.E. | Positive Controlb | Mean % inhibition | S.D. |
|---|---|---|---|---|---|---|
| Cav1.2 | 64.3 | 5.8 | 4.1 | Nifedipine (1 μM) | 87.7 | 0.6 |
| Cav3.2 | 48.8 | 0.5 | 0.4 | Nickel (100 μM) | 88.3 | 0.2 |
| HCN2 | 41.3 | 0.6 | 0.4 | Zatebradine (100 μM) | 81.2 | 1.5 |
| HCN4 | 7.3 | 1.1 | 0.8 | Zatebradine (100 μM) | 86.5 | 3.6 |
| hERG | 4.2 | 7.3 | 5.2 | E-4031 (0.5 μM) | 98.5 | 0.2 |
| Kir2.1 | 1.5 | 0.6 | 0.5 | Barium (100 μM) | 88.8 | 7.1 |
| Kir3.1/3.4 | −1.1 | 4.3 | 3.0 | Barium (300 μM) | 75.9 | 8.3 |
| Kir6.2/SUR2A | −3.8 | 3.1 | 2.2 | Glybenclamide (1 μM) | 98.3 | 0.5 |
| Kv1.5 | 98.5 | 0.1 | 0.0 | 4-AP (2000 μM) | 87.0 | 0.2 |
| Kv4.3 | 3.9 | 3.6 | 2.5 | Flecainide (100 μM) | 73.9 | 2.0 |
| KvLQT/mink | 14.8 | 1.2 | 0.9 | Chromanol 293B (30 μM) | 94.9 | 0.7 |
| Nav1.5 tonic | 8.7 | 3.5 | 2.5 | Lidocaine (2000 μM) | 68.0 | 6.7 |
| Nav1.5 phasic | 18.8 | 0.4 | 0.3 | Lidocaine (2000 μM) | 68.0 | 6.7 |
CHO or HEK293 single cells were exposed to 10 μM jaspamide for 5 min at room temperature.
CHO or HEK293 single cells were exposed to previously determined optimal concentrations of the positive control for 5 min at room temperature
In vitro assays were performed to determine if jaspamide is cardiotoxic.
Jaspamide inhibited Kv1.5 cardiac ion channel activity by 98.5%.
Jaspamide decreased beat amplitude and increased beat rate in human cardiomyocytes.
Assay data from the known cardiotoxicant mitoxantrone were compared to jaspamide.
These results may explain the acute in vivo toxicity (in rat, dog) with jaspamide.
3.2 Jaspamide and Mitoxantrone Decrease Cardiomyocyte Viability
A dose- and time-dependent decrease in cell index occurred rapidly in cells treated with jaspamide (Figure 1a). A decrease of ≥ 10% in cell index was noted at concentrations ≥ 1 M after 1 h exposure (Figure 1b). This jaspamide-induced decrease in cell index paralleled a time-dependent decrease in the concentration of drug required to reduce the starting cell index by 50% (IC50). The IC50 decreased rapidly during the first 12 h of treatment (more than 10-fold from 19.0 to 1.7 μM), then slowly over the remainder of treatment (to 1.0 μM at 72 h; Figure 1b).
Fig. 1.

Cardiomyocyte viability after exposure to jaspamide and mitoxantrone. Human iPSC cardiomyocytes were treated for 72 h with drug concentrations ranging from 30 nM to 30 μM. Viability is expressed in terms of cell index vs. time, or a single ATP measurement after completion of impedance recording. Dynamic monitoring of cell index change is shown for cells exposed to vehicle and jaspamide (a) or mitoxantrone (c). Cell index for each well was normalized to baseline value prior to dosing (marked by the arrow). Dose-response analysis of jaspamide (b) and mitoxantrone (d) is shown at selected time points. The IC50 was calculated from the change in cell index or ATP content expressed as a percentage of the time-matched vehicle group. n = 7 wells/concentration for jaspamide and six for mitoxantrone in three E-plates seeded with cells from three different lots.
Of note, the ATP content measured at 72 h jaspamide treatment did not match the dramatic decrease noted in cell index, which suggests decreased attachment and/or altered morphology rather than decreased viability. The IC50 for ATP was extrapolated as 57.0 μM (Figure 1b).
Exposure to mitoxantrone resulted in a dose- and time-dependent decrease in cell index which occurred at a relatively slower onset than jaspamide. After 1 h exposure, only concentrations ≥ 10 M caused a decrease in cell index by ≥ 10% (Figure 1c). The IC50 at 1 h was extrapolated as 127 M, vs. 19 M by jaspamide (Figure 1d). However, the shift in IC50 during the first 12 h of treatment with mitoxantrone occurred rapidly (~ 41-fold from 127 to 3.1 M), and was similar to jaspamide in that it shifted more slowly over the rest of treatment (to 0.5 M at 72 h). Importantly, the decrease in ATP at 72 h correlated with the decrease in cell index. The IC50 for ATP was calculated as 0.8 μM (Figure 1d).
3.3 Jaspamide and Mitoxantrone Alter Cardiomyocyte Function
As illustrated by the images of representative cardiomyocyte rhythmic beat tracings (Figure 2a and 2d), jaspamide and mitoxantrone induced time- and dose-dependent changes in both beat rate and beat amplitude. For jaspamide (Figure 2b), a statistically significant increase in beat rate was noted at concentrations ≥ 0.3 μM at 1 h exposure. At 6 h, the beat rate increased significantly at low concentrations (1 and 3 μM), but decreased significantly at high concentrations (10 and 30 μM). At exposures > 6 h, a dose-dependent decrease in beat rate was noted, and no beating was detected at concentrations ≥ 3 μM. Mitoxantrone caused a different dynamic change in beat rate (Figure 2e). A statistically significant decrease in beat rate was noted at 30 μM at 1 h exposure, and at both 10 and 30 μM at 6 h. At ≥ 24 h, the beat rate was increased significantly at low concentrations (0.1 and 0.3 μM), but decreased significantly or the beating stopped completely at high concentrations (≥ 3 μM). Mitoxantrone also induced delayed repolarization-mediated type arrhythmic beats at concentrations ≥ 3 μM immediately prior to the disappearance of detectable beatings (Figure 2d).
Fig. 2.

Beating waveform analysis. Representative beating traces were selected to show time and dose-dependent changes in beating waveforms of cardiomyocytes exposed to jaspamide (a) and mitoxantrone (d). The impedance-captured beating exhibited a downward deflection upon the contraction of human iPSC cardiomyocytes, which rapidly returned to baseline level during relaxation. The baseline level in (a) declined rapidly in the well exposed to 30 μM jaspamide, reflecting the rapid decrease in cell index. The reduced beat amplitude and transiently increased beat rate were well-manifested. Summarized data of beat rate and amplitude for jaspamide are presented in (b–c), and for mitoxantrone in (e–f). Blue arrows indicate tachycardia or fibrillation-like arrhythmia, and the red arrow indicates delayed-repolarization mediated-like arrhythmia. * P ≤ 0.05 vs. the time-matched vehicle group. n = 7 wells/concentration for jaspamide and six for mitoxantrone in three E-plates seeded with cells from three different lots.
Both jaspamide and mitoxantrone induced a dose- and time-dependent decrease in beat amplitude (Figure 2c and 2f). Jaspamide caused a statistically significant decrease in beat amplitude at ≥ 3 μM at 1 h, and at ≥ 0.1 μM at 6 h. At exposure times ≥ 24 h, beat amplitude decreased at ≥ 0.1 μM, with no beating detected at ≥ 3 μM. Exposure to mitoxantrone resulted in slightly different changes in beat amplitude than jaspamide. At 1 h exposure, a concentration of 30 μM was required to detect a statistically significant decrease in beat amplitude. At ≥ 6 h, concentrations of ≥ 0.1 μM caused a statistically significant decrease in beat amplitude. At ≥ 24 h, beating was not detected in all wells exposed to ≥ 10 μM.
3.4 Positive ion channel blockers do not alter cardiomyocyte viability
None of the positive control reference drugs significantly changed either the cell index or ATP content at the range of concentrations tested. Figure 3 shows the representative beating traces obtained at selected times after drug treatment. As expected, E-4031 (1 nM to 1 μM), TTX (0.03 to 30 μM) and nifedipine (3 nM to 3 μM) affected beat amplitude and beat rate in a manner identical to that reported previously (Guo et al., 2011). S9947 (0.3 to 30 μM) resulted in a time- and dose-dependent decrease in the beat rate of cardiomyocytes. An immediate reduction in beat rate and/or complete beat arrest was observed at 10 and 30 μM upon S9947 application. Beat rate recovered during the 72 h drug exposure.
Fig. 3.
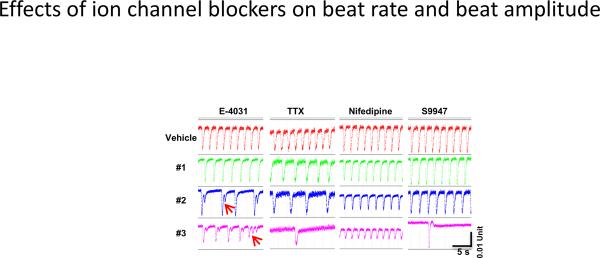
Representative beating traces which illustrate the effects of E-4031 (0.01, 0.03, 0.1 M), TTX (1, 3, 10 M), nifedipine (0.03, 0.1, 0.3 M) and S9947 (3, 10, 30 M). #1 to #3, dose concentrations from low to high. The red arrow indicates the delayed-repolarization mediated-like arrhythmia. Beating traces for E-4031 and TTX were selected at 30 min after drug treatment, and for nifedipine and S9947 at 72 h after drug exposure.
3.5 Multiparameter imaging analysis confirms the structural cytotoxicity of jaspamide and mitoxantrone
The representative fluorescence images in Figure 4 illustrate the effects of jaspamide and mitoxantrone on cell density, membrane permeability, mitochondrial transmembrane potential and cell morphology. Cells treated with the vehicle for 72 h remained in a nice monolayer (Figure 4A). The nuclei (blue) were often located in the center of the cell with mitochondria (red) distributed evenly across the entire plasma area. Exposure to 3 M mitoxantrone resulted in a significant loss of viable cell numbers and plasma area. Mitochondrial staining was reduced but permeability dye (green) staining increased. Cells became spindle-shaped as a result of plasma shrinkage (Figure 4B). Exposure to 30 M mitoxantrone resulted in a loss of ~90% of the cells. The remaining cells lost almost all plasma area and became clumped together. Cells were stained weakly with mitochondrial transmembrane dye and moderately with permeability dye (Figure 4C).
Fig. 4.
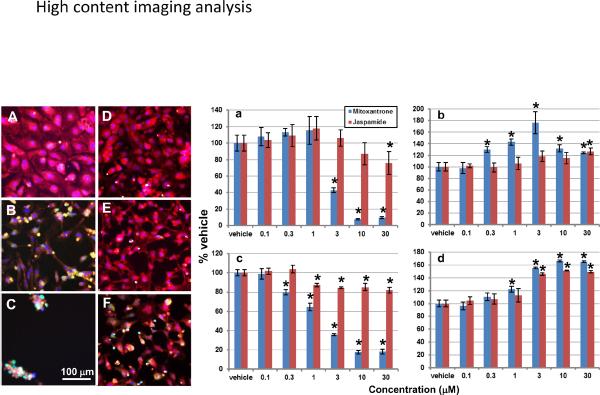
Multiparameter fluorescence imaging analysis. Cells were treated with vehicle (A), mitoxantrone at 3 (B) and 30 M (C), or jaspamide at 0.3 (D), 3 (E) and 30 M (F) for 72 h, then stained with nuclei (blue), membrane permeability (green) and mitochondrial membrane potential (red) dyes. Summarized data show the drug effect on the viable cell density (a), membrane permeability (b), mitochondrial membrane potential (c) and cytotoxicity index (d). * P ≤ 0.05 vs. the time-matched vehicle group. n = 6 wells/concentration in three E-plates seeded with cells from three different lots.
Jaspamide induced a dose-dependent reduction in cell size (Figure 4D–4F). As a result of plasma shrinkage at 0.3 and 3 M (Figure 4D and 4E), mitochondria (red) were concentrated in certain regions of cells. At 30 M (Figure 4F), jaspamide reduced mitochondrial transmembrane potential staining and increased permeability dye staining.
Data summarized from three plates (n = 6 wells/concentration) were tabulated in Figure 4a–d. Both jaspamide and mitoxantrone induced a dose-dependent decrease in viable cell number and mitochondrial transmembrance potential, and a dose-dependent increase in the membrane permeability and cytotoxicity index, albeit, mitoxantrone caused a larger change than jaspamide. The IC50 for cell number and mitochondrial transmembrance potential was extrapolated as 65 and 97 M, respectively, for jaspamide, but was 2.4 and 1.8 M, respectively, for mitoxantrone. For the membrane permeability and cytotoxicity indices, a statistically significant increase was noted only at 30 M and 3–30 M, respectively, for jaspamide; whereas it was 0.3–30 M and 1–30 M for mitoxantrone (Figure 4b and 4d).
4. Discussion
The in vitro studies summarized herein investigated the effect of jaspamide on cardiac ion channel activity and cardiomyocyte viability and function. Jaspamide was cytotoxic to human cardiomyocytes as shown by a time- and dose-dependent decrease in cell index (which shifted from an IC50 of 19 μM to 1 μM over 72 h). The IC50 for cardiomyocyte injury is within the range of jaspamide concentrations required for anti-tumor efficacy against sensitive cancer cell lines. Similar to the weak effect on PC-3 cancer cell cytotoxicity as determined by ATP level (Senderowicz et al., 1995), jaspamide at 30 μM only caused an ~40% reduction in ATP content in cardiomyocytes treated for 72 h, suggesting that the mechanism of cardiotoxicity may be related to the pharmacologic mechanism or at least overlaps. In either event, it is unlikely that mitochondria are the primary target of either the pharmacologic or toxicologic effects of jaspamide. This conclusion was supported by multiparameter imaging data. Jaspamide induced a remarkable dose-dependent decrease in cardiomyocyte size, but only moderately decreased viable cell number and mitochondrial transmembrane potential. The IC50 for these two parameters was estimated at 65 and 97 M, respectively, which was very close to that calculated from the ATP content of cardiomyocytes in E-plates (72 M), but significantly different from the IC50 calculated from the cell index change (1 M). In contrast, the IC50's correlate for these parameters for mitoxantrone, an anthracenedione antineoplastic drug known to cause irreversible cardiomyopathy and heart failure (Slørdal et al., 2006). The IC50 was 0.5, 0.8, 2.4 and 1.8 M for the decrease in cell index, ATP content, cell number and mitochondrial transmembrane potential, respectively, by mitoxantrone. Importantly, the lowest concentration that induced a statistically significant increase in cytotoxicity index was 0.3 M for mitoxantrone and 3 M for jaspamide. Both were in line with the IC50 for cell index, confirming their structural cardiotoxic effect. These results demonstrate that a change in cardiomyocyte morphology and/or attachment, rather than a massive cell death similar to that induced by mitoxantrone, was the primary cause of the remarkable decrease in cell index by jaspamide. Indeed, our recent experiments revealed that jaspamide effectively disrupted F-actin in cardiomyocytes in the nanomolar range (data to be published later).
Jaspamide affects the binding of actin to the cytoskeleton (Bubb et al., 1994; Senderowicz et al., 1995). In rabbit skeletal muscle in vitro, jaspamide (0.5–7.4 μM) induced actin polymerization disrupting the actin cytoskeleton (Bubb et al., 1994). Maximal actin disruption was noted in PC-3 prostate carcinoma cells after exposure to 80 nM jaspamide for 24 h (Senderowicz et al., 1995). Numerous structure-activity relationship studies conducted with jaspamide analogs indicate that actin disruption plays a role in the cytotoxic effect of jaspamide (Tannert et al., 2010; Gala et al., 2007, 2008, 2009; Marimganti et al., 2005). The inactivity of jaspamide analogs is possibly due to structural modifications on jaspamide at the actin binding site (Tannert et al., 2010; Waldmann et al., 2008; Gala et al. 2007, 2008, 2009; Marimganti et al., 2005). We found that the cell index IC50 (1 M) for jaspamide-exposed cardiomyocytes after 72 h exposure was well above the jaspamide concentration (80 nM) required for maximal actin disruption in PC-3 cells.
Jaspamide at 10 μM inhibited (98.5%) Kv1.5 ion channel activity in CHO cells. Kv1.5, a voltage-gated potassium channel expressed in human atria, is a primary component of the Ikur ultra-rapid delayed rectifier current, and regulates atrial repolarization (Olson et al., 2006; Karczewski et al., 2009). Loss-of-function mutations in this ion channel are associated with human atrial fibrillation (Olson et al., 2006). Jaspamide also inhibited the calcium channels Cav1.2 and Cav3.2, and the HCN2 potassium channel, but to a much lesser extent than the Kv1.5 channel. Calcium cardiac ion channel function has not been shown to be dependent upon an intact cytoskeleton (Nerbonne and Kass, 2005). In contrast, Kv ion channel function in cells, including myocardium, may be regulated via interactions with the actin cytoskeleton (Nerbonne and Kass, 2005). For example, in hypertrophied rat ventricular myocytes, phalloidin (which stabilizes actin) markedly reduced cellular action potential duration (Yang et al., 2003; Nerbonne and Kass, 2005). An intact actin cytoskeleton is also required for current modulation of cloned Kv1.5 channels expressed in Xenopus laevis oocytes (Nerbonne and Kass, 2005; Mason et al., 2002), and for the expression of Kv1.5 channels in HEK cells (Maruoka et al., 2000; Nerbonne and Kass, 2005). Since jaspamide binds actin, and Kv1.5 ion channel function is dependent upon an intact actin cytoskeleton (Nerbonne and Kass, 2005), Kv1.5 cardiac ion channel activity may be disrupted in intact cells by jaspamide.
Our cardiac ion channel results suggest that atrial fibrillation may occur upon in vivo exposure to jaspamide. In our in vitro studies, jaspamide affected the functionality of human cardiac myocytes by decreasing beat amplitude and causing biphasic changes in beat rate. These effects were not mimicked by any of the selective ion channel blockers (E-4031, TTX and nifedipine), especially the highly potent Kv1.5/Ikur channel blocker S9747 (Bachmann et al., 2001) tested in this study, but were well-correlated with a concentration-dependent decrease in cell index over time, suggesting that the observed tachycardia or fibrillation-type arrhythmias were primarily the result of cell damage rather than a direct effect upon ion channels. Thus far, our data support the hypothesis that jaspamide toxicity is due to a direct cytotoxic effect on cardiomyocytes. The use of a human stem cell derived cardiomyocyte model to conduct in vitro explorations of mechanism has shown promise and may provide a means to screen the direct effects of investigational agents on cardiac myocyte viability and function.
Acknowledgment
The authors would like to acknowledge Cheryl Heidelberger and Luke Coyle for their technical assistance with this project.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Anson BD, Kolaja KL, Kamp TJ. Opportunities for use of human cells in predictive toxicology. Clin. Pharmacol. Ther. 2011;89:754–758. doi: 10.1038/clpt.2011.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachmann A, Gutcher I, Kopp K, Brendel J, Bosch RF, Busch AE, Gögelein H. Characterization of a novel Kv1.5 channel blocker in Xenopus oocytes, CHO cells, human and rat cardiomyocytes. Naunyn Schmiedebergs Arch. Pharmacol. 2001;364:472–478. doi: 10.1007/s002100100474. [DOI] [PubMed] [Google Scholar]
- Bubb MR, Senderowicz AM, Sausville EA, Duncan KLK, Korn E. Jasplakinolide, a cytotoxic natural product, induces actin polymerization and competitively inhibits the binding of phalloidin to F-actin. J. Biol. Chem. 1994;269:14869–14871. [PubMed] [Google Scholar]
- Bubley G, Balk S. Treatment of androgen-independent prostate cancer. Oncologist. 1996;1:30. [PubMed] [Google Scholar]
- Crews P, Manes LV, Boehler M. Jasplakinolide, a cyclodepsipeptide from the marine sponge, Jaspis sp. Tetrahedron Lett. 1986;27:2797–2800. [Google Scholar]
- Fabian I, Shur I, Bleiberg I, Rudi A, Kashman Y, Lishman M. Growth modulation and differentiation of acute myeloid leukemia cells by jaspamide. Exp. Hematol. 1995;23:583–587. [PubMed] [Google Scholar]
- Gala F, D'Auria MV, De Marino S, Zollo F, Smith CD, Copper JE, Zampellaa A. New jaspamide derivatives with antimicrofilament activity from the sponge Jaspis splendans. Tetrahedron. 2007;63:5212–5219. [Google Scholar]
- Gala F, D'Auria MV, De Marino S, Sepe V, Zollo F, Smith CD, Copper JE, Zampellaa A. Jaspamides H-L, new actin-targeting depsipeptides from the sponge Jaspis splendans. Tetrahedron. 2008;64:7127–7130. [Google Scholar]
- Gala F, D'Auria MV, De Marino S, Sepe V, Zollo F, Smith CD, Keller SN, Zampellaa A. Jaspamides M-P: new tryptophan modified jaspamide derivatives from the sponge Jaspis splendans. Tetrahedron. 2009;65:51–56. [Google Scholar]
- Guo L, Abrams RM, Babiarz JE, Cohen JD, Kameoka S, Sanders MJ, Chiao E, Kolaja KL. Estimating the risk of drug-induced proarrhythmia using human induced pluripotent stem cell-derived cardiomyocytes. Toxicol. Sci. 2011;123:281–289. doi: 10.1093/toxsci/kfr158. [DOI] [PubMed] [Google Scholar]
- Karczewski J, Kiss L, Kane SA, Koblan KS, Lynch RJ, Spencer RH. High-throughput analysis of drug binding interactions for the human cardiac channel, Kv1.5. Biochem. Pharmacol. 2009;77:177–185. doi: 10.1016/j.bcp.2008.09.035. [DOI] [PubMed] [Google Scholar]
- Ma J, Guo L, Fiene SJ, Anson BD, Thomson JA, Kamp TJ, Kolaja KL, Swanson BJ, January CT. High purity human-induced pluripotent stem cell-derived cardiomyocytes: electrophysiological properties of action potentials and ionic currents. Am. J. Physiol. Heart Circ. Physiol. 2011;301:H2006–H2017. doi: 10.1152/ajpheart.00694.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marimganti S, Yasmeen S, Fischer D, Maier ME. Synthesis of jasplakinolide analogues containing a novel w-amino acid. Chem. Eur. J. 2005;11:6687–6700. doi: 10.1002/chem.200500319. [DOI] [PubMed] [Google Scholar]
- Maruoka ND, Steele DF, Au BP-Y, Dan P, Zhang X, Moore EDW, Fedida D. α-actinin-2 couples to cardiac Kv1.5 channels, regulating current density and channel localization in HEK cells. FEBS Letters. 2000;473:188–194. doi: 10.1016/s0014-5793(00)01521-0. [DOI] [PubMed] [Google Scholar]
- Mason HS, Latten MJ, Godoy LD, Horowitz B, Keny on JL. Modulation of Kv1.5 currents by protein kinase A, tyrosine kinase, and protein tyrosine phosphatase requires an intact cytoskeleton. Mol. Pharmacol. 2002;61:285–293. doi: 10.1124/mol.61.2.285. [DOI] [PubMed] [Google Scholar]
- Nerbonne JM, Kass RS. Molecular physiology of cardiac repolarization. Physiol. Rev. 2005;205:1205–1233. doi: 10.1152/physrev.00002.2005. [DOI] [PubMed] [Google Scholar]
- Olson TM, Alekseev AE, Liu XK, Park S, Zingman LV, Bienengraeber M, Sattiraju S, Ballew JD, Jahangir A, Terzic A. Kv1.5 channelopathy due to KCNA5 loss-of-function mutation causes human atrial fibrillation. Human Molec. Genetics. 2006;15:2185–2191. doi: 10.1093/hmg/ddl143. [DOI] [PubMed] [Google Scholar]
- Senderowicz AM, Kaur G, Sainz E, Charmaine L, Inman WD, Rodriguez J, Crews P, Malspeis L, Grever MR, Sausville EA, Duncan KLK. Jasplakinolide's inhibition of the growth of prostate carcinoma cells in vitro with disruption of the actin cytoskeleton. J. Natl. Cancer Inst. 1995;87:46–51. doi: 10.1093/jnci/87.1.46. [DOI] [PubMed] [Google Scholar]
- Schindler-Horvat J, Fairchild DG, Hassler C, Tomaszewski JE, Donohue SJ, Tyson CA. Toxicity of jasplakinolide (NSC 613009) in rats and dogs. Proc. Amer. Assoc. Cancer Res. 1998;39:597. (A4063) [Google Scholar]
- Slørdal L, Spigset O. Heart failure induced by non-cardiac drugs. Drug Saf. 2006;29:567–86. doi: 10.2165/00002018-200629070-00003. [DOI] [PubMed] [Google Scholar]
- Stingl J, Andersen RJ, Emerman T. In vitro screening of crude extracts and pure metabolites obtained from marine invertebrates for the treatment of breast cancer. Cancer Chemother. Pharmacol. 1992;30:401. doi: 10.1007/BF00689969. [DOI] [PubMed] [Google Scholar]
- Takeuchi H, Ara G, Sausville EA, Teicher B. Jasplakinolide: interaction with radiation and hyperthermia in human prostate carcinoma and Lewis lung carcinoma. Cancer Chemother. Pharmacol. 1998;42:491–496. doi: 10.1007/s002800050850. [DOI] [PubMed] [Google Scholar]
- Tannert R, Milroy L-G, Ellinger B, Hu T-S, Arndt H-D, Waldmann H. Synthesis and structure-activity correlation of natural-product inspired cyclodepsipeptides stabilizing F-actin. J. Am. Chem. Soc. 2010;132:3063–3077. doi: 10.1021/ja9095126. [DOI] [PubMed] [Google Scholar]
- Waldmann H, Hu T-S, Renner S, Menninger S, Tannert R, Oda T, Arndt HD. Total synthesis of chondramide C and its binding mode to F-actin. Angew. Chem. Int. Ed. Engl. 2008;47:6473–6477. doi: 10.1002/anie.200801010. [DOI] [PubMed] [Google Scholar]
- Xi B, Yu N, Wang X, Xu X, Abassi YA. The application of cell-based label-free technology in drug discovery. Biotechnol. J. 2008;3:484–495. doi: 10.1002/biot.200800020. [DOI] [PubMed] [Google Scholar]
- Yang X, Salas PJ, Pham TV, Wasserlauf BJ, Smets MJ, Myerburg RJ, Gelband H, Hoffman BF, Bassett AL. Cytoskeletal actin microfilaments and the transient outward K+ current in hypertrophied rat ventriculocytes. J. Physiol. 2003;541:411–421. doi: 10.1113/jphysiol.2002.019562. [DOI] [PMC free article] [PubMed] [Google Scholar]