

Abstract
Acetate supplementation attenuates neuroglia activation in a rat model of neuroinflammation by a mechanism associated with an increase in brain acetyl-CoA, an alteration in histone acetylation, and reduction of interleukin (IL)-1β expression. We propose that reduced astroglial activation occurs by disrupting astrocyte-derived inflammatory signaling and cytokine release. Using primary astroglial cultures, we found that LPS (0–25 ng/ml, 4 hr) increased tumor necrosis factor (TNF-α) and IL-1β in a concentration-dependent manner, which was reduced by treatment with sodium acetate (12 mM). LPS did not alter H3K9 acetylation or IL-6 levels, whereas acetate treatment increased H3K9 acetylation by 2-fold and decreased basal levels of IL-6 by 2-fold. Acetate treatment attenuated the LPS-induced increase in TNF- mRNA, but did not reverse the mRNA levels of other pro-inflammatory cytokines. By contrast, LPS decreased TGF-β1 and IL-4 protein and TGF-β1 mRNA, all of which was reversed with acetate treatment. Further, we found that acetate treatment completely reversed LPS-induced phosphorylation of MAPK p38 and decreased basal levels of phosphorylated extracellular signal-regulated kinases1/2 (ERK1/2) by 2-fold. Acetate treatment also reversed LPS-elevated NF-κB p65, CCAAT/enhancer-binding protein beta protein levels, and reduced basal levels of phosphorylated NF-κB p65 at serine 536. These results suggest that acetate treatment has a net anti-inflammatory effect in LPS-stimulated astrocytes that is largely associated with a disruption in MAPK and NF-κB signaling.
Keywords: MAPK, NF-κB, astrocyte, cytokines, acetate, inflammation
Introduction
Acetylation is a predominant post-translational modification that occurs on a wide range of histone and non-histone proteins. Acetylation of histones changes gene expression (Strahl and Allis, 2000), while acetylation of non-histone proteins effects the subcellular localization, DNA binding, transcriptional activity, protein-protein interaction, and stability of acetylated protein (Polevoda and Sherman, 2002; Glozak et al., 2005). Acetylation reactions are dependent on the formation of acetyl-CoA that is formed from acetate and catalyzed by acetyl-CoA synthetases (Fujino et al., 2001; Ariyannur et al., 2010). Oral glyceryl triacetate, used to induce acetate supplementation, increases brain acetyl-CoA levels and induces a pattern of site- and time-specific brain histone hyperacetylation in rats (Soliman and Rosenberger, 2011). Long-term acetate supplementation in a rat model of neuroinflammation attenuates LPS-induced neuroglial activation, the loss of cholinergic immunoreactivity (Reisenauer et al., 2011), interleukin (IL)-1β expression, and increases histone H3 at lysine 9 (H3K9) acetylation (Soliman et al., 2012b). In a rat model of Lyme neuroborreliosis, acetate supplementation reduces microglia activation and IL-1β levels suggesting that its anti-inflammatory effect is not limited to toll-like receptor 4 activation but rather disruption in glial-derived inflammatory signaling (Brissette et al., 2012). Acetate supplementation is also neuroprotective in a rat model of head trauma (Arun et al., 2010a) and a tremor model of Canavan’s disease (Arun et al., 2010b).
Microglia, the primary resident immune cells of the central nervous system, upon exposure to different stresses differentiate into a more reactive phenotype with increased secretion of inflammatory mediators and enhanced phagocytic activity (Streit et al., 1999; Olson and Miller, 2004; Hanisch and Kettenmann, 2007; Ransohoff and Perry, 2009; Lehnardt, 2010). In inflammatory conditions, astrocytes undergo reactive astrogliosis characterized by astrocyte hypertrophy, upregulation of intermediate filaments like vimentin and glial fibrillary acidic protein (GFAP), altered molecular expression profile, and scar formation (Pekny and Nilsson, 2005; Sofroniew, 2009; Kang and Hebert, 2011). Although essential for proper tissue healing, excessive astrocyte reactivity contributes to the inflammatory response by increasing pro-inflammatory cytokine release (Dong and Benveniste, 2001; Gorina et al., 2009; Gorina et al., 2011) and is thus recognized as a potential therapeutic target (Hamby and Sofroniew, 2010).
The release of cytokines in brain serves as communication signals among different cell types and contributes to bystander neuronal lysis when uncontrolled (Tian et al., 2012). The pro-inflammatory cytokines tumor necrosis factor-alpha (TNF-α), IL-1β, and IL-6 are involved in the pathogenesis of neurodegenerative diseases including acute cerebral ischemia (Denes et al., 2010), Alzheimer’s disease (Shaftel et al., 2008; Johnston et al., 2011), Parkinson’s disease (Qian et al., 2010), multiple sclerosis (Merson et al., 2010) and traumatic brain injury (Helmy et al., 2011). In contrast, the anti-inflammatory cytokines transforming growth factor-beta1 (TGF-β1), IL-4 and IL-10 reduce the injury potential by downregulating the pro-inflammatory cytokines and stimulate tissue repair (Ledeboer et al., 2000; Vitkovic et al., 2001). Therefore, equilibrium is maintained between the pro- and anti-inflammatory cytokines where the interaction between these opposing molecular groups determines the outcome of the immune response. Excessive pro-inflammatory cytokines are linked to neurodegeneration, while excessive anti-inflammatory cytokines are associated with susceptibility to systemic infections (Munoz et al., 1991; Kasai et al., 1997).
With regard to the maintenance of cytokine balance, acetate treatment in LPS-stimulated microglia reverses the protein levels of pro-inflammatory cytokines IL-1β, TNF-α and IL-6 and increases the transcription of the anti-inflammatory cytokines TGF-β1 and IL-4 (Soliman et al., 2012a). This shift in cytokine balance is correlated with a reversal of LPS-induced H3K9 hypoacetylation and NF-κB p65 phosphorylation and protein levels, and the induction of NF-κB p65 hyperacetylation at lysine 310. Acetate treatment has only a transient effect on LPS-induced MAPK p38 phosphorylation, has no effect on JNK phosphorylation and increases ERK1/2 phosphorylation only in the presence of LPS. Therefore, stimulating acetyl-CoA metabolism in microglia has a net anti-inflammatory effect correlated to an increase in the of histone and non-histone targets that is largely independent of MAPK signaling (Soliman et al., 2012a).
CCAAT/enhancer-binding proteins (C/EBP) are a class of transcription factors (C/EBPα–C/EBPζ) which have a role in the regulation of cellular proliferation, differentiation, metabolism, and inflammation. The activity and expression of C/EBPβ are induced by inflammatory stimuli LPS and pro-inflammatory cytokines, whereas C/EBPα is inhibited by these stimuli. Target genes regulated by the C/EBP family include interferon-γ, IL-1β, IL-6, TNF-α, cyclooxygenase-2 and nitric oxide synthase, among many others. C/EBPβ-deficient mice show impaired expression of serum amyloid proteins, complement C3 component, and TNF-α (Poli et al., 1990; Tanaka et al., 1995; Poli, 1998; Ramji and Foka, 2002).
To determine whether an increase in acetyl-CoA metabolism alters inflammatory signaling in astrocytes, we measured the effect acetate treatment had on LPS-stimulation using similar experimental conditions to that used with the microglia. In addition, because C/EBPβ activation positively regulates pro-inflammatory cytokine release to understand the differential effect of acetate on TNF-α versus IL-1β and IL-6 mRNA, we examined the effect acetate treatment had on C/EBPβ protein level. We found that acetate treatment induced H3K9 hyperacetylation, reduced LPS-induced increases in IL-1β and TNF-α protein but not mRNA, NF-κB p65 and C/EBPβ protein levels, and phosphorylated MAPK p38. Acetate treatment also decreased the basal levels of phosphorylated ERK1/2, IL-6, and phosphorylated NF-κB p65 at serine 536. LPS however did not alter H3K9 acetylation, IL-6 level, or MAPK JNK and ERK1/2 phosphorylation. These data suggest that acetate treatment has a net anti-inflammatory effect in LPS-stimulated astrocytes that is largely associated with the disruption of MAPK and NF-κB signaling.
Methods
Materials
LPS (Escherichia Coli 055:B5) was purchased from Sigma (St. Louis, MO) antibodies against total histone H3, acetylated H3K9, phosphorylated p38 (Thr180/Tyr182), p38, phosphorylated JNK (Thr183/Tyr185, Thr221/Tyr223), phosphorylated ERK1/2 (Th202/Tyr204, Thr185/Tyr187) and ERK1/2 were from Millipore (Billerica, MA), an antibody towards C/EBPβ was purchased from Santa Cruz (Santa Cruz, CA), and JNK and NF-κB p65 antibodies were purchased from Cell Signaling Technology Incorporated (Danvers, MA). Rabbit polyclonal antibodies to IL-1β, IL-6, TNF-α, TGF-β1, IL-4, IL-10 and acetyl-CoA synthetase were from Abcam (Cambridge, MA), anti-GFAP rabbit antibody was from Sigma (St. Louis, MO), a biotinylated anti-rabbit antibody, Vectasheild mounting medium containing propidium iodide, Elite Vectastain ABC Avidin and Vector VIP chromogen were purchased from Vector Laboratories (Burlingame, CA). All Western blot supplies and a goat anti-rabbit horse radish peroxidase (HRP)-linked antibody and iScript cDNA synthesis kits were purchased from Bio-Rad Laboratories (Hercules, CA). Reverse and forward IL-1β, IL-6, TNF-α, IL-4, IL-10, TGF-β1 and β-actin primers from SA Biosciences (Frederick, MD), FastStart Universal SYBR Green Master from Roche Applied Science (Indianapolis, IN), TRIzol® reagent from Life Technologies (Grand Island, NY), DMEM–F-12 media from Invitrogen (Grand Island, NY), and buffering reagents and other chemicals from EMD Biosciences (Gibbstown, NJ). DNase I and DNase I reaction buffer were purchased from New England Biolabs (Ipswich, MA), and a DeadEnd™ fluorometric TUNEL assay kit was from Promega (Madison, WI).
Primary astroglial culture
Astrocyte cultures were derived from C57BL/6 mouse brains as described previously (Dhawan et al., 2012). The purity of the astrocyte cell cultures was found to be 91 ± 3% as determined by immunostaining using anti-GFAP antibody (1:1000). Prior to stimulating the cells (3 hr), the media were changed to serum-free media. Plates were divided into 4 different groups; a group treated with 12 mM sodium chloride (NaCl) as a control group, another group treated with 12 mM sodium acetate, a third group treated with both 6.25 ng/ml LPS and 12 mM NaCl, and a fourth group treated with both 6.25 ng/ml LPS and 12 mM sodium acetate (n = 6 per group). The control group was treated with 12 mM NaCl to control for osmolarity changes found in treatment with 12 mM sodium acetate. The sodium acetate concentration used here is similar to that used with LPS-stimulated microglia, and is the highest acetate concentration that does not cause significant cell death while maximizing cellular acetyl-CoA levels (Soliman et al., 2012a). For dose-response studies, plates were divided in 6 different groups treated with LPS in the following concentrations: 25, 12.5, 6.25, 3.125, 1.56, or 0 ng/ml (n = 3). After 4 hr, the media was collected and stored at −20° C, and the cells were lysed in either TRIzol® reagent for quantitative real-time polymerase chain reaction (qrt-PCR) analysis or ice cold RIPA lysis buffer (150 mM sodium chloride, Triton X-100, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulphate, 50 mM Tris, pH 8.0) for Western blot analysis and stored at −80° C until used.
Western blot analysis
Western blots were performed as described previously (Soliman et al., 2012a). The antibody concentrations used were 1:500 for total H3 and acetyl-CoA synthetase, 1:1000 for each of acetylated H3K9, IL-1β, IL-6, TNF-α, TGF-β1, IL-4, IL-10, p38, phosphorylated p38, JNK, phosphorylated JNK and C/EBPβ, and 1:3000 for α-tubulin. All Western blot data are expressed as the ratio of the optical density of the respective protein to the optical density of α-tubulin, except acetylated H3K9 (normalized to total histone H3), and phosphorylated MAPK p38, JNK and ERK1/2 (normalized to total MAPK p38, JNK and ERK1/2, respectively).
Quantitative real-time polymerase chain reaction
mRNA extraction, quantification, and cDNA synthesis and amplification were performed as described previously (Soliman et al., 2012a). The expression of IL-1β, IL-6, TNF-α, IL-4, IL-10, and TGF-β1 transcripts amplified was normalized to the expression of β-actin.
Lactate dehydrogenase assay
Cellular release of lactate dehydrogenase (LDH) used to measure cell viability was measured using a commercial nonradioactive assay kit (Clontech Inc.), according to the manufacturer’s guidelines. Absorbance measurements were taken at 490 nm.
TUNEL assay
Apoptosis was evaluated using fluorescin-12-deoxy-uracil triphosphate (d-UTP) by direct visualization with confocal microscopy (Carl Zeiss Microscopy, Peabody, MA). The procedure was performed according to the manufacturers’ instructions. Briefly, primary astrocytes were grown to approximately 95% confluence on poly-L-lysine-coated glass coverslips, treated as described above, immersion fixed in 4% paraformaldehyde for 25 min at 4° C, washed with phosphate-buffered saline ((PBS 137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 2 mM KH2PO4, and pH 7.4) at room temperature, permeabilized by immersion in 0.2% Triton X-100 for 5 min at room temperature, and rinsed twice with PBS at room temperature. coverslips were then incubated with equilibrium buffer, nucleotide mix, and recombinant terminal deoxynucleotidyl transferase enzyme at 37° C for 1 hr, and washed with PBS. The coverslips were embedded in Vectashield mounting medium containing propidium iodide then visualized using a confocal microscope (n = 5). Positive controls were generated by treating parallel groups with 50 units/mL DNase I (n = 4).
Statistical analysis
One-way analysis of variance (ANOVA) followed by Tukey’s post-hoc test using GraphPad InStat software (Version 3.06 for Windows, San Diego, CA). All values are represented as the mean fold change relative to control ± SD with significance set at p 0.05. Figures were prepared using SigmaPlot for Windows, version 10.0, Build 10.0.1.25.
Results
Temporal effect of acetate treatment and LPS on histone acetylation
To determine the duration of acetate treatment required to achieve H3K9 hyperacetylation pattern in vitro similar to that found in vivo (Soliman et al., 2012a), we treated primary astrocytes with 12 mM sodium acetate for 1, 2 and 4 hr (Fig 1A and B). Cell lysates were used for Western blot analysis to measure acetylated H3K9, total histone H3 (Fig 1A) which were detected as protein bands corresponding to the molecular weight of 17 kDa. We found that acetate treatment increased H3K9 acetylation at 1 hr which remained increased out to 4 hr (Fig 1B). To insure protein expression following treatment, we used 4 hr as the treatment duration for all the experiments. To determine the optimal LPS concentration required to produce the same percentage of H3K9 hypoacetylation in vitro, using a serial dilution of LPS (0–25 ng/ml), we treated primary astrocytes for 4 hr (Fig 1Cand D ). Cell lysates were used for Western blot analysis to measure acetylated H3K9, total histone H3 and the pro-inflammatory cytokines pro-IL-1β, IL-6, and TNF-α, which were detected as protein bands corresponding to the molecular weights of 17, 17, 32, 25 and 23 kDa, respectively (Fig 1C). Interestingly, LPS did not alter H3K9 acetylation or IL-6 protein levels and increased pro-IL-1β and TNF-α protein levels in a concentration-dependent manner (Fig 1D–G). Based on these data, LPS at a concentration of 6.25 ng/ml was used in all experiments based on the increase of both pro-IL-1β and TNF-α protein levels, and was similar to the concentration used for the treatment of primary and BV-2 microglia (Soliman et al., 2012a).
Fig. 1.
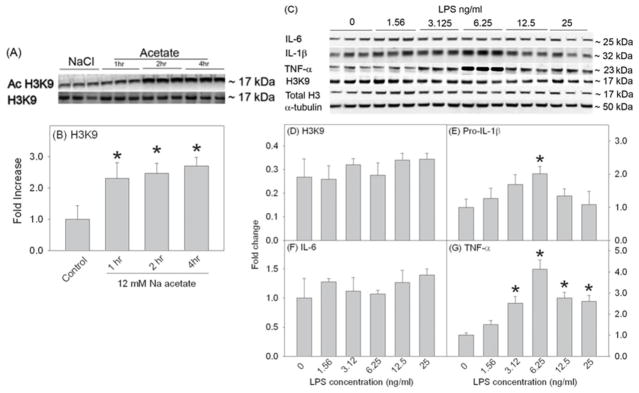
Time-dependent acetate treatment-induced H3K9 hyperacetylation and dose-response study showing the effects of different LPS concentrations (0–25 ng/ml, 4 hr) on H3K9 acetylation and the expression of pro-inflammatory cytokines in primary mouse astrocyte cell cultures. Panels A and C show representative images of the Western blots. Panel B shows the averaged proportion of H3K9 normalized to total H3 (n = 3) after 1, 2 and 4 hr of treatment with 12 mM sodium acetate. Panels D, E, F and G show the optical densities of H3K9 normalized to total H3 and the pro-inflammatory cytokines pro-IL-1β, IL-6, and TNF-α normalized to the loading control α-tubulin (n = 3). All values represent the mean fold change relative to control ± SD. Statistical significance (* = compared to LPS 0 ng/ml) was set at p ≤ 0.05, as determined by One Way ANOVA followed by Tukey’s post-hoc test.
Acetate treatment causes H3K9 hyperacetylation without inducing cytotoxicity in LPS-stimulated primary astrocytes
In order to determine the effect of acetate treatment on H3K9 acetylation in LPS-challenged astrocytes, we treated primary astrocytes with 6.25 ng/ml LPS in the presence and absence of 12 mM sodium acetate, and compared the results to cells treated with 12 mM NaCl as a control group. Using whole cell lysates for Western blot analysis, we found that primary astrocytes express acetyl-CoA synthetase, the enzyme which converts acetate to acetyl-CoA, as protein bands corresponding to the molecular weight of 79 kDa (Fig 2A). The expression levels of ACS were not different between groups (Fig 2B). In addition, while LPS did not alter H3K9 acetylation, acetate supplementation increased H3K9 acetylation by 2-fold in the presence and absence of the LPS challenge (Fig 2C). This finding differs from that found using microglia where LPS reduces H3K9 acetylation by 50% (Soliman et al., 2012a). Measuring cellular release of LDH as an index of cell death showed no difference in cell survival between groups (Fig 3A). To assess the possibility of treatment inducing apoptosis, a TUNEL assay was performed to determine DNA fragmentation. These experiments showed no DNA fragmentation or apoptosis occurring with any of the treatments (Fig 3B). These data suggest that changes found following LPS challenge and/or acetate treatment are not due to changes in cell survival or the induction of apoptosis. Further, acetate treatment induced H3K9 hyperacetylation in astrocytes similar to that found in vivo using oral glyceryl triacetate in a rat model of neuroinflammation (Soliman et al., 2012b) and in LPS-stimulated microglia in vitro (Soliman et al., 2012a), without altering acetyl-CoA synthetase protein levels.
Fig. 2.

Histone acetylation in primary mouse astrocyte cell cultures stimulated for 4 hr with LPS 6.25 ng/ml and/or 12 mM sodium acetate, with 12 mM NaCl as control. Panel A shows representative images of the Western blots. Panels B and C show the optical densities of acetyl-CoA synthetase enzyme normalized to the loading control α-tubulin, and acetylated H3K9 normalized to total H3, respectively. All values represent the mean fold change relative to control ± SD. Statistical significance (* = compared to NaCl, n = 4, 5, 4, and 5) was set at p ≤ 0.05, as determined by One Way ANOVA followed by Tukey’s post-hoc test.
Fig. 3.

Cell viability in primary mouse astrocyte cell cultures stimulated for 4 hr with LPS 6.25 ng/ml and/or 12 mM sodium acetate, with 12 mM NaCl as control. Panel A shows the quantification of the ratio of secreted LDH in the media to total cellular LDH. All values represent the mean fold change relative to control ± SD (n = 6). Statistical significance was set at p 0.05 as determined by One Way ANOVA followed by Tukey’s post-hoc test. Panel B shows confocal microscopic images of primary astrocyte cultures used for TUNEL assay (n = 5), with fluorescin-dUTP-labeled fragmented DNA of apoptotic cells (green), the nuclei counterstained by propidium iodide (red), and co-localization of the fragmented DNA and nuclei (yellow). Positive control group received treatment with DNase I 50 Units/ml (n = 4).
Acetate reverses LPS-induced increases in the pro-inflammatory cytokine proteins, but not mRNA
To determine the ability of acetate to reverse pro-inflammatory cytokine production in vitro similar to that found in whole brain in vivo (Soliman et al., 2012b) and in LPS-stimulated microglia in vitro (Soliman et al., 2012a), cell lysates were analyzed using Western blot to probe for pro-IL-1β, IL-6 and TNF-α (Fig 4A). We found that LPS increased pro-IL-1β, TNF-α production, but not IL-6, by about 3.5 and 2.5-fold, respectively (Fig 4B, D and F). Acetate treatment only partially attenuated LPS-induced increases of pro-IL-1β (Fig 4B), completely reversed TNF-α (Fig 4F), and decreased IL-6 basal levels by about 40% (Fig 4D). Quantifying mRNA using qrt-PCR showed that LPS increased the mRNA levels of the three pro-inflammatory cytokines measured which were not altered by acetate treatment (Fig 4C and E) with the exception of TNF-α mRNA which was only partially attenuated (Fig 4G).
Fig. 4.

Pro-inflammatory cytokines pro-IL-1β, IL-6, and TNF-α in primary mouse astrocyte cell cultures stimulated for 4 hr with LPS 6.25 ng/ml and/or 12 mM sodium acetate, with 12 mM NaCl as control. Panel A shows representative images of the Western blots. Panels B, D and F show the optical densities of the pro-inflammatory cytokines pro-IL-1β, IL-6 and TNF-α, respectively, normalized to the loading control α-tubulin (n = 6). Panels C, E and G show the changes in the mRNA levels of the pro-inflammatory cytokines IL-1β, IL-6 and TNF-α, quantified by qrt-PCR and normalized to β-actin (n = 6). All values represent the mean fold change relative to control ± SD. Statistical significance (a = compared to NaCl, and b = compared to LPS) was set at p ≤ 0.05, as determined by One Way ANOVA followed by Tukey’s post-hoc test.
Acetate modulates the expression of the anti-inflammatory cytokines
Anti-inflammatory cytokines are produced during the inflammatory response as a part of self-checking mechanisms to control the inflammatory response. We measured the effect of acetate treatment on the expression levels of the anti-inflammatory cytokines TGF-β1, IL-4, and IL-10 in LPS-stimulated primary astrocytes (Fig 5A), that were detected as protein bands corresponding to the molecular weights of around 25, 30 and 18 kDa, respectively. Acetate treatment reversed the LPS-induced reduction in the TGF-β1 mRNA and protein, and IL-4 protein (Fig 5B, C and D) and upregulated IL-4 mRNA by 3-fold and protein by 1.3-fold (Fig 5D and E). Interestingly, IL-10 mRNA was increased by about 4-fold with acetate treatment, while the protein levels were decreased by 2-fold, which were not altered by LPS (Fig 5F and G). We speculate that this may be due to either interference with mRNA translation, increased protein turnover, or enhanced secretion into the extracellular milieu. Regardless, these data suggest that acetate modulates pro- and anti-inflammatory cytokine in primary astrocytes towards a more anti-inflammatory state similar to that found with microglia (Soliman et al., 2012a).
Fig. 5.

Anti-inflammatory cytokines TGF-β1, IL-4, and IL-10 in primary mouse astrocyte cell cultures stimulated for 4 hr with LPS 6.25 ng/ml and/or 12 mM sodium acetate, with 12 mM NaCl as control. Panel A shows representative images of the Western blots. Panels B, D and F show the optical densities of the anti-inflammatory cytokines TGF-β1, IL-4, and IL-10, respectively, normalized to the loading control α-tubulin (n = 6). Panels C, E and G show the changes in the mRNA levels of the anti-inflammatory cytokines TGF-β1, IL-4, and IL-10, quantified by qrt-PCR and normalized to β-actin (n = 6). All values represent the mean fold change relative to control ± SD. Statistical significance (a = compared to NaCl, and b = compared to LPS) was set at p ≤ 0.05, as determined by One Way ANOVA followed by Tukey’s post-hoc test.
Acetate reverses LPS-induced MAPK p38 phosphorylation and decreases ERK1/2 phosphorylation
MAPK inflammatory signaling is involved in neuroinflammation and pro-inflammatory cytokine production. Specific lysine acetylation activates a MAPK phosphatase-1, which deactivates MAPK. Based on this, we measured the effect of acetate on LPS-induced MAPK phosphorylation. Whole cell lysates were used for Western blot analysis, and p38, phosphorylated p38, JNK, phosphorylated JNK, ERK1/2, and phosphorylated ERK1/2 were detected as protein bands corresponding with the molecular weights of around 38, 38, 46 and 54, 46 and 54, 42 and 46, and 42 and 46 kDa, respectively (Fig 6A). LPS increased p38 phosphorylation by 1.5-fold which was returned to control level with acetate treatment (Fig 6B). Neither LPS nor acetate had an effect on JNK phosphorylation (Fig 6C). While LPS did not alter ERK1/2 phosphorylation, the basal levels of phosphorylated ERK1/2 were decreased by acetate treatment in the presence and absence of LPS by almost 2-fold (Fig 6D). These data demonstrate that acetate and LPS selectively modulate MAPK phosphorylation in primary astrocytes which can potentially explain the anti-inflammatory effect of acetate treatment in astrocytes. This response differs from that found in microglia where LPS increases MAPK p38 and JNK phosphorylation but is not altered by acetate treatment, and acetate increases phosphorylated ERK1/2 only in the presence of LPS (Soliman et al., 2012a).
Fig. 6.
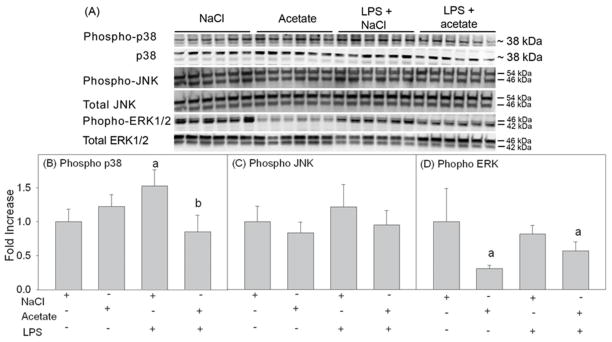
Phosphorylation state of MAPK p38, JNK and ERK1/2 in primary mouse astrocyte cell cultures stimulated for 4 hr with LPS 6.25 ng/ml and/or 12 mM sodium acetate, with 12 mM NaCl as control. Panel A shows representative images of the Western blots. Panels B, C and D show the optical densities of phosphorylated MAPK p38, JNK and ERK1/2 normalized to the loading controls MAPK p38, JNK and ERK1/2, respectively (n = 6). All values represent the mean fold change relative to control ± SD. Statistical significance (a = compared to NaCl and b = compared to LPS) was set at p ≤ 0.05, as determined by One Way ANOVA followed by Tukey’s post-hoc test.
Acetate reverses LPS-induced elevated NF-κB p65 protein level, and decreases the basa levels of p65 phosphorylation at serine 536
NF-κB is a major factor in neuroinflammation and the biosynthesis of pro-inflammatory mediators. NF-κB p65 subunit is regulated by a wide range of post-translational modifications which have diverse functional consequences (Huang et al., 2010). To quantify the effect that acetate treatment and LPS had on NF-κB signaling, we quantified the total protein levels and selected post-translational modification of NF-κB p65 in total cell lysates using Western blot analysis. Total NF-κB p65 protein, acetylated p65 at lysine 310, and phosphorylated p65 at serine residues 468 and 536 were detected as protein bands corresponding to the molecular weight 65 kDa (Fig 7A). We found that LPS increased the protein levels of NF-κB p65 by 1.5-fold, and acetate treatment reduced it back to control levels (Fig 7B). Moreover, acetate treatment reduced the basal level of phosphorylated p65 at serine 536 (Fig 7D). Neither LPS nor acetate treatment altered acetylated p65 at lysine 310 or phosphorylated p65 at serine 468 (Fig 7C and E). These data suggest that acetate treatment can alter NF-κB p65 protein and post-translational modification in LPS-challenged primary astrocytes.
Fig. 7.

Changes in the protein level and phosphorylation and acetylation states of NF-κB p65 in primary mouse astrocyte cell cultures stimulated for 4 hr with LPS 6.25 ng/ml and/or 12 mM sodium acetate, with 12 mM NaCl as control. Panel A shows representative images of the Western blots. Panel B shows the optical density of total NF-κB p65 normalized to the loading control α-tubulin. Panels C, D and E show the optical densities of acetylated p65 at lysine 310, phosphorylated p65 at S536, and phosphorylated p65 at serine 468, respectively (n = 6). All values represent the mean fold change relative to control ± SD. Statistical significance (a = compared to NaCl and b = compared to LPS) was set at p ≤ 0.05, as determined by One Way ANOVA followed by Tukey’s post-hoc test.
Acetate reverses LPS-induced elevated C/EBPβ protein level
Because C/EBPβ is involved in the regulation and expression of certain pro-inflammatory cytokines in astrocytes (Ramji and Foka, 2002), we measured the effect that acetate treatment had on C/EBPβ protein levels. Using total cell lysates for Western blot analysis, C/EBPβ was detected as protein bands corresponding to the molecular weight of 51 kDa (Fig 8A). LPS treatment increased C/EBPβ by 2-fold, which was decreased by acetate treatment to control levels (Fig 8B), while acetate treatment alone did not alter C/EBPβ protein levels. This data, together with the changes in MAPK phosphorylation and NF- κB phosphorylation and protein levels suggest that acetate treatment in primary astrocyte cell cultures is largely associated with a disruption in MAPK signaling which may result in the attenuation of TNF-α expression.
Fig. 8.

level of C/EBPβ in primary mouse astrocytes cultures stimulated for 4 hr with LPS 6.25 ng/ml and/or 12 mM sodium acetate, with 12 mM NaCl as control. Panel A shows representative images of the Western blots. Panel B shows the optical density of C/EBPβ normalized to the loading control α-tubulin (n = 6). All values represent the mean fold change relative to control ± SD. Statistical significance (a = compared to NaCl) was set at p ≤ 0.05, as determined by One Way ANOVA followed by Tukey’s post-hoc test.
Discussion
Astrocytes have a number of physiological functions including regulation of blood flow, fluid, ion and transmitter homeostasis, modulation of synaptic functions, and supporting the blood brain barrier (Sofroniew and Vinters, 2010). The immune functions of the astrocytes are recognized as astrocytes are an abundant source of pro-inflammatory cytokines and other inflammatory mediators in brain injury and infection (Dong and Benveniste, 2001; Gorina et al., 2009; Sofroniew and Vinters, 2010) and reactive astrogliosis is a pathological component of numerous neurological disorders (Hamby and Sofroniew, 2010; Sofroniew and Vinters, 2010). In this study, we demonstrated that acetate treatment induced H3K9 hyperacetylation, decreased ERK1/2 phosphorylation, IL-6, and NF-κB p65 phosphorylation at serine 536, and reversed the LPS-induced MAPK p38 phosphorylation, elevated NF-κB p65 and C/EBPβ protein levels and pro-inflammatory cytokines IL-1β and TNF-α in primary astrocytes in vitro. In addition, acetate treatment reversed LPS-induced reduction in TGF-β1 and IL-4. These results suggest that acetate treatment has a net anti-inflammatory effect in LPS-stimulated astrocytes that is largely associated with a disruption in MAPK and NF-κB signaling.
The differential effect of acetate treatment on mRNA and protein levels suggests that the reduction in pro-inflammatory cytokines is due to a disruption in mRNA translation rather than gene transcription, or enhanced protein turnover as discussed earlier (Soliman et al., 2012a). Acetate-treatment in vivo induces a number of site-specific histone H3 and H4 acetylation changes (Soliman and Rosenberger, 2011; Soliman et al., 2012b). However, the acetylation of only H3K9 is decreased by inflammation and increased with acetate supplementation (Soliman et al., 2012b). In addition, H3K9 hypoacetylation is associated with neuroinflammation and glial activation (Zhang et al., 2008; Govindarajan et al., 2011; Silva et al., 2012). Therefore, in this in vitro astrocyte model as well as in microglia (Soliman et al., 2012a), we focused on H3K9 acetylation that is altered by increasing the acetyl-CoA metabolism. Since H3K9 hyperacetylation is associated with enhanced gene expression (Rice and Allis, 2001), it is likely that acetate treatment-induced H3K9 hyperacetylation is linked to the upregulation of the anti-inflammatory cytokines TGF-β1 and IL-4 transcription. Nevertheless, it is possible that acetate alters gene expression by inducing the acetylation of non-histone transcription factors like transcription factor (TF)IIEβ and TFIIF (Imhof et al., 1997), or other site-specific DNA-binding factors subject to acetylation (Bannister and Miska, 2000). H3K9 is modified also by methylation which is associated with repressed gene transcription in contrast to H3K9 acetylation (Rice and Allis, 2001). Methylation and acetylation mutually influence one another. For example, hypoacetylated chromatin is enriched in methylated H4, whereas chromatin areas with increased H4 acetylation contain more methylated H3 (Annunziato et al., 1995). The exact functional outcome that histone methylation and acetylation regarding the control of gene expression and the individual genes involved is not known. However, these results suggest that post-translational acetylation of histones following acetate treatment is associated with a shift in cytokine balance to a more anti-inflammatory state. LPS did not alter H3K9 acetylation in astrocytes unlike that found in a rat model of neuroinflammation (Soliman et al., 2012b) and in LPS-stimulated primary and BV-2 microglia (Soliman et al., 2012a). The lack of an effect on H3K9 acetylation with LPS may be attributed to a cell-type specific effect of an increase in acetyl-CoA metabolism in the brain. Nevertheless, LPS stimulation of astrocytes increases pro-inflammatory cytokine production which agrees with previous reports (Lu et al., 2010; Gorina et al., 2011).
Acetate has long been used as a substrate to study glial metabolism (Thoren et al., 2005; Wyss et al., 2009; Wyss et al., 2011), particularly because it is preferentially utilized by astrocytes (Muir et al., 1986; Waniewski and Martin, 1998; Hosoi et al., 2009). Peripherally-derived acetate crosses the blood brain barrier by simple diffusion, and the rate-limiting step of acetate utilization is its activation by acetyl-CoA synthetase to form acetyl-CoA (Oldendorf, 1973; Terasaki, 1992). The transport of acetate into astrocytes is through a carrier that has similar biochemical properties to monocarboxylate transporters (MCT) (Garcia et al., 1994b; Garcia et al., 1994a). MCT1 and 4 are expressed in astrocytes while MCT2 is expressed in the neurons (Broer et al., 1997; Pierre et al., 2000), which can differ based on age (Rafiki et al., 2003). The presence of different MCT isoforms in astrocytes and neurons may explain the differential utilization of acetate by astrocytes (Waniewski and Martin, 1998). Acetate-derived acetyl-CoA can be used as a substrate for ketone bodies, fatty acids and cholesterol biosynthesis, and oxidation in Krebs cycle for energy generation (McGarry and Foster, 1980; Des Rosiers et al., 1991; Deutsch et al., 2002; Fukao et al., 2004). Acetyltransferases use acetyl-CoA as acetyl donor for post-translational acetylation reactions on lysine and arginine residues which can lead to structural and functional consequences in proteins. Non-histone substrates include transcription factors, nuclear import factors, and cytoskeletal proteins (Polevoda and Sherman, 2002; Glozak et al., 2005). The functional consequences of acetylation depend on where exactly within the protein acetylation takes place. For example, NF-κB p65 acetylation at lysines 218, 221 and 310 increases nuclear localization, while acetylation at 122 and 123 reduces the binding affinity of p65 to DNA which promotes IκB-p65 interaction and nuclear export (Chen et al., 2002; Huang et al., 2010). We have demonstrated that acetate treatment decreases the basal levels of NF-κB p65 phosphorylation at serine 536 while not altering p65 phosphorylation at serine 468 or acetylation at lysine 310. In this regard, phosphorylation of serine 536 lowers the affinity of p65 to IκBα leading to NF- κB translocation into the nucleus and enhanced activity (Bohuslav et al., 2004; Buss et al., 2004). Because p65 phosphorylation at serine 536 stimulates subsequent acetylation at lysine 310 (Chen et al., 2005; Hoberg et al., 2006), may explain why acetate does not increase acetylation at lysine 310 in astrocytes, unlike that found in microglia (Soliman et al., 2012a). Because acetate supplementation did not reduce the mRNA expression of IL-6 and IL-1β which are regulated by NF-κB and C/EBPβ, we speculate that acetate-mediated changes in NF-κB p65 and C/EBPβ protein levels are not be paralleled by equivalent reduction in the function of these transcription factors. This underscores the need for future studies to determine the functional effect of acetate-induced changes in NF-κB p65 phosphorylation in terms of IκB binding, nuclear translocation, DNA binding, and transcriptional activity. The goal of which is to determine whether the acetate-induced reduction in NF-κB p65 protein and phosphorylation and C/EBPβ protein levels are associated with a parallel decrease in their transcriptional activity. Alternatively, the lack of effect of acetate treatment on the transcription of pro-inflammatory cytokines IL-6 and IL-1β may potentially be due to adaptive compensation of MAPK, NF-κB p65, and C/EBPβ by other NF-κB and C/EBP isoforms or transcription factors including nuclear factor of activated T cells (NFAT) (Fernandez et al., 2007) and activated protein-1 (AP-1) (Schonthaler et al., 2011).
MAPK phosphatase-1 is activated by certain lysine acetylation which leads to inactivation of MAPK signaling pathway, providing an important link between acetylation and phosphorylation in the regulation of neuroinflammation (Cao et al., 2008). In this study, we showed that acetate reduces LPS-induced MAPK p38 phosphorylation and basal level phosphorylation of ERK1/2 which may be attributable to acetylation of MAPK phosphatase-1. The effect of acetate on MAPK activation is important given the role that MAPK signaling has in neuroinflammation. In this regard, interferon-β reduces traumatic spinal cord injury-induced ERK hyperphosphorylation that is associated with functional recovery (Ito et al., 2009). Inhibition of MAPK p38 and ERK reduces edema and the inflammatory mediator matrix metalloproteinase-9 in brain trauma (Mori et al., 2002), the infarct size (Sugino et al., 2000) and iNOS, TNF-α, and cyclooxygenase-2 expression (Piao et al., 2003) in ischemia, and reduces neurological deficits following transient (Legos et al., 2001) and permanent (Barone et al., 2001) ischemia.
Glial communication plays a crucial role in sustaining a neuroinflammatory response that can lead to neuronal bystander death if left unchecked (Streit et al., 1999; Hamby and Sofroniew, 2010; Tian et al., 2012). This crosstalk can involve beneficial effects. For example, astrocytic factors upregulate microglia-derived anti-oxidants and thus reduce microglial reactive oxygen species generation (Min et al., 2006). Likewise, astrocyte-conditioned media rapidly reduce interferon-γ-stimulated microglia-derived inflammatory mediators such as iNOS (Kim et al., 2010) and stimulates microglial release of the pro-survival mediator brain-derived neurotropic factor (Yang et al., 2012). Downregulating the astrocytes-derived inflammatory response protects neurons from the potential of excessive uncontrolled pro-inflammatory cytokines (Hamby and Sofroniew, 2010). In this regard, acetate treatment reduces pro-inflammatory cytokine levels in a rat model of neuroinflammation (Soliman et al., 2012b), in LPS-stimulated microglia (Soliman et al., 2012a), as well as in primary astrocyte cultures (the current study), which may explain the attenuation of LPS-induced glial activation observed with acetate supplementation in vivo (Reisenauer et al., 2011).
We have observed many neuroglial cell type-specific similarities and differences in terms of their response to acetate treatment and LPS challenge. Similarities include acetate treatment reducing LPS-induced microglial and astrocyte activation in vivo, increasing H3K9 acetylation, upregulating anti-inflammatory cytokine IL-4 and TGF-β1 mRNA, and returning LPS-induced NF-κB p65 protein levels to control levels in both cell types in vitro. Moreover, acetate treatment reduces pro-inflammatory cytokines IL-1β, IL-6 and TNF-α protein but not mRNA in both cell types, suggesting that treatment decreases pro-inflammatory cytokine levels by possibly interfering with translation of mRNA rather than modulating transcription, or by stimulating protein turnover. Differences between the astroglial and microglia responses include LPS reducing H3K9 acetylation, increasing IL-6, and increasing JNK phosphorylation only in microglia. Moreover, whereas MAPK p38 phosphorylation is increased by LPS both in microglia and astrocytes, it is reduced by acetate treatment at 4 hr in astrocytes cell cultures only. Acetate treatment also increases NF-κB p65 acetylation at lysine 310 and reverses LPS-induced hyperphosphorylation at serine 468 only in microglial cell culture and decreases basal levels of phosphorylation at 536 only in astrocyte cell cultures. ERK1/2 phosphorylation increases by acetate treatment only in the presence of LPS in microglia cultures, and is reduced beyond control levels with acetate treatment in astrocyte cultures (Reisenauer et al., 2011; Soliman et al., 2012a; Soliman et al., 2012b). All of which demonstrate clear cell type-specific responses to acetate treatment despite the similar overall anti-inflammatory outcome. This is further supported by other reports showing the distinctive responses of astrocytes and microglia to injury, infection, inflammation and other anti-inflammatory agents (Lee et al., 2010; Lu et al., 2010).
Because acetate treatment reverses the LPS-induced MAPK p38 phosphorylation and decreases the basal levels of phosphorylated ERK1/2 in astrocyte and not in microglial cultures, we speculate that interruption of MAPK signaling has a more dominant role in the anti-inflammatory effect of acetate treatment in astrocytes. Conversely, because H3K9 acetylation is reduced by LPS and reversed by acetate treatment in microglia, it is possible that the epigenetic mechanisms are more influential in microglia. This premise is promising for the potential of selectively downregulating MAPK signaling in only one cell type without impeding its physiological functions in other cell types, as opposed to universal inhibitors of MAPK signaling. This adds to the advantage of acetate treatment being well-tolerated and with minimal observed biochemical abnormalities both in animals (Mathew et al., 2005) and humans (Madhavarao et al., 2009; Segel et al., 2011). The discrepancy in the glial responses to acetate treatment, which partially reduces LPS-mediated microglial activation while completely reducing astrocyte activation in vivo, may be attributed to astrocytes being more efficient at the uptake and/or utilization of acetate.
In summary, acetate treatment in astrocytes results in a net anti-inflammatory effect manifested by downregulating pro-inflammatory cytokine release, upregulating the expression of anti-inflammatory cytokines, and disrupting MAPK p38 signaling. These correlate with enhanced H3K9 acetylation and decreased NF-κB p65 protein level and phosphorylation that are known to be associated with anti-inflammatory outcomes. To further understand the mechanisms underlying the anti-inflammatory effect of acetate treatment, future studies are needed to determine the effect of acetate treatment on NF-κB functionality in terms of nuclear translocation, DNA binding and transcriptional activity in both astrocytes and microglia.
Acknowledgments
This publication was made possible by Grant Number 5P20RR017699 from the National Center for Research Resources (NCRR), a component of the National Institutes of Health (NIH). We would like to thank Dr. Bhanu Dasari for his assistance in using the confocal microscope in the Edward C. Carlson Image and Image Analysis Core Facility.
Footnotes
Conflict of interest:
The authors declare that they have no conflict of interest.
References
- Annunziato AT, Eason MB, Perry CA. Relationship between methylation and acetylation of arginine-rich histones in cycling and arrested HeLa cells. Biochemistry. 1995;34:2916–2924. doi: 10.1021/bi00009a023. [DOI] [PubMed] [Google Scholar]
- Ariyannur PS, Moffett JR, Madhavarao CN, Arun P, Vishnu N, Jacobowitz DM, Hallows WC, Denu JM, Namboodiri AM. Nuclear-cytoplasmic localization of acetyl coenzyme a synthetase-1 in the rat brain. J Comp Neurol. 2010;518:2952–2977. doi: 10.1002/cne.22373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arun P, Ariyannur PS, Moffett JR, Xing G, Hamilton K, Grunberg NE, Ives JA, Namboodiri AM. Metabolic acetate therapy for the treatment of traumatic brain injury. J Neurotrauma. 2010a;27:293–298. doi: 10.1089/neu.2009.0994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arun P, Madhavarao CN, Moffett JR, Hamilton K, Grunberg NE, Ariyannur PS, Gahl WA, Anikster Y, Mog S, Hallows WC, Denu JM, Namboodiri AM. Metabolic acetate therapy improves phenotype in the tremor rat model of Canavan disease. J Inherit Metab Dis. 2010b;33:195–210. doi: 10.1007/s10545-010-9100-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bannister AJ, Miska EA. Regulation of gene expression by transcription factor acetylation. Cell Mol Life Sci. 2000;57:1184–1192. doi: 10.1007/PL00000758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barone FC, Irving EA, Ray AM, Lee JC, Kassis S, Kumar S, Badger AM, White RF, McVey MJ, Legos JJ, Erhardt JA, Nelson AH, Ohlstein EH, Hunter AJ, Ward K, Smith BR, Adams JL, Parsons AA. SB 239063, a second-generation p38 mitogen-activated protein kinase inhibitor, reduces brain injury and neurological deficits in cerebral focal ischemia. J Pharmacol Exp Ther. 2001;296:312–321. [PubMed] [Google Scholar]
- Bohuslav J, Chen LF, Kwon H, Mu Y, Greene WC. p53 induces NF-kappaB activation by an IkappaB kinase-independent mechanism involving phosphorylation of p65 by ribosomal S6 kinase 1. J Biol Chem. 2004;279:26115–26125. doi: 10.1074/jbc.M313509200. [DOI] [PubMed] [Google Scholar]
- Brissette CA, Houdek HM, Floden AM, Rosenberger TA. Acetate supplementation reduces microglia activation and brain interleukin-1beta levels in a rat model of Lyme neuroborreliosis. J Neuroinflammation. 2012;9:249. doi: 10.1186/1742-2094-9-249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broer S, Rahman B, Pellegri G, Pellerin L, Martin JL, Verleysdonk S, Hamprecht B, Magistretti PJ. Comparison of lactate transport in astroglial cells and monocarboxylate transporter 1 (MCT 1) expressing Xenopus laevis oocytes. Expression of two different monocarboxylate transporters in astroglial cells and neurons. J Biol Chem. 1997;272:30096–30102. doi: 10.1074/jbc.272.48.30096. [DOI] [PubMed] [Google Scholar]
- Buss H, Dorrie A, Schmitz ML, Hoffmann E, Resch K, Kracht M. Constitutive and interleukin-1-inducible phosphorylation of p65 NF-{kappa}B at serine 536 is mediated by multiple protein kinases including I{kappa}B kinase (IKK)-{alpha}, IKK{beta}, IKK{epsilon}, TRAF family member-associated (TANK)-binding kinase 1 (TBK1), and an unknown kinase and couples p65 to TATA-binding protein-associated factor II31-mediated interleukin-8 transcription. J Biol Chem. 2004;279:55633–55643. doi: 10.1074/jbc.M409825200. [DOI] [PubMed] [Google Scholar]
- Cao W, Bao C, Padalko E, Lowenstein CJ. Acetylation of mitogen-activated protein kinase phosphatase-1 inhibits Toll-like receptor signaling. J Exp Med. 2008;205:1491–1503. doi: 10.1084/jem.20071728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen LF, Mu Y, Greene WC. Acetylation of RelA at discrete sites regulates distinct nuclear functions of NF-kappaB. Embo J. 2002;21:6539–6548. doi: 10.1093/emboj/cdf660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen LF, Williams SA, Mu Y, Nakano H, Duerr JM, Buckbinder L, Greene WC. NF-kappaB RelA phosphorylation regulates RelA acetylation. Mol Cell Biol. 2005;25:7966–7975. doi: 10.1128/MCB.25.18.7966-7975.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denes A, Thornton P, Rothwell NJ, Allan SM. Inflammation and brain injury: acute cerebral ischaemia, peripheral and central inflammation. Brain Behav Immun. 2010;24:708–723. doi: 10.1016/j.bbi.2009.09.010. [DOI] [PubMed] [Google Scholar]
- Des Rosiers C, David F, Garneau M, Brunengraber H. Nonhomogeneous labeling of liver mitochondrial acetyl-CoA. J Biol Chem. 1991;266:1574–1578. [PubMed] [Google Scholar]
- Deutsch J, Rapoport SI, Rosenberger TA. Coenzyme A and short-chain acyl-CoA species in control and ischemic rat brain. Neurochem Res. 2002;27:1577–1582. doi: 10.1023/a:1021614422668. [DOI] [PubMed] [Google Scholar]
- Dhawan G, Floden AM, Combs CK. Amyloid-beta oligomers stimulate microglia through a tyrosine kinase dependent mechanism. Neurobiol Aging. 2012;33:2247–2261. doi: 10.1016/j.neurobiolaging.2011.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong Y, Benveniste EN. Immune function of astrocytes. Glia. 2001;36:180–190. doi: 10.1002/glia.1107. [DOI] [PubMed] [Google Scholar]
- Fernandez AM, Fernandez S, Carrero P, Garcia-Garcia M, Torres-Aleman I. Calcineurin in reactive astrocytes plays a key role in the interplay between proinflammatory and anti-inflammatory signals. J Neurosci. 2007;27:8745–8756. doi: 10.1523/JNEUROSCI.1002-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujino T, Kondo J, Ishikawa M, Morikawa K, Yamamoto TT. Acetyl-CoA synthetase 2, a mitochondrial matrix enzyme involved in the oxidation of acetate. J Biol Chem. 2001;276:11420–11426. doi: 10.1074/jbc.M008782200. [DOI] [PubMed] [Google Scholar]
- Fukao T, Lopaschuk GD, Mitchell GA. Pathways and control of ketone body metabolism: on the fringe of lipid biochemistry. Prostaglandins Leukot Essent Fatty Acids. 2004;70:243–251. doi: 10.1016/j.plefa.2003.11.001. [DOI] [PubMed] [Google Scholar]
- Garcia CK, Li X, Luna J, Francke U. cDNA cloning of the human monocarboxylate transporter 1 and chromosomal localization of the SLC16A1 locus to 1p13.2–p12. Genomics. 1994a;23:500–503. doi: 10.1006/geno.1994.1532. [DOI] [PubMed] [Google Scholar]
- Garcia CK, Goldstein JL, Pathak RK, Anderson RG, Brown MS. Molecular characterization of a membrane transporter for lactate, pyruvate, and other monocarboxylates: implications for the Cori cycle. Cell. 1994b;76:865–873. doi: 10.1016/0092-8674(94)90361-1. [DOI] [PubMed] [Google Scholar]
- Glozak MA, Sengupta N, Zhang X, Seto E. Acetylation and deacetylation of non-histone proteins. Gene. 2005;363:15–23. doi: 10.1016/j.gene.2005.09.010. [DOI] [PubMed] [Google Scholar]
- Gorina R, Font-Nieves M, Marquez-Kisinousky L, Santalucia T, Planas AM. Astrocyte TLR4 activation induces a proinflammatory environment through the interplay between MyD88-dependent NFkappaB signaling, MAPK, and Jak1/Stat1 pathways. Glia. 2011;59:242–255. doi: 10.1002/glia.21094. [DOI] [PubMed] [Google Scholar]
- Gorina R, Santalucia T, Petegnief V, Ejarque-Ortiz A, Saura J, Planas AM. Astrocytes are very sensitive to develop innate immune responses to lipid-carried short interfering RNA. Glia. 2009;57:93–107. doi: 10.1002/glia.20738. [DOI] [PubMed] [Google Scholar]
- Govindarajan N, Agis-Balboa RC, Walter J, Sananbenesi F, Fischer A. Sodium butyrate improves memory function in an Alzheimer’s disease mouse model when administered at an advanced stage of disease progression. J Alzheimers Dis. 2011;26:187–197. doi: 10.3233/JAD-2011-110080. [DOI] [PubMed] [Google Scholar]
- Hamby ME, Sofroniew MV. Reactive astrocytes as therapeutic targets for CNS disorders. Neurotherapeutics. 2010;7:494–506. doi: 10.1016/j.nurt.2010.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanisch UK, Kettenmann H. Microglia: active sensor and versatile effector cells in the normal and pathologic brain. Nat Neurosci. 2007;10:1387–1394. doi: 10.1038/nn1997. [DOI] [PubMed] [Google Scholar]
- Helmy A, De Simoni MG, Guilfoyle MR, Carpenter KL, Hutchinson PJ. Cytokines and innate inflammation in the pathogenesis of human traumatic brain injury. Prog Neurobiol. 2011;95:352–372. doi: 10.1016/j.pneurobio.2011.09.003. [DOI] [PubMed] [Google Scholar]
- Hoberg JE, Popko AE, Ramsey CS, Mayo MW. IkappaB kinase alpha-mediated derepression of SMRT potentiates acetylation of RelA/p65 by p300. Mol Cell Biol. 2006;26:457–471. doi: 10.1128/MCB.26.2.457-471.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosoi R, Matsuyama Y, Hirose S, Koyama Y, Matsuda T, Gee A, Inoue O. Characterization of (14)C-acetate uptake in cultured rat astrocytes. Brain Res. 2009;1253:69–73. doi: 10.1016/j.brainres.2008.11.068. [DOI] [PubMed] [Google Scholar]
- Huang B, Yang XD, Lamb A, Chen LF. Posttranslational modifications of NF-kappaB: another layer of regulation for NF-kappaB signaling pathway. Cell Signal. 2010;22:1282–1290. doi: 10.1016/j.cellsig.2010.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imhof A, Yang XJ, Ogryzko VV, Nakatani Y, Wolffe AP, Ge H. Acetylation of general transcription factors by histone acetyltransferases. Curr Biol. 1997;7:689–692. doi: 10.1016/s0960-9822(06)00296-x. [DOI] [PubMed] [Google Scholar]
- Ito M, Natsume A, Takeuchi H, Shimato S, Ohno M, Wakabayashi T, Yoshida J. Type I interferon inhibits astrocytic gliosis and promotes functional recovery after spinal cord injury by deactivation of the MEK/ERK pathway. J Neurotrauma. 2009;26:41–53. doi: 10.1089/neu.2008.0646. [DOI] [PubMed] [Google Scholar]
- Johnston H, Boutin H, Allan SM. Assessing the contribution of inflammation in models of Alzheimer’s disease. Biochem Soc Trans. 2011;39:886–890. doi: 10.1042/BST0390886. [DOI] [PubMed] [Google Scholar]
- Kang W, Hebert JM. Signaling pathways in reactive astrocytes, a genetic perspective. Mol Neurobiol. 2011;43:147–154. doi: 10.1007/s12035-011-8163-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasai T, Inada K, Takakuwa T, Yamada Y, Inoue Y, Shimamura T, Taniguchi S, Sato S, Wakabayashi G, Endo S. Anti-inflammatory cytokine levels in patients with septic shock. Res Commun Mol Pathol Pharmacol. 1997;98:34–42. [PubMed] [Google Scholar]
- Kim JH, Min KJ, Seol W, Jou I, Joe EH. Astrocytes in injury states rapidly produce anti-inflammatory factors and attenuate microglial inflammatory responses. J Neurochem. 2010;115:1161–1171. doi: 10.1111/j.1471-4159.2010.07004.x. [DOI] [PubMed] [Google Scholar]
- Ledeboer A, Breve JJ, Poole S, Tilders FJ, Van Dam AM. Interleukin-10, interleukin-4, and transforming growth factor-beta differentially regulate lipopolysaccharide-induced production of pro-inflammatory cytokines and nitric oxide in co-cultures of rat astroglial and microglial cells. Glia. 2000;30:134–142. doi: 10.1002/(sici)1098-1136(200004)30:2<134::aid-glia3>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- Lee S, Zhao YQ, Ribeiro-da-Silva A, Zhang J. Distinctive response of CNS glial cells in oro-facial pain associated with injury, infection and inflammation. Mol Pain. 2010;6:79. doi: 10.1186/1744-8069-6-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legos JJ, Erhardt JA, White RF, Lenhard SC, Chandra S, Parsons AA, Tuma RF, Barone FC. SB 239063, a novel p38 inhibitor, attenuates early neuronal injury following ischemia. Brain Res. 2001;892:70–77. doi: 10.1016/s0006-8993(00)03228-5. [DOI] [PubMed] [Google Scholar]
- Lehnardt S. Innate immunity and neuroinflammation in the CNS: the role of microglia in Toll-like receptor-mediated neuronal injury. Glia. 2010;58:253–263. doi: 10.1002/glia.20928. [DOI] [PubMed] [Google Scholar]
- Lu X, Ma L, Ruan L, Kong Y, Mou H, Zhang Z, Wang Z, Wang JM, Le Y. Resveratrol differentially modulates inflammatory responses of microglia and astrocytes. J Neuroinflammation. 2010;7:46. doi: 10.1186/1742-2094-7-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madhavarao CN, Arun P, Anikster Y, Mog SR, Staretz-Chacham O, Moffett JR, Grunberg NE, Gahl WA, Namboodiri AM. Glyceryl triacetate for Canavan disease: a low-dose trial in infants and evaluation of a higher dose for toxicity in the tremor rat model. J Inherit Metab Dis. 2009;32:640–650. doi: 10.1007/s10545-009-1155-3. [DOI] [PubMed] [Google Scholar]
- Mathew R, Arun P, Madhavarao CN, Moffett JR, Namboodiri MA. Progress toward acetate supplementation therapy for Canavan disease: glyceryl triacetate administration increases acetate, but not N-acetylaspartate, levels in brain. J Pharmacol Exp Ther. 2005;315:297–303. doi: 10.1124/jpet.105.087536. [DOI] [PubMed] [Google Scholar]
- McGarry JD, Foster DW. Regulation of hepatic fatty acid oxidation and ketone body production. Annu Rev Biochem. 1980;49:395–420. doi: 10.1146/annurev.bi.49.070180.002143. [DOI] [PubMed] [Google Scholar]
- Merson TD, Binder MD, Kilpatrick TJ. Role of cytokines as mediators and regulators of microglial activity in inflammatory demyelination of the CNS. Neuromolecular Med. 2010;12:99–132. doi: 10.1007/s12017-010-8112-z. [DOI] [PubMed] [Google Scholar]
- Min KJ, Yang MS, Kim SU, Jou I, Joe EH. Astrocytes induce hemeoxygenase-1 expression in microglia: a feasible mechanism for preventing excessive brain inflammation. J Neurosci. 2006;26:1880–1887. doi: 10.1523/JNEUROSCI.3696-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori T, Wang X, Aoki T, Lo EH. Downregulation of matrix metalloproteinase-9 and attenuation of edema via inhibition of ERK mitogen activated protein kinase in traumatic brain injury. J Neurotrauma. 2002;19:1411–1419. doi: 10.1089/089771502320914642. [DOI] [PubMed] [Google Scholar]
- Muir D, Berl S, Clarke DD. Acetate and fluoroacetate as possible markers for glial metabolism in vivo. Brain Res. 1986;380:336–340. doi: 10.1016/0006-8993(86)90231-3. [DOI] [PubMed] [Google Scholar]
- Munoz C, Carlet J, Fitting C, Misset B, Bleriot JP, Cavaillon JM. Dysregulation of in vitro cytokine production by monocytes during sepsis. J Clin Invest. 1991;88:1747–1754. doi: 10.1172/JCI115493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldendorf WH. Carrier-mediated blood-brain barrier transport of short-chain monocarboxylic organic acids. Am J Physiol. 1973;224:1450–1453. doi: 10.1152/ajplegacy.1973.224.6.1450. [DOI] [PubMed] [Google Scholar]
- Olson JK, Miller SD. Microglia initiate central nervous system innate and adaptive immune responses through multiple TLRs. J Immunol. 2004;173:3916–3924. doi: 10.4049/jimmunol.173.6.3916. [DOI] [PubMed] [Google Scholar]
- Pekny M, Nilsson M. Astrocyte activation and reactive gliosis. Glia. 2005;50:427–434. doi: 10.1002/glia.20207. [DOI] [PubMed] [Google Scholar]
- Piao CS, Kim JB, Han PL, Lee JK. Administration of the p38 MAPK inhibitor SB203580 affords brain protection with a wide therapeutic window against focal ischemic insult. J Neurosci Res. 2003;73:537–544. doi: 10.1002/jnr.10671. [DOI] [PubMed] [Google Scholar]
- Pierre K, Pellerin L, Debernardi R, Riederer BM, Magistretti PJ. Cell-specific localization of monocarboxylate transporters, MCT1 and MCT2, in the adult mouse brain revealed by double immunohistochemical labeling and confocal microscopy. Neuroscience. 2000;100:617–627. doi: 10.1016/s0306-4522(00)00294-3. [DOI] [PubMed] [Google Scholar]
- Polevoda B, Sherman F. The diversity of acetylated proteins. Genome Biol. 2002;3:0006.1–0006.6. doi: 10.1186/gb-2002-3-5-reviews0006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poli V. The role of C/EBP isoforms in the control of inflammatory and native immunity functions. J Biol Chem. 1998;273:29279–29282. doi: 10.1074/jbc.273.45.29279. [DOI] [PubMed] [Google Scholar]
- Poli V, Mancini FP, Cortese R. IL-6DBP, a nuclear protein involved in interleukin-6 signal transduction, defines a new family of leucine zipper proteins related to C/EBP. Cell. 1990;63:643–653. doi: 10.1016/0092-8674(90)90459-r. [DOI] [PubMed] [Google Scholar]
- Qian L, Flood PM, Hong JS. Neuroinflammation is a key player in Parkinson’s disease and a prime target for therapy. J Neural Transm. 2010;117:971–979. doi: 10.1007/s00702-010-0428-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rafiki A, Boulland JL, Halestrap AP, Ottersen OP, Bergersen L. Highly differential expression of the monocarboxylate transporters MCT2 and MCT4 in the developing rat brain. Neuroscience. 2003;122:677–688. doi: 10.1016/j.neuroscience.2003.08.040. [DOI] [PubMed] [Google Scholar]
- Ramji DP, Foka P. CCAAT/enhancer-binding proteins: structure, function and regulation. Biochem J. 2002;365:561–575. doi: 10.1042/BJ20020508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ransohoff RM, Perry VH. Microglial physiology: unique stimuli, specialized responses. Annu Rev Immunol. 2009;27:119–145. doi: 10.1146/annurev.immunol.021908.132528. [DOI] [PubMed] [Google Scholar]
- Reisenauer CJ, Bhatt DP, Mitteness DJ, Slanczka ER, Gienger HM, Watt JA, Rosenberger TA. Acetate supplementation attenuates lipopolysaccharide-induced neuroinflammation. J Neurochem. 2011;117:264–274. doi: 10.1111/j.1471-4159.2011.07198.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice JC, Allis CD. Histone methylation versus histone acetylation: new insights into epigenetic regulation. Curr Opin Cell Biol. 2001;13:263–273. doi: 10.1016/s0955-0674(00)00208-8. [DOI] [PubMed] [Google Scholar]
- Schonthaler HB, Guinea-Viniegra J, Wagner EF. Targeting inflammation by modulating the Jun/AP-1 pathway. Ann Rheum Dis. 2011;70(Suppl 1):i109–112. doi: 10.1136/ard.2010.140533. [DOI] [PubMed] [Google Scholar]
- Segel R, Anikster Y, Zevin S, Steinberg A, Gahl WA, Fisher D, Staretz-Chacham O, Zimran A, Altarescu G. A safety trial of high dose glyceryl triacetate for Canavan disease. Mol Genet Metab. 2011;103:203–206. doi: 10.1016/j.ymgme.2011.03.012. [DOI] [PubMed] [Google Scholar]
- Shaftel SS, Griffin WS, O’Banion MK. The role of interleukin-1 in neuroinflammation and Alzheimer disease: an evolving perspective. J Neuroinflammation. 2008;5:7. doi: 10.1186/1742-2094-5-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva PF, Garcia VA, da Dornelles AS, Silva VK, Maurmann N, Portal BC, Ferreira RD, Piazza FC, Roesler R, Schroder N. Memory impairment induced by brain iron overload is accompanied by reduced H3K9 acetylation and ameliorated by sodium butyrate. Neuroscience. 2012;200:42–49. doi: 10.1016/j.neuroscience.2011.10.038. [DOI] [PubMed] [Google Scholar]
- Sofroniew MV. Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci. 2009;32:638–647. doi: 10.1016/j.tins.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sofroniew MV, Vinters HV. Astrocytes: biology and pathology. Acta Neuropathol. 2010;119:7–35. doi: 10.1007/s00401-009-0619-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soliman ML, Rosenberger TA. Acetate supplementation increases brain histone acetylation and inhibits histone deacetylase activity and expression. Mol Cell Biochem. 2011;352:173–180. doi: 10.1007/s11010-011-0751-3. [DOI] [PubMed] [Google Scholar]
- Soliman ML, Puig KL, Combs CK, Rosenberger TA. Acetate reduces microglia inflammatory signaling in vitro. J Neurochem. 2012a;123:555–567. doi: 10.1111/j.1471-4159.2012.07955.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soliman ML, Smith MD, Houdek HM, Rosenberger TA. Acetate supplementation modulates brain histone acetylation and decreases interleukin-1beta expression in a rat model of neuroinflammation. J Neuroinflammation. 2012b;9:51. doi: 10.1186/1742-2094-9-51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403:41–45. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- Streit WJ, Walter SA, Pennell NA. Reactive microgliosis. Prog Neurobiol. 1999;57:563–581. doi: 10.1016/s0301-0082(98)00069-0. [DOI] [PubMed] [Google Scholar]
- Sugino T, Nozaki K, Takagi Y, Hattori I, Hashimoto N, Moriguchi T, Nishida E. Activation of mitogen-activated protein kinases after transient forebrain ischemia in gerbil hippocampus. J Neurosci. 2000;20:4506–4514. doi: 10.1523/JNEUROSCI.20-12-04506.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka T, Akira S, Yoshida K, Umemoto M, Yoneda Y, Shirafuji N, Fujiwara H, Suematsu S, Yoshida N, Kishimoto T. Targeted disruption of the NF-IL6 gene discloses its essential role in bacteria killing and tumor cytotoxicity by macrophages. Cell. 1995;80:353–361. doi: 10.1016/0092-8674(95)90418-2. [DOI] [PubMed] [Google Scholar]
- Terasaki T. Studies on the mechanism of drug distribution in tissues. Yakugaku Zasshi. 1992;112:887–905. doi: 10.1248/yakushi1947.112.12_887. [DOI] [PubMed] [Google Scholar]
- Thoren AE, Helps SC, Nilsson M, Sims NR. Astrocytic function assessed from 1-14C-acetate metabolism after temporary focal cerebral ischemia in rats. J Cereb Blood Flow Metab. 2005;25:440–450. doi: 10.1038/sj.jcbfm.9600035. [DOI] [PubMed] [Google Scholar]
- Tian L, Ma L, Kaarela T, Li Z. Neuroimmune crosstalk in the central nervous system and its significance for neurological diseases. J Neuroinflammation. 2012;9:155. doi: 10.1186/1742-2094-9-155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitkovic L, Maeda S, Sternberg E. Anti-inflammatory cytokines: expression and action in the brain. Neuroimmunomodulation. 2001;9:295–312. doi: 10.1159/000059387. [DOI] [PubMed] [Google Scholar]
- Waniewski RA, Martin DL. Preferential utilization of acetate by astrocytes is attributable to transport. J Neurosci. 1998;18:5225–5233. doi: 10.1523/JNEUROSCI.18-14-05225.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyss MT, Magistretti PJ, Buck A, Weber B. Labeled acetate as a marker of astrocytic metabolism. J Cereb Blood Flow Metab. 2011;31:1668–1674. doi: 10.1038/jcbfm.2011.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyss MT, Weber B, Treyer V, Heer S, Pellerin L, Magistretti PJ, Buck A. Stimulation-induced increases of astrocytic oxidative metabolism in rats and humans investigated with 1-11C-acetate. J Cereb Blood Flow Metab. 2009;29:44–56. doi: 10.1038/jcbfm.2008.86. [DOI] [PubMed] [Google Scholar]
- Yang H, Feng GD, Liang Z, Vitale A, Jiao XY, Ju G, You SW. In vitro beneficial activation of microglial cells by mechanically-injured astrocytes enhances the synthesis and secretion of BDNF through p38MAPK. Neurochem Int. 2012;61:175–186. doi: 10.1016/j.neuint.2012.04.020. [DOI] [PubMed] [Google Scholar]
- Zhang B, West EJ, Van KC, Gurkoff GG, Zhou J, Zhang XM, Kozikowski AP, Lyeth BG. HDAC inhibitor increases histone H3 acetylation and reduces microglia inflammatory response following traumatic brain injury in rats. Brain Res. 2008;1226:181–191. doi: 10.1016/j.brainres.2008.05.085. [DOI] [PMC free article] [PubMed] [Google Scholar]