

Abstract
Four different libraries of overall twenty three N3-substituted thymidine (dThd) analogues, including eleven 3-carboranyl thymidine analogues (3CTAs), were synthesized. The latter are potential agents for Boron Neutron Capture Therapy (BNCT) of cancer. Linker between the dThd scaffold and the m-carborane cluster at the N3-position of the 3CTAs contained amidinyl-(3e and 3f), guanidyl-(7e-7g), tetrazolylmethyl-(9b1/2-9d1/2), or tetrazolyl groups (11b1/2-11d1/2) to improve human thymidine kinase 1 (hTK1) substrate characteristics and water solubilities compared with 1st generation 3CTAs, such as N5 and N5-2OH. The amidinyl- and guanidyl-type N3-substitued dThd analogues (3a-3f and 7a-7g) had hTK1 phosphorylation rates of <30% relative to that of dThd, the endogenous hTK1 substrate, whereas the tetrazolyl-type N3-substitued dThd analogues 9a, 9b1/2-9d1/2 and 11a, 11b1/2-11d1/2) had relative phosphorylation rates (rPRs) of >40%. Compounds 9a, 9b1/2-9d1/2 and 11a, 11b1/2-11d1/2 were subjected to in-depth enzyme kinetics studies and the obtained rkcat/Km (kcat/Km relative to that of dThd) ranged from 2.5-26%. The tetrazolyl-type N3-substitued dThd analogues 9b1/2 and 11d1/2 were the best substrates of hTK1 with rPRs of 52.4% and 42.5% and rkcat/Km values of 14.9% and 19.7% respectively. In comparison, the rPR and rkcat/Km values of N5-2OH in this specific study were 41.5% and 10.8%, respectively. Compounds 3e and 3f were >1,900 and >1,500 times, respectively, better soluble in PBS (pH 7.4) than N5-2OH whereas solubilities for 9b1/2-9d1/2 and 11b1/2-11d1/2 were only 1.3 – 13 times better.
Keywords: Thymidine, m-Carborane, Boron Neutron Capture Therapy (BNCT), human Thymidine Kinase 1 (hTK1), Thymidine conjugates
1. Introduction
Thymidine (dThd) bioconjugates with carborane clusters (3-carboranyl dThd analogues or 3CTAs) have been studied extensively for Boron Neutron Capture Therapy (BNCT), which is a radio-chemotherapeutic modality for cancer treatment [1,2]. These bioconjugates were designed for selective uptake and retention in tumor cells by kinase mediated trapping (KMT) through 5′-monophosphorylation by human thymidine kinase 1 (hTK1), an enzyme of the salvage pathways of DNA precursor synthesis with high activity levels only in rapidly proliferating cells [1,3]. So far, KMT has been employed primarily in positron emission tomography (PET) of tumors using 3′-deoxy-3′-[18F]fluorothymidine [18F-FLT] as the imaging agent [4].
First generation 3CTAs, such as N5 and N5-2OH (Fig. 1), were found to be substrates of hTK1 in enzyme kinetic studies [5-9]. The in vitro uptake of N5 and N5-2OH in L929 TK (+) cells was 5-10 fold higher than in L929 TK (−) cells [6]. Biodistribution studies with N5-2OH in rats bearing intracranial RG2 glioma resulted in 10 fold higher boron concentrations in the tumor than in normal brain (27.6 μg B/g tumor vs. 2.6 μg B/g tumor) [8]. BNCT with rats bearing intracranial RG2 glioma that had received N5-2OH prior to irradiation increased survival times by a factor two compared with BNCT of rats bearing RG2 glioma that had not received N5-2OH [8].
Figure 1.
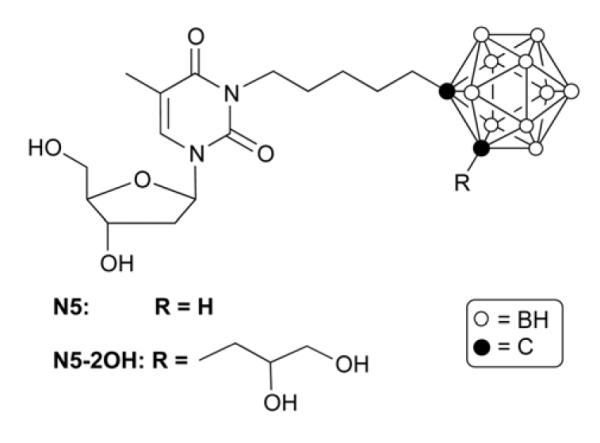
Structures of N5 and N5-2OH.
These biological studies clearly established the therapeutic potential for 3CTAs. On the other hand, the same studies [5-9] revealed two shortcomings of N5 and N5-2OH: 1) The lipophilic nature of N5-2OH caused low water-solubility, which necessitated the use of organic solvents, such as DMSO, to solubilize the agent for biological studies. 2) Compared with endogenous dThd, N5 and N5-2OH show suboptimal hTK1 substrate characteristics.
X-ray crystallographic data with various TK1-like enzymes indicated that hydrogen bonds between the N3-H, C2=O, and C4=O of dThd and the amide backbone of the so-called “lasso-loop” [10,11] of the enzymes are crucial for substrate binding [10-16]. In the case of hTK1 co-crystallized with dThd triphosphate, N3-H and C2=O of the dThd scaffold interacted with Val 172 and Val 174 of the lasso-loop [10,13]. Similar studies with Bacillus anthracis TK (BaTK) indicated a hydrogen bond network between Tyr 179 and Arg 157 of the lasso-loop and N3-H and C4=O of dThd [1]. In the case of Ureaplasma urealyticum TK (UuTK), hydrogen bonds occurred between N3-H and C4=O of dThd and Ile 178 and Lys 180 of the lasso-loop, respectively [10,11]. In all of these cases, N3-H and C2=O and/or C4=O acted as proton donors and acceptors, respectively. Sequra-Peña et al. studied in depth the structural changes in Thermotoga maritima thymidine kinase (TmTK) as a result of dThd and ATP binding [14]. Apparently, interactions similar to those discussed above contributed to dramatic conformational changes in the lasso-loop resulting in the conversion of an open (apo) form to a closed (holo) form of the enzyme, which locks dThd tightly into the substrate binding pocket [12,14]. In the cases of N5 and N5-2OH, the N3-proton is replaced by a bulky carboranyl alkyl chain. Thus, hydrogen bonds crucial for the induction of closed conformations of TK1-like enzymes are missing [1]. We hypothesize that this may limit the abilities of N5 and N5-2OH to interact with the substrate-binding site of the enzyme. In addition, computational studies indicated that N5-2OH cannot be docked effectively into the substrate binding pockets of holo TK1-like enzymes [5] whereas docking was possible in the case of an apo form. This suggests that the presence of the bulky carborane cluster may also interfere with the induction of a completely closed enzyme confirmation. Since the final binding confirmation of TK1-like enzymes with N5-2OH and other 3CTAs is unknown, computer-aided structural design is not an effective tool to support structure-activity-relationship (SAR) studies in pursuit of improved 3CTAs.
A bulky carboranyl substituent at N3 is the trademark of any 3CTA and this cannot be changed. On the other hand, it is conceivable that the missing hydrogen bond interaction between NH-3 and C=O, in part, can be compensated by introducing functional groups in the vicinity of N3 of the 3CTA scaffold having the ability to re-introduce hydrogen bonding. Thus, this paper discusses the synthesis, hTK1 substrate characteristics, and the results of water solubility measurements of four novel classes of N3-substituted dThd analogues: N3-amidinyl- (3a-3f), N3-guanidyl- (7a-7g), N3-(1-tetrazolylmethyl)- (9a, 9b1/2-9d1/2), and N3-tetrazolyl-dThd analogues (11a, 11b1/2-11d1/2) [Schemes 1-4]. The amidinyl- and guanidyl functions in the spacer between the dThd scaffold and the m-carborane cage of target compounds 3a-3f and 7a-7g serve as proton donors. They may restore hydrogen bond interactions with proton accepting atoms in amino acid side chains as well as in the peptide back bone of the lasso loop of hTK1 thereby inducing a more closed enzyme conformation. This, in turn, may render these types of dThd analogues more tightly bound to the active site. As a result, target compounds 3a-3f and 7a-7g may have better enzyme substrate characteristics in comparison with N5-2OH. In addition, the basic nitrogen atoms in these structures should be protonated under physiological conditions, which will improve their water-solubilities. In contrast to the amidinyl- and guanidyl groups in target compounds 3a-3f and 7a-7g, the tetrazolyl moieties in target compounds 9a, 9b1/2 9d1/2 and 11a, 11b1/2-11d1/2 have proton accepting ability only. Nevertheless, they may still interact with various proton donating atoms in amino acid side chains (e.g. Arg 165, Lys 170, Lys 180) and in the peptide back bone of the lasso-loop thereby facilitating a closed enzyme conformation. Lesnikowski et al. recently described the synthesis of several N3-substituted triazolylcarboranyl dThd analogues [17,18] that may have similar TK1-binding properties than the tetrazolyl-type N3-substitued dThd analogues (9a, 9b1/2-9d1/2 and 11a, 11b1/2-11d1/2) described in this paper.
Scheme 1.

Synthesis of N3-amidine-type dThd derivatives (3a-3f).
Scheme 4.

Synthesis of N3-tetrazolyl-type dThd derivatives (11a, 11b1/2-11d1/2).
2. Results and discussion
2.1. Chemistry
Four different libraries of N3-substituted dThd analogues were synthesized in two general steps. In the first step, 3-(cyanomethyl)thymidine (2, Scheme 1) and 3′,5′-(di-tert.-butyldimethylsilyl)-3-cyanothymidine (5, Scheme 2) were synthesized by the reaction of dThd and 3′,5′-di-TBDMS-protected dThd, with iodoactonitrile and cyanogen bromide, respectively. In the second step, these reactive nitriles were treated with various m-carboranylalkyl- [19], alkyl-, and arylamines to yield amidinyl- and guanidyl-type N3-substitued dThd analogues (3a-3f and 7a-7g, Schemes 1 and 2). Compounds 2 and 5 were also treated with sodium azide to produce the tetrazolyl-type dThd analogues 8 and 10 (Schemes 3 and 4), which were subsequently treated with various m-carboranylalkyl-[19] and alkyliodides to yield the tetrazolyl-type target compounds 9a, 9b1/2-9d1/2 and 11a, 11b1/2-11d1/2 (Schemes 3 and 4). m-closo-Carborane was used for the synthesis of all carboranyl target compounds (3e-3f, 7e-7g, 9b1/2-9d1/2 and 11b1/2-11d1/2) because it is more resistant to basic degradation than o-closo-carborane [19], which could occur particularly during the synthesis and biological evaluation of the amidine- and guanidine containing compounds 3e, 3f and 7e-7g.
Scheme 2.

Synthesis of N3-guanidine-type dThd derivatives (7a-7g).
Scheme 3.

Synthesis of N3-tetrazolylmethyl-type dThd derivatives (9a, 9b1/2-9d1/2).
3-(Cyanomethyl)thymidine (2) was synthesized in 78% yield by selective N-alkylation at the 3-position of dThd with iodoacetonitrile (Scheme 1) in presence of potassium carbonate in DMF/acetone (1/1, v/v) at 60 °C for 6 h. Compound 2 was purified by silica gel column chromatography. For the synthesis of the non-carboranyl amidine-type dThd analogues 3a-3d, compound 2 and alkyl- or arylamines were heated in a closed pressure reaction vessel (Quark Glass) in the presence of hexafluoroisopropanol (HFIP) overnight at 95 °C. The use of HFIP for this type of reaction has been previously reported by Snider and O’Hare [22]. The use of methanol, tert.-butanol, tetrahydrofuran, and acetonitrile as solvents did not lead to product formation in this study [22]. The reaction mixtures were added to water and extracted with ethyl acetate. The aqueous layers were separated, evaporated, and purified by RP-18 HPLC. Yields ranged from 24% to 81%.
For the synthesis of the m-carboranylalkyl target compounds 3e and 3f, the same method proved unsuccessful and a modified strategy was developed. Equimolar quantities of 2, m-carboranyl alkyl amine [19], and 1 mL of HFIP were placed in a round bottom flask and heated overnight at 120 °C without condenser. Heating resulted in complete solvent evaporation and formation of 3e and 3f in yields of 7% and 10%, respectively, after purification by RP-18 HPLC.
The synthesis of N3-guanidine-type dThd analogues (7a-7g, Scheme 2) was carried out with 3′,5′-di-TBDMS-protected dThd (4) [20] as the starting material, because initial attempts to treat dThd with cyanogen bromide in presence of potassium carbonate resulted in <5% yield of the desired product, 3-cyanothymidine. Reaction of 4 with cyanogen bromide in presence of triethylamine (TEA) in acetone at 0 °C gave 3′,5′-(di-tert.-butyldimethylsilyl)-3-cyanothymidine (5) in 90% yield after column chromatographic purification [21].
Compounds 7a-7g were synthesized by treating 5 with the amines shown in Scheme 2 in presence of HFIP in a closed pressure reaction vessel (Quark Glass) [22]. The reactions of 5 with propyl- and butyl amine were carried out in an ice bath for 5 h to give 6a and 6b, respectively, in > 95% yield whereas phenyl- and benzyl amines required overnight reaction at room temperature to yield 6c and 6d, respectively, in > 90% yield. The reaction of 1-(2-aminoethyl)-m-carborane [19], 1-(3-aminopropyl)-m-carborane [19], and 1-(4-aminobutyl)-m-carborane [19] with 5 was carried out overnight at 95 °C to afford 6e, 6f, and 6g, respectively, each in about 30-40% yield. Compounds 6a-6g were dissolved in trifluoroacetic acid (TFA)/dichloromethane (10 %, v/v) and stirred at room temperature for 1 h producing 7a-7g in 40-82% yield after purification by RP-18 HPLC. Both N3-amidinyl- (3a-3f) and guanidyl-type (7a-7g) dThd analogues were found to be unstable at room temperature (over several days) and were therefore stored at −5 °C. However, even under these storage conditions partial to complete degradation was observed after a ~ 1 year period.
The synthesis of tetrazolylmethyl- and tetrazolyl-type target compounds 9a, 9b1/2-9d1/2 and 11a, 11b1/2-11d1/2 is described in Schemes 3 and 4, respectively. These compounds were prepared using a click chemistry approach [23]. The reactive nitrile group of 2 was treated with sodium azide in the presence of zinc bromide (ZnBr2) in a mixture of water and isopropanol (2:1, v/v) at 90 °C for three days to produce 8 in 90% yield (Scheme 3). In case of 5, the same reaction conditions led to a mixture of 3-(tetrazol-5-yl)thymidine (10, Scheme 4) along with 3′- or 5′-mono-TBDMS and 3′,5′-(di-TBDMS)-3-(tetrazol-5-yl)thymidine. Deprotection of remaining TBDMS-groups was accomplished in situ by adding TFA in dichloromethane (10%, v/v) to the reaction mixture for 1 h at room temperature to give 10 in 44% overall yield (Scheme 4). Both 8 and 10 were purified by RP-18 HPLC.
Compound 8 and 10 were N-alkylated at the tetrazolyl ring with 1-iodopropane, 1-(2-iodoethyl)-m-carborane [19], 1-(3-iodopropyl)-m-carborane [19], and 1-(4-iodobutyl)-m-carborane [19] in the presence of potassium carbonate in a mixture of acetone and dimethylformamide (50%, v/v) at 60 °C overnight to produce target compounds 9a and 11a as well as 9b1/2-9d1/2 and 11b1/2-11d1/2 as isomeric (Nb-alkylated and Nc-alkylated) mixtures (Schemes 3 and 4). In the case of the propyl analogues 9a and 11a it was possible to isolate the Nc-isomers as pure compounds by RP-18 HPLC with 59 % to 71% yield, respectively. Unfortunately, it was not possible to separate the isomers of the carboranylalkyl derivatives 9b1/2-9d1/2 and 11b1/2-11d1/2. Overall, the Nc-isomers were the dominating species in these mixtures with 70-95% (Schemes 3 and 4). The isomers were characterized by means of 1H-NMR. The chemical shifts for the Nb-CH2 protons ranged from 4.44-4.62 ppm, whereas those for the Nc-CH2 protons ranged from 4.54-4.82 ppm. A downfield shift of 0.15-0.53 ppm for Nc-CH2 protons vs. the Nb-CH2 protons was also reported by Sveshnikov and Nelson [24] for various alkyltetrazoles. The observed Nc:Nb isomeric ratios were in accordance with those reported previously for other alkylation reactions at tetrazole rings [25]. In addition, 1H-1H NOESY NMR of the mixture of 9b1 and 9b2 showed a NOE cross-peak between the triplet for Nb-CH2 (δ 4.52) and the singlet for N3-CH2 (5.35 ppm). In contrast, no cross peak was observed between the signals of Nc-CH2 and N3-CH2 (see Supplementary material). This result was expected based on a shorter interproton distance between Nb-CH2 and N3-CH2 (2.5 Å) than between Nc-CH2 and N3-CH2 (4.9 Å) (calculated with Chem3D Pro [Chambridge Soft, Cambridge, MA, USA]). It unambiguously confirmed the isomer assignment.
2.2. Enzymatic studies
Compounds 3a-3f, 7a-7g, 8, 9a, 9b1/2-9d1/2, 10, and 11a, 11b1/2-11d1/2 were evaluated in phosphoryl transfer assays (PTAs) with recombinant hTK1 as described, previously with minor modifications [26-29]. Thymidine and N5-2OH were used as reference compounds. The obtained phosphorylation rates are expressed as percent of the phosphorylation rate of dThd, which was set to 100% (rPRs). Such comparison is used to give an estimation of the structure activity relationships of substrate analogues compared to the activity of the natural substrate, dThd. Analogues with relatively high PRs were further selected for in depths kinetic studies (apparent kcat/Km values) to determine the apparent catalytic efficiency of hTK1 for these substrates.
Most of the amidinyl-type dThd analogues (3a-3f) and all of the guanidyl-type dThd analogues (7a-7g) proved to be unstable under phosphoryl transfer assay conditions, as evidenced by the production of phosphorylated side products. Fig. 2A shows the monophosphate formation of the carboranyl compounds of both libraries (3e, 3f, 7e-7g). For compounds 7e-7g, additional spots are visible at the level of dThd monophosphate (dTMP). This indicates that these compounds may have been partially degraded to dThd during the assay, which was then converted to dTMP. A Low Resolution-Electron Spray Ionization (LR-ESI) mass spectra of an enzyme assay mixture containing compound 7f and 31P-ATP showed a signal for the monophosphate of 7f(Calcd. for [M-PO3H2+H]+: 549.3; Found: m/z 549.3) and a signal for dTMP (Calcd for [M+H]+: 323.1; Found: m/z 323.1, see Supplementary material). The observed MS data support the formation of dTMP during the enzyme assay as a result of degradation processes. Enzyme assay mixtures containing compounds 3e and 3f did not appear to produce significant amounts of dTMP as a side product. However, it is conceivable that in the case of both compounds, dTMP may not be visible because of low concentrations
Figure 2.

Phosphorylation rates of dThd, N5-2OH and N3-substituted dThd analogues IPwith hTK1. Assay products were separated by PEI–cellulose TLC and quantified by β-radiography. (A). Compounds 3e, 3f and 7e-7g; (a) dThd monophosphate (dTMP), (b) compound-monophosphates (N5-2OH, 3e, 3f and 7e-7g) and (c) monophosphorylated degradation products, presumably dTMP. (B). Compounds 8, 9a, 9b1/2-9d1/2, (a) dTMP and (b) compound-monophosphates (8, 9a, 9b1/2-9d1/2). (C). Compounds 10, 11a, 11b1/2-11d1/2, (a) dTMP and (b) compound-monophosphates (11a, 11b1/2-11d1/2)
Compounds with alkylamidinyl- and alkylguanidyl substituents (3a, 3b, 7a, 7b) had rPRs of 0-3%, whereas those with arylamidinyl- and arylguanidyl substituents (3c, 3d, 7c, 7d) had slightly higher rPRs of 2-17% (Table 1). However, for compounds 3a-3d and 7a-7d, 20-100% of the formed phosphorylated material appeared to be the dTMP side product. Compounds with m-carboranylalkylamidinyl substituents 3e and 3f had rPRs of 3% and 6% whereas the m-carboranylalkylguanidyl compounds 7e-7g had overall slightly higher rPRs with 16-27% but also showed the formation of the dTMP side product with rPRs of 6-12% (Table 1 and Fig. 2A). Overall, the rPRs of both the amidinyl-type and the guanidyl-type N3-substitued dThd analogues were lower than that of N5-2OH (rPR of 46%).
Table 1.
Relative hTK1 phosphorylation rates (rPRs) of N3-substituted dThd analogues
| Compounds | rPR (%) ± SD1 | Compounds | rPR (%) ± SD1 |
|---|---|---|---|
| dThd | 100 | 7f | 15.5 ± 1.9 |
| N5-2OH | 46.3 ± 14.0 | 7g | 27.3 ± 3.0 |
| 3a | 1.9 ± 0.5 | 8 | 39.5 ± 3.6 |
| 3b | 2.3 ± 0.4 | 9a | 55.7 ± 5.2 |
| 3c | 11.3 ± 0.9 | 9b1/2 | 52.4 ± 5.8 |
| 3d | 16.8 ± 0.8 | 9c1/2 | 58.2 ± 1.2 |
| 3e | 3.0 ± 0.2 | 9d1/2 | 63.7 ± 3.4 |
| 3f | 5.6 ± 0.5 | 10 | 0.0 ± 0.0 |
| 7a | 0.0 ± 0.0 | 11a | 57.6 ± 8.5 |
| 7b | 1.2 ± 0.9 | 11b1/2 | 55.6 ± 3.2 |
| 7c | 1.9 ± 1.3 | 11c1/2 | 42.9 ± 5.1 |
| 7d | 14.9 ± 2.1 | 11d1/2 | 42.5 ± 5.8 |
| 7e | 16.3 ± 1.0 |
Data represent the means of three replicates ± SD.
In contrast to the amidinyl- (3a-3f) and guanidyl-type (7a-7g) dThd analogues, the tetrazolylmethyl-type (8, 9a, 9b1/2-9d1/2) and the tetrazolyl-type dThd analogues (11a, 11b1/2-11d1/2) were stable under phosphoryl transfer assay conditions (Table 1, Fig. 2B and Fig. 2C). With the exception of compound 10, all tetrazolyl-type dThd analogues displayed relatively higher rPRs with 40-64%. The signals for monophosphate product of 10 may actually overlap with ATP related spots. The tetrazolylmethyl-type dThd analogues with propyl- and butyl spacer between carborane and tetrazole (9c1/2, 9d1/2) appeared to be somewhat better phosphorylated than their tetrazolyl-type counterparts (11c1/2, 11d1/2) (58-64% vs. 42-43%).
The tetrazolyl-type dThd analogues 9a, 9b1/2-9d1/2 and 11a, 11b1/2-11d1/2 were selected for in depths kinetic studies (apparent kcat/Km values) based on their rPRs, which were higher than that of N5-2OH. Both dThd and N5-2OH were used as reference compounds. These studies were conducted with varying substrate concentrations (0.4-300 μM) (see Experimental Section for details). The rkcat/Km value for N5-2OH using the current recombinant hTK1 preparation with a specific activity of 441 nmol dThd monophosphate formation per min and mg of enzyme was lower compared with the value obtained with a different enzyme preparation used in a previous study (10.8% vs 35.8%) [9]. Narayanasamy et al. [26] reported a specific activity of 640 nmol dThd monophosphate formation per min and mg of enzyme for a different recombinant hTK1 used in a previous study with 3CTAs. This suggests that specific activities between enzyme preparations vary significantly and that this, in turn, may cause variations in the rkcat/Km values of 3CTAs. The rkcat/Km values for compound 9a, 9c1/2, 9d1/2, and 11b1/2 were comparable with that of N5-2OH (7.6-12.9% vs. 10.8%). Compounds 9b1/2 (14.9%), 11a (26.0%) and 11d1/2 (19.7%) had notably higher rkcat/Km values indicating that these compounds are better substrates of hTK1 than N5-2OH.
Based on the isomeric composition of the tetrazoyl target compounds 9a, 9b1/2-9d1/2 and 11a, 11b1/2-11d1/2 (Schemes and 4), it is reasonable to assume that their observed enzymatic activities are primarily related to the Nc-isomers.
2.3. Solubility studies
The solubilities of carboranyl compounds that appeared to be stable during enzyme assays (3e,3f, 9b1/2-9d1/2, and 11b1/2-11d1/2) were determined in phosphate-buffered saline (PBS) at 25 °C in comparison with N5-2OH using thermodynamic solubility measurement assay conditions (Table 3) [27,28]. The carboranylethyl dThd analogue 3e and its propyl homologue 3f were > 1900 and >1500 times, respectively, better soluble in PBS than N5-2OH. The solubility patterns for the tetrazolylmethyl-(9b1/2 and tetrazolyl- (11d1/2-9d1/2) dThd analogues decreased with increasing chain length. Compounds 9b1/2-9d1/2 were ~3-13 times better soluble in PBS than N5-2OH whereas compounds 11b1/2 and 11c1/2 were ~2-4 times better soluble. The solubility profile of 11d1/2 was similar to that of N5-2OH. Overall, the tetrazolylmethyl derivatives were slightly better soluble than their corresponding tetrazolyl counterparts.
Table 3.
Solubilities of the N5-2OH, 3e, 3f, 9b1/2-9d1/2 and 11b1/2-11d1/2 in PBS at pH 7.4 at 25 °C.
| Compounds | PBS pH 7.41 | Compounds | PBS pH 7.41 |
|---|---|---|---|
| N5-2OH | 7-10 |g/mL | 9d1/2 | 33.9 ± 1.4 |g/mL |
| 3e | >19.5 mg/mL | 11b1/2 | 42.6 ± 1.0 |g/mL |
| 3f | >15 mg/mL | 11c1/2 | 25.4 - 26.3 |g/mL2 |
| 9b1/2 | 133.1 ± 5.2 |g/mL | 11d1/2 | 13.3 - 13.8 |g/mL* |
| 9c1/2 | 84.2 ± 4.1 |g/mL |
Standard deviations are based on three experiment
Data represent duplicate measurements
3. Summary and Conclusions
In summary, several N3-substituted dThd analogues containing amidinyl-(3a-3f), guanidyl-(7a-7g), tetrazolylmethyl-(8, 9a, 9b1/2-9d1/2), tetrazolyl groups (10, 11a, 11b1/2-11d1/2) were successfully synthesized. The amidinyl- and guanidyl-type dThd analogues (3a-3f and 7a-7g) did not possess the necessary stability under aqueous conditions. The excellent solubilities of 3e and 3f in PBS were expected because of the presence of basic amidinyl groups in these structures, which should be protonated at physiological pH (7.4). Unfortunately, the solubilities of the tetrazolyl-type dThd analogues in PBS were only moderately better than that of N5-2OH. The tetrazolylmethyl- (8, 9a, 9b1/2-9d1/2) and tetrazolyl-type dThd analogues (10 11a, 11b1/2-11d1/2) had comparable or somewhat higher rPRs (40-64%) than that of, N5-2OH whereas those of the amidine- (3a-3f) and guanidine-type (7a-7g) dThd analogues had lower rPRs with 2-12% and 0-27%, respectively. The rkcat/Km values for the tetrazolylcarboranyl dThd analogues with relatively high rPRs (9a, 9b1/2-9d1/2, 11a, 11b1/2-11d1/2) ranged from 7.6-26% in comparison to N5-2OH (10.8%). Compounds 11d1/2 (rkcat/Km = 19.7%) and 9b1/2 (rkcat/Km = 14.9%) were ~1.8 and ~1.4 times, respectively, better substrates of hTK1 than N5-2OH.
The rkcat/Km values obtained for compounds 9b1/2 and 11d1/2 indicate that proton accepting groups in the spacer between carborane cluster and dThd scaffold have the potential to improve hTK1 substrate characteristics. In addition, basic functions in the spacer seem to enhance aqueous solubility, as demonstrated at the example of compounds 3e and 3f. Unfortunately, the validity of enzymatic data obtained for amidinyl- and guanidyl-type dThd analogues (3a-3f and 7a-7g) is limited due to lack of stability under aqueous and routine storage conditions. Preliminary studies in our laboratories indicated that inserting more than two methylene groups between the dThd scaffold and basic amino group containing functions in the spacer prevents decomposition under aqueous conditions. As discussed in the introduction, a major shortcoming is lack of detailed structural information that can assist in delineating the basic molecular interactions between hTK1 and 3CTAs.
4. Experimental protocol
4.1. Chemistry
1H- and 13C NMR spectra were obtained on a Bruker DRX 400 at The Ohio State University College of Pharmacy (400 MHz for 1H and 100 MHz for 13C and 128 MHz for 11B). 1H-1H NOESY NMR was obtained on a Bruker DRX 800 at The Ohio State University Campus Chemical Instrument Center. Chemical shifts (δ) are reported in ppm from internal deuterated chloroform, acetone, and methanol. Coupling constants are reported in Hz. 13C NMR spectra are fully decoupled. Low- and High Resolution–Electrospray ionization (LR/HR-ESI) mass spectra were obtained on a Micromass LCT spectrometer at The Ohio State University Campus Chemical Instrumentation Center, Columbus, OH. For all carborane-containing compounds, the mass of the most intensive peak of the isotopic pattern was reported (80% boron-11 to 20% boron-10 distribution). Measured patterns agreed with calculated patterns. Silica gel 60 (0.063-0.200 mm) used for gravity column chromatography and silica gel 60 (0.015-0.049 mm) used for flash column chromatography were purchased from Dynamic Adsorbents Inc. (3280 Peachtree Corners Circle, E, Norcross, GA 30092, USA). Reagent-grade solvents were used for column chromatography. Precoated glass-backed TLC plates with silica gel 60 F254 (0.25-mm layer thickness) from Dynamic Adsorbents (Norcross, GA) were used for TLC. General compound visualization for TLC was achieved by UV light. Carborane-containing compounds were selectively visualized by spraying the plate with a 0.06% PdCl2/1% HCl solution and heating at 120 °C, which caused the slow (15-45 s) formation of a gray spot due to the reduction of Pd2+ to Pd0. Anhydrous solvents, such as dichloromethane, were purchased directly either from Acros Organics (Morris Plains, NJ) or from Sigma Aldrich (Milwaukee, WI). Pressure reaction vessels were purchased from Quark Glass (NJ, USA). Other chemicals and solvents were purchased from standard commercial suppliers. Tetrahydrofuran (THF) and benzene were distilled from sodium and benzophenone indicator under argon. m-Carborane was purchased from Katchem Ltd. (Prague, Czech Republic). Cyanogen bromide (purchased from Sigma Aldrich) is a highly toxic compound. The Material Safety Data Sheet (MSDS) should be consulted before usage. [γ-32P]ATP (specific activity: 3000Ci/mmol) and [methyl-3H]dThd (specific activity: 83.7 Ci/mmol) were obtained from Perkin Elmer, Waltham, MA. Unless specified otherwise, all reactions were carried out under argon atmosphere. Preparative HPLC was performed with a Gemini 5μ C18 column (21.20 mm × 250 mm, 5 μm particle size) supplied by Phenomenex Inc. CA, USA on a Hitachi HPLC system (L-2130) with a Windows based data acquisition and Hitachi Diode array detector (L-2455). Purification was accomplished using a water (0.1% TFA)/methanol (0.1% TFA) gradient at 7 mL/min flow rate (method: 100:0 to 50:50 over 70 min followed by 50:50 to 0:100 over 40 min). Analytical HPLC was carried out with a Gemini 5μ C18 110A Column (250 × 4.6 mm) supplied by Phenomenex Inc. CA, USA using the HPLC system above. Two different gradients [water (0.1% TFA)/acetonitrile (0.1% TFA) and water (0.1% TFA)/methanol (0.1% TFA)] were used at 1 mL/min flow rate (method: 100:0 to 75:25 over 20 min followed by 75:25 to 100:0 over 10 min). See Supplementary material for more details on compound purification and analysis.
4.1.1. 3-(Cyanomethyl)thymidine (2)
Thymidine (10 g, 41.3 mmol) and potassium carbonate (17 g, 123 mmol) were dissolved in acetone/DMF (100 mL, 1/1, v/v) and iodoacetonitile (6.5 mL, 90 mmol) was added. The reaction mixture was refluxed for 6 h, then added to water (750 mL), and extracted with ethyl acetate (250 mL × 4). The organic layer was separated, dried using anhydrous magnesium sulfate (100 g), evaporated under reduced pressure, and purified by silica gel column chromatography using dichloromethane and methanol as the solvent system. The compound was eluted at 4% methanol. Rf = 0.70 (10% methanol in dichloromethane), yield = 9.1 g, 78% as yellow solid, (m.p. 52 °C); 1H NMR (400 MHz, CD3OD, δ ppm): 7.86 (s, 1H, H-6), 6.25 (t, J = 6.6 Hz, 1H, 1′H), 4.80 (s, 2H, CH2-CN), 4.33-4.41 (m, 1H, 3′H), 3.87-3.95 (m, 1H, 4′H), 3.78 (dd, J = 2.8 and 12.0 Hz, 2H, 5″H), 3.70 (dd, J = 3.1 and 12.0 Hz, 2H, 5′H), 2.23-2.33 (m, 1H, 2″H), 2.12-2.22 (m, 1H, 2′H), 1.89 (s, 3H, 5-CH3). 13C NMR (CD3OD, 100 MHz, δ ppm): 163.29 (C-4 C=O), 150.62 (C-2 C=O), 136.77 (C-6), 115.46 (CN), 110.30 (C-5), 88.27 (C-1′), 86.79 (C-4′), 71.40 (C-3′), 62.16 (C-5′), 41.09 (C-2′), 28.92 (N3-CH2-CN), 13.05 (5-CH3); HR-MS (ESI-MS) for C12H15N3NaO5. Calcd: 304.0909. Found: m/z 304.0888 (M+Na)+.
4.1.2. General procedure for the synthesis of 3-[2-Imino-2-(propylamino)ethyl]thymidine (3a), 3-[2-Imino-2-(butylamino)ethyl]thymidine (3b), 3-[2-Imino-2- (phenylamino)ethyl]thymidine (3c), and 3-[2-Imino-2-(benzylamino)ethyl]thymidine (3d)
Compound 2 (280 mg, 1 mmol) and amine (1.1 mmol) were dissolved in hexafluoroisopropanol (1 mL) in a closed pressure vessel (Quark Glass).22 The reaction mixture was heated at 95 °C overnight, suspended in water (80 mL), and extracted with ethyl acetate (50 mL × 3). The aqueous layer was evaporated under reduced pressure and the residue purified by preparative HPLC. The percentage purities of the synthesized compounds were > 95%.
4.1.2.1. 3-[2-Imino-2-(propylamino)ethyl]thymidine (3a)
Retention time (analytical HPLC) = 9.1 min, yield = 80 mg, 24% (white amorphous solid); 1H NMR (400 MHz, CD3OD, δ ppm): 7.97 (s, 1H, H-6), 6.31 (t, J = 6.6 Hz, 1H, 1′H), 4.87 (s, 2H, N3-CH2), 4.38-4.48 (m, 1H, 3′H), 3.95 (dd, J = 3.2 and 6.4 Hz, 1H, 4′H), 3.82 (dd, J = 3.0 and 12.0 Hz, 1H, 5″H), 3.75 (dd, J = 3.5 and 12.0 Hz, 1H, 5′H), 3.25 (t, J = 7.2 Hz, 2H, =NH-CH2), 2.18-2.35 (m, 2H, 2′H and 2″H), 1.94 (s, 3H, 5-CH3), 1.60-1.71 (m, 2H, =NH-CH2-CH2), 0.99 (t, J = 7.4 Hz, 3H, propyl-CH3). 13C NMR (CD3OD, 100 MHz, δ ppm): 165.18 (C-4 C=O), 165.08 (HN=C-NH), 152.35 (C-2 C=O), 137.57 (C-6), 110.77 (C-5), 89.18 (C-1′), 87.51 (C-4′), 72.28 (C-3′), 62.86 (C-5′), 45.33 (N3-CH2), 42.50 (=NH-CH2), 41.43 (C-2′), 22.04 (=NH-CH2-CH2), 13.15 (5-CH3), 11.52 (propyl-CH3); HR-MS (ESI-MS) for C15H25N4O5. Calcd: 341.1825. Found: m/z 341.1799 (M+H)+.
4.1.2.2. 3-[2-Imino-2-(butylamino)ethyl]thymidine (3b)
Retention time (analytical HPLC) = 10 min, yield = 160 mg, 45% (white amorphous solid); 1H NMR (400 MHz, CD3OD, δ ppm): 7.98 (s, 1H, H-6), 6.32 (t, J = 6.5 Hz, 1H, 1′H), 4.88 (s, 2H, N3-CH2), 4.37-4.47 (m, 1H, 3′H), 3.90-4.00 (m, 1H, 4′H), 3.83 (dd, J = 2.5 and 11.9 Hz, 1H, 5″H), 3.76 (dd, J = 3.4 and 11.9 Hz, 1H, 5′H), 3.25-3.35 (m, 2H, Methanol-CH3 and NH-CH2), 2.20-2.38 (m, 2H, 2′H and 2″H), 1.96 (s, 3H, 5-CH3), 1.58-1.72 (m, 2H, NH-CH2-CH2), 1.36-1.50 (m, 2H, butyl-CH2-CH3), 0.99 (t, J = 7.2 Hz, 3H, butyl-CH3). 13C NMR (CD3OD, 100 MHz, δ ppm): 165.00 (C-4 C=O), 152.27 (C-2 C=O), 137.50 (C-6), 110.70 (C-5), 89.09 (C-1′), 87.44 (C-4′), 72.20 (C-3′), 62.79 (C-5′), 43.47 (N3-CH2), 42.42 (NH-CH2), 41.35 (C-2′), 30.63 (NH-CH2-CH2), 20.95 (butyl-CH2-CH3), 13.98 (butyl-CH3), 13.08 (5-CH3); HR-MS (ESI-MS) for C16H27N4O5. Calcd: 355.1981. Found: m/z 355.1985 (M+H)+.
4.1.2.3. 3-[2-Imino-2-(phenylamino)ethyl]thymidine (3c)
Retention time (analytical HPLC) = 7.6 min, yield = 290 mg, 79% (white amorphous solid); 1H NMR (400 MHz, CD3OD, δ ppm): 8.00 (s, 1H, H-6), 7.45-7.60 (m, 3H, phenyl-Ar-H), 7.34 (d, J = 7.4 Hz, 2H, phenyl-Ar-H), 6.35 (t, J = 6.5 Hz, 1H, 1′H), 5.07 (dd, J = 16.5 and 23.4 Hz, 2H, N3-CH2), 4.38-4.48 (m, 1H, 3′H), 3.95 (dd, J = 3.2 and 6.4 Hz, 1H, 4′H), 3.83 (dd, J = 3.0 and 12.0 Hz, 1H, 5″H), 3.76 (dd, J = 3.4 and 12.0 Hz, 1H, 5′H), 2.20-2.38 (m, 2H, 2′H and 2″H), 1.98 (s, 3H, 5-CH3). 13C NMR (CD3OD, 100 MHz, δ ppm): 166.13 (C-4 C=O), 165.09 (HN=C-NH), 152.35 (C-2 C=O), 137.61 (phenyl-Ar-C), 135.04 (C-6), 131.56, 130.49, 126.83 (phenyl-Ar-C), 110.75 (C-5), 89.14 (C-1′), 87.52 (C-4′), 72.21 (C-3′), 62.80 (C-5′), 42.66 (N3-CH2), 41.38 (C-2′), 13.08 (5-CH3); HR-MS (ESI-MS) for C18H23N4O5. Calcd: 375.1668. Found:. m/z 375.1677 (M+H)+.
4.1.2.4. 3-[2-Imino-2-(benzylamino)ethyl]thymidine (3d)
Retention time (analytical HPLC) = 11.8 min, yield = 310 mg, 81% (white amorphous solid); 1H NMR (400 MHz, CD3OD, δ ppm): 7.92 (s, 1H, H-6), 7.20-7.40 (m, 5H, benzyl-Ar-H), 6.31 (t, J = 6.5 Hz, 1H, 1′H), 4.65 (s, 2H, N3-CH2), 4.42 (s, 3H, 3′H and NH-CH2), 3.90-3.97 (m, 1H, 4′H), 3.83 (dd, J = 3.0 and 12.0 Hz, 1H, 5″H), 3.75 (dd, J = 3.4 and 12.0 Hz, 1H, 5′H), 2.20-2.38 (m, 2H, 2′H and 2″H), 1.95 (s, 3H, 5-CH3). 13C NMR (CD3OD, 100 MHz, δ ppm): 168.79 (HN=C-NH), 164.19 (C-4 C=O), 151.30 (C-2 C=O), 138.66 (benzyl-Ar-C), 135.90 (C-6), 128.53, 127.45, 127.20 (benzyl-Ar-C), 109.53 (C-5), 87.87 (C-1′), 86.28 (C-4′), 71.02 (C-3′), 61.73 (C-5′), 43.35 (N3-CH2), 43.12 (benzyl-CH2), 40.38 (C-2′), 12.22 (5-CH3); HR-MS (ESI-MS) for C19H25N4O5. Calcd: 389.1825. Found: m/z 389.1808 (M+H)+.
4.1.3. General procedure for the synthesis of 3-[2-Imino-2-{2-(closo-1,7-carboranyl)ethylamino}ethyl]thymidine (3e) and 3-[2-Imino-2-{2-(closo-1,7-carboranyl)propylamino}ethyl]thymidine (3f)
Carboranyl alkyl amine (1 mmol) and 2 (280 mg, 1 mmol) were dissolved in hexafluoroisopropanol (1 mL) in a round bottom flask and heated at 120 °C under argon atmosphere overnight to allow the solvent to evaporate. The reaction mixture was suspended in water (80 mL) and extracted with ethyl acetate (50 mL × 3). The aqueous layer was concentrated under reduced pressure and the residue purified by preparative HPLC. The percentage purities of the synthesized compounds were > 95%.
4.1.3.1. 3-[2-Imino-2-{2-(closo-1,7-carboranyl)ethylamino}ethyl]thymidine (3e)
Retention time = (analytical HPLC) 14.8 min, yield = 32 mg, 7% (white wax); 1H NMR (400 MHz, CD3OD, δ ppm): 7.96 (s, 1H, H-6), 6.30 (t, J = 6.6 Hz, 1H, 1′H), 4.84 (s, 2H, N3-CH2), 4.37-4.45 (m, 1H, 3′H), 3.90-3.96 (m, 1H, 4′H), 3.81 (dd, J = 2.9 and 12.0 Hz, 1H, 5″H), 3.75 (dd, J = 3.3 and 12.0 Hz, 1H, 5′H), 3.59 (s, 1H, Carboranyl H), 3.28-3.38 (m, 2H, CD3-OD and NH-CH2), 2.35 (t, J = 7.7 Hz, 2H, Carborane-CH2-), 2.20-2.30 (m, 2H, 2′H and 2″H), 1.94 (s, 3H, 5-CH3). 13C NMR (CD3OD, 100 MHz, δ ppm): 165.47 (C-4 C=O), 165.05 (HN=C-NH), 152.33 (C-2 C=O), 137.61 (C-6), 110.83 (C-5), 89.18 (C-1′), 87.52 (C-4′), 74.00 (Carboranyl C), 72.29 (C-3′), 62.88 (C-5′), 57.50 (Carboranyl CH), 43.07 (N3-CH2), 42.60 (NH-CH2), 41.42 (C-2′), 34.98 (Carborane-CH2-), 13.17 (5-CH3); HR-MS (ESI-MS) for C16H33B10N4O5. Calcd: 469.3454. Found: m/z 469.3476 (M+H)+.
4.1.3.2. 3-[2-Imino-2-{3-(closo-1,7-carboranyl)propylamino}ethyl]thymidine (3f)
Retention time = (analytical HPLC) 15.2 min, yield = 48 mg, 10% (white wax); 1H NMR (400 MHz, CD3OD, δ ppm): 7.98 (s, 1H, H-6), 6.34 (t, J = 6.6 Hz, 1H, 1′H), 4.88 (s, 2H, N3-CH2), 4.44-4.52 (m, 1H, 3′H), 3.94-4.00 (m, 1H, 4′H), 3.84 (dd, J = 3.0 and 12.0 Hz, 1H, 5″H), 3.77 (dd, J = 3.4 and 11.9 Hz, 1H, 5′H), 3.55 (s, 1H, Carboranyl H), 3.25 (t, J = 6.7 Hz, 2H, NH-CH2), 2.22-2.36 (m, 2H, 2′H and 2″H), 2.04-2.16 (m, 2H, Carborane-CH2-), 1.97 (s, 3H, 5-CH3), 1.66-1.78 (m, 2H, Carborane-CH2-CH2-). 13C NMR (CD3OD, 100 MHz, δ ppm): 164.32 (C-4 C=O), 163.88 (HN=C-NH), 151.20 (C-2 C=O), 136.44 (C-6), 109.69 (C-5), 88.03 (C-1′), 86.35 (C-4′), 72.26 (Carboranyl C), 71.13 (C-3′), 61.73 (C-5′), 56.031 (Carboranyl CH), 48.86 (NH-CH2-), 41.63 (N3-CH2), 41.45 (NH-CH2), 40.31 (C-2′), 33.70 (NH-CH2-CH2), 28.07 (Carborane-CH2-), 12.05 (5-CH3); HR-MS (ESI-MS) for C17H35B10N4O5. Calcd: 483.3611. Found: m/z 483.3611 (M+H)+.
4.1.4. 3′,5′-(Di-tert.-butyldimethylsilyl)-3-cyanothymidine (5)
3′,5′-(Di-tert.-butyldimethylsilyl)thymidine (4) [20] (10 g, 21.2 mmol) and cyanogen bromide (3 M solution in dichloromethane; 14 mL, 42 mmol) were dissolved in acetone (50 mL). Triethylamine (2.75 mL, 20 mmol) was added at 0 °C and the reaction mixture was stirred for 2 h at this temperature [21]. Water (750 mL) was added and the resulting mixture was extracted with ethyl acetate (250 mL × 4). The organic layer was separated, dried using anhydrous magnesium sulfate (100 g), and evaporated. The residue was purified by silica gel column chromatography using hexanes and dichloromethane as solvent system. The compound was eluted at 50% dichloromethane in hexane. Rf: 0.63 (100% dichloromethane) yield = 8.5 g, 81% (white solid, m.p. 93 °C); 1H NMR (400MHz, CDCl3, δ ppm): 7.63 (s, 1H, H-6), 6.27 (q, J = 5.7 and 7.8 Hz, 1′H, 1H), 4.35-4.45 (m, 1H, 3′H), 3.96-4.06 (m, 1H, 4′H), 3.90 (dd, J = 2.4 and 11.5 Hz, 1H, 5″H), 3.78 (dd, J = 2.2 and 11.5 Hz, 1H, 5′H), 2.32 (ddd, J = 2.4, 5.7 and 13.1 Hz, 1H, 2″H), 1.99-2.10 (m, 1H, 2′H), 1.98 (s, 3H, 5-CH3), 0.90 and 0.93 (two s, 18H, ≡C-CH3), 0.08-0.14 (m, 12H, Si-CH3). 13C NMR (CDCl3, 100 MHz, δ ppm): 159.82 (C-4 C=O), 147.82 (C-2 C=O), 136.51 (C-6), 109.81 (C-5), 102.75 (C≡N), 88.99 (C-1′), 86.98 (C-3′), 72.73 (C-4′), 63.42 C-5′), 41.96 (C-2′), 26.32 and 26.11 (two ≡C-CH3), 18.80 and 18.38 (two Si-C≡), 13.32 (5-CH3), -4.44 and -4.99 (two Si-CH3). IR: (N3-CN) 2264.9 cm−1. HR-MS (ESI-MS) for C23H41N3NaO5Si2. Calcd: 518.2482. Found: m/z 518.2462 (M+Na)+.
4.1.5. General procedure for the synthesis of 3-(Propylcarbamimidoyl)-3′,5′-(di-tert.-butyldimethylsilyl)thymidine (6a), and 3-(Butylcarbamimidoyl)-3′,5′-(di-tert.-butyldimethylsilyl)thymidine (6b) [22]
Compound 5 (100 mg, 0.21 mmol) was dissolved in hexafluoroisopropanol (200 μL) at 0 °C. n-Propyl or n-butyl amine (0.4 mmol) was added and the reaction mixture was stirred for 5 h at 0 °C. The aqueous layer was evaporated under reduced pressure and the residue purified by preparative HPLC.
4.1.5.1. 3-(Propylcarbamimidoyl)-3′,5′-(di-tert.-butyldimethylsilyl)thymidine (6a)
Rf: 0.40 (100% dichloromethane), Retention time (preparative HPLC) = 46-52 min, yield = 111 mg, 95% (white amorphous solid); 1H NMR (400 MHz, CDCl3, δ ppm): 7.47 (s, 1H, H-6), 6.28 (dd, J = 5.7 and 7.9 Hz, 1′H, 1H), 4.40-4.46 (m, 1H, 3′H), 3.91 (q, J = 2.3 Hz, 1H, 4′H), 3.84 (dd, J = 11.4 and 2.6 Hz, 1H, 5″H), 3.73 (dd, J = 11.4 and 2.3 Hz, 1H, 5′H), 3.16 (t, J = 7.0 Hz, 2H, NH-CH2), 2.23 (ddd, J = 13.1, 5.7 and 2.5 Hz, 1H, 2″H), 1.93-2.03 (m, 1H, 2′H), 1.89 (s, 3H, 5-CH3), 1.56-1.69 (m, 2H, NH-CH2-CH2), 0.95 (t, J = 7.4 Hz, 2H, propyl-CH3), 0.85 and 0.90 (two s, 18H, ≡C-CH3), 0.08 and 0.04 (two s, 12H , Si-CH3). 13C NMR (CDCl3, 100 MHz, δ ppm): 162.82 (C-4 C=O), 150.02 (C-2 C=O), 135.58 (C-6), 111.35 (C-5), 88.87 (C-1′), 86.41 (C-3′), 73.18 (C-4′), 63.88 (C-5′), 46.61 (C-2′), 42.27 (NH-CH2), 26.82 and 26.60 (≡C-CH3), 23.26 (NH-CH2-CH2), 19.29 and 18.86 (two Si-C≡), 13.83 (5-CH3), 12.49 (propyl-CH3), -3.96 and -4.47 (two Si-CH3). HR-MS (ESI-MS) for C26H51N4O5Si2. Calcd: 555.3398. Found: m/z 555.3376 (M+H)+.
4.1.5.2. 3-(Butylcarbamimidoyl)-3′,5′-(di-tert.-butyldimethylsilyl)thymidine (6b)
Rf: 0.45 (100% dichloromethane), Retention time (preparative HPLC) = 62-70 min, yield = 110 mg, 95% (white amorphous solid); C1H NMR (400 MHz, CD3OD, δ pp0m): 7.66 (s, 1H, H-6), 6.21 (dd, J = 5.8 and 8.0 Hz, 1′H, 1H), 4.40-4.54 (m, 1H, 3′H), 3.93 (dd, J = 3.1 and 5.8 Hz, 1H, 4′H), 3.68-3.90 (m, 2H, 5″H and 5′H), 3.42 (t, J = 7.0 Hz, 2H, NH-CH2), 2.10-2.58 (m, 2H, 2″H and 2′H), 1.91 (s, 3H, 5-CH3), 1.60-1.70 (m, 2H, NH-CH2-CH2-), 1.39-1.40 (m, 2H, butyl-CH2-CH3), 0.95 (t, J = 7.3 Hz, 2H, butyl-CH3), 0.89 and 0.92 (two s, 18H, ≡C-CH3), 0.09 and 0.11 (two s, 12H , Si-CH3). 13C NMR (CDCl3, 100 MHz, δ ppm): 162.48 (C-4 C=O), 153.79 (=N-C-NH), 149.68 (C-2 C=O), 137.66 (C-6), 111.12 (C-5), 88.57 (C-1′), 87.17 (C-3′), 73.77 (C-4′), 64.22 (C-5′), 44.21 (NH-CH2), 41.72 (C- 2′), 30.45(NH-CH2-CH2), 26.52 and 26.29 (≡C-CH3), 20.81 (butyl-CH2-CH3), 19.32 and 18.88 (two Si-C≡), 13.91 (5-CH3), 12.84 (butyl-CH3), −4.59 and −5.18 (two Si-CH3). HR-MS (ESI-MS) for C27H53N4O5Si2. Calcd: 569.3554. Found: m/z 569.3551 (M+H)+ and for C27H52N4NaO5Si2. Calcd: 591.3374. Found: m/z 591.3370 (M+Na)+.
4.1.6. General procedure for the synthesis of 3-(Phenylcarbamimidoyl)-3′,5′-(di-tert.-butyldimethylsilyl)thymidine (6c), and 3-(Benzylcarbamimidoyl)-3′,5′-(di-tert.- butyldimethylsilyl)thymidine (6d) [22]
Compound 5 (100 mg, 0.21 mmol) was dissolved in hexafluoroisopropanol (200 μL), aniline or benzyl amine (0.4 mmol) was added, and the reaction mixture was stirred for overnight at room temperature. The aqueous layer was evaporated under reduced pressure and the residue purified by preparative HPLC.
4.1.6.1. 3-(Phenylcarbamimidoyl)-3′,5′-(di-tert.-butyldimethylsilyl)thymidine (6c)
Rf: 0.70 (1% methanol in dichloromethane), Retention time (preparative HPLC) = 62-72 min, yield = 110 mg, 93% (white amorphous solid); 1H NMR (400 MHz, CD3OD, δ ppm): 7.61 (s, 1H, H-6), 7.20-7.40 (s, 2H, phenyl H), 6.90-7.20 (br s, 3H, phenyl H), 6.15-6.35 (m, 1H, 1′H), 4.48 (m, 1H, 3′H), 3.93-3.4.00 (m, 1H, 4′H), 3.90 (dd, J = 2.5 and 11.5 Hz, 1H, 5″H), 3.83 (dd, J = 1.8 and 11.5 Hz, 1H, 5′H), 2.10-2.30 (m, 2H, 2′H and 2″H), 1.92 (s, 3H, 5-CH3), 0.96 and 0.95 (two s, 18H, ≡C-CH3) 0.15 and 0.16 (two s, 12H, Si-CH3). 13C NMR (CD3OD, 100 MHz, δ ppm): 163.69 (C-4 C=O), 150.69 (=N-C-NH), 147.64 (C-2 C=O), 136.55 (C-6), 130.30, 125.08, 123.14 (Aromatic-C), 111.16 (C-5), 89.36 (C-1′), 86.94 (C-4′), 73.65 (C-3′), 64.15 (C-5′), 41.96 (C-2′), 26.55 and 26.35 (two ≡C-CH3), 19.33 and 18.91 (two Si-C≡), 13.15 (5-CH3), −4.40, −4.54, −5.14 (d, J = 9.8 Hz, Si-CH3), −5.18 (d, J = 3.0 Hz, Si-CH3); HR-MS (ESI-MS) for C29H49N4O5Si2. Calcd: 589.3241. Found: m/z 589.3251 (M+H)+.
4.1.6.2. 3-(Benzylcarbamimidoyl)-3′,5′-(di-tert.-butyldimethylsilyl)thymidine (6d)
Rf: 0.75 (1% methanol in dichloromethane), Retention time (preparative HPLC) = 47-52 min, yield = 120 mg, >99% (white amorphous solid); 1H NMR (400 MHz, CD3OD, δ ppm): 7.74 (d, J = 1.2 Hz, 1H, H-6), 7.35-7.53 (m, 5H, phenyl H), 6.29 (dd, J = 5.9 and 7.8 Hz, 1H, 1′H), 4.73 (s, 2H, NH-CH2-), 4.49-4.53 (m, 1H, 3′H), 4.00 (dd, J = 3.2 and 5.8 Hz, 1H, 4′H), 3.92 (dd, J = 3.3 and 11.5 Hz, 1H, 5″H), 3.85 (dd, J = 3.2 and 11.5 Hz, 1H, 5′H), 2.29 (ddd, J = 2.7, 5.9 and 13.3 Hz, 1H, 2′H), 2.16-2.24 (m, 1H, 2″H), 1.84 (s, 3H, 5-CH3), 0.96 and 0.98 (two s, 18H, ≡C-CH3), 0.15 and 0.17 (two s, 12H, Si-CH3). 13C NMR (CD3OD, 100 MHz, † ppm): 162.58 (C-4 C=O), 154.19 (=N-C-NH), 149.78 (C-2 C=O), 137.80 (C-6), 134.77, 130.15, 129.64, 128.86 (Aromatic-C), 111.19 (C-5), 89.67 (C-1′), 87.26 (C-4′), 73.86 (C-3′), 64.28 (C-5′), 47.98 (NH-CH2-), 41.78 (C-2′), 26.51 and 26.28 (two ≡C-CH3), 19.34 and 18.90 (two Si-C≡), 12.84 (5-CH3), −4.56 (d, J = 9.8 Hz, Si-CH3), −5.18 (d, J = 3.0 Hz, Si-CH3); HR-MS (ESI-MS) for C30H51N4O5Si2. Calcd: 603.3398. Found: m/z 603.3331 (M+H)+ and for C30H50N4NaO5Si2. Calcd: 625.3217. Found: m/z 625.3253 (M+Na)+.
4.1.7. General procedure for the synthesis of 3-[2-(closo-1,7-Carboranyl)ethylcarbamimidoyl]-3′,5′-(di-tert.-butyldimethylsilyl)thymidine (6e), 3-[3-(closo-1,7-Carboranyl)propylcarbamimidoyl]-3′,5′-(di-tert.-butyldimethylsilyl)thymidine (6f), and 3-[4-(closo-1,7-Carboranyl)butylcarbamimidoyl]-3′,5′-(di-tert.- butyldimethylsilyl)thymidine (6g) [22]
Compound 11 (100 mg, 0.21 mmol) was dissolved in hexafluoroisopropanol (200 μL) and closo-1,7-carboranyl)alkyl amine (0.4 mmol) was added. The reaction mixture was stirred overnight at 90 °C. The aqueous layer was evaporated under reduced pressure and the residue purified by preparative HPLC.
4.1.7.1. 3-[2-(closo-1,7-Carboranyl)ethylcarbamimidoyl]-3′,5′-(di-tert.-butyldimethylsilyl)thymidine (6e)
Rf: 0.85 (1% methanol in dichloromethane), Retention time (preparative HPLC) = 42-47 min, yield = 44 mg, >32% (white amorphous solid); 1H NMR (400 MHz, CD3OD, δ ppm): 7.70 (s, 1H, H-6), 6.24 (t,J = 6.6 Hz, 1′H, 1H), 4.45-4.55 (m, 1H, 3′H), 3.93-4.00 (m, 1H, 4′H), 3.89 (dd, J = 2.8 and 11.5 Hz, 1H, 5″H), 3.83 (dd, J = 2.5 and 11.5 Hz, 1H, 5′H), 3.60 (Carbranyl-H), 3.50 (t, J = 7.6 Hz, 2H, NH-CH2), 2.40 (t, J = 7.6 Hz, 2H, Carborane-CH2), 2.11-2.30 (m, 2H, 2′H and 2″H), 1.94 (s, 3H, 5-CH3), 0.93 and 0.96 (two s, 18H, ≡C-CH3), 0.13 and 0.15 (two s, 12H, Si-CH3). 13C NMR (CD3OD, 100 MHz,δ ppm): 162.59 (C-4 C=O), 153.93 (=N-C-NH), 149.79 (C-2 C=O), 137.86 (C-6), 111.28 (C-5), 89.73 (C-1′), 87.34 (C-4′), 73.89 (Carboranyl-C/C-3′), 64.36 (C-5′), 57.54 (Carboranyl-CH), 43.96 (NH-CH2), 41.85 (C-2′), 35.11 (Carborane-CH2), 26.64 and 26.41 (two ≡C-CH3), 19.45 and 19.01 (two Si-C≡), 12.95 (5-CH3), −4.46 and −5.04 (two Si-CH3); HR-MS (ESI-MS) for C27H59B10N4O5Si2. Calcd: 683.5027. Found: m/z 683.5076 (M+H)+.
4.1.7.2. 3-[3-(closo-1,7-Carboranyl)propylcarbamimidoyl]-3′,5′-(di-tert.-butyldimethylsilyl)thymidine (6f)
The reaction conditions described above resulted in a mixture of 3-[3-(closo-1,7-carboranyl)propylcarbamimidoyl]thymidine (7f) along with 3′/5′-mono-TBDMS- and 3′,5′-di-TBDMS-3-[3-(closo-1,7-carboranyl)propylcarbamimidoyl] dThd (6f). Product formation was confirmed by MS using a small quantity of the reaction mixture. Deprotection of remaining TBDMS-groups was accomplished in situ by adding trifluoroacetic acid in dichloromethane (10 mL, 10%, v/v) to the reaction mixture for 4 h at room temperature to give 7f in 29% overall yield. Rf: 0.85 (1% methanol in dichloromethane), HR-MS (ESI-MS) for 6f (C22H47B10N4O5Si). Calcd: 697.5184. Found: m/z 697.5167 (M+H)+. HR-MS (ESI-MS) for 3′/5′-mono-TBDMS-3-[3-(closo-1,7-carboranyl)propylcarbamimidoyl]thymidine (C22H47B10N4O5Si). Calcd: 583.4319. Found: m/z 583.3486 (M+H)+.
4.1.7.3. 3-[4-(closo-1,7-Carboranyl)butylcarbamimidoyl]-3′,5′-(di-tert.-butyldimethylsilyl)thymidine (6g)
Rf: 0.90 (1% methanol in dichloromethane), Retention time (preparative HPLC) = 49-55 min, yield = 55 mg, > 38% (white amorphous solid); 1HNMR (400 MHz, CD13OD, δ ppm): 7.72 (s, 1H, H-6), 6.25 (t, J = 6.6 Hz, 1′H, 1H), 4.45-4.55 (m, 1H, 3′H), 4.10 (q, J = 7.0 Hz, 1H, 4′H), 3.90 (dd, J = 2.5 and 11.5 Hz, 1H, 5″H), 3.84 (dd,J = 2.0 and 11.5 Hz, 1H, 5′H), 3.40-3.60 (m, 3H, Carbranyl-H and NH-CH2), 2.96-3.20 (m, 2H, Carborane-CH2), 2.10-2.30 (m, 2H, 2′H and 2″H) 1.96-2.10 (m, 2H, NH-CH2CH2), 1.96 (s, 3H, 5-CH3), 1.40-1.60 (m, 2H, Carborane-CH2CH2), 0.93 and 0.96 (two s, 18H, ≡C-CH3), 0.13 and 0.15 (two s, 12H, Si-CH ). 133 C NMR (CD3OD, 100MHz, δ ppm): 162.53 (C-4 C=O), 154.09 (=N-C-NH), 149.78 (C-2 C=O), 137.84 (C-6), 111.26 (C-5), 89.80 (C-1′), 87.37 (C-4′), 77.59 (Carboranyl-C), 74.03 (C-3′), 64.42 (C-5′), 57.02 (Carboranyl-CH), 46.23 (NH-CH2), 41.88 (C-2′), 37.71 (Carborane-CH2), 30.51(NH-CH2-CH2), 28.25 (Carborane-CH2-CH2), 28.38 and 28.61 (two ≡C-CH3), 19.44 and 19.00 (two Si-C≡), 12.93 (5-CH3), −4.38 and −5.05 (two Si-CH3); HR-MS (ESI-MS) for C29H63B10N4O5Si2. Calcd: 711.5340. Found: m/z 711.5376 (M+H)+.
4.1.8. General procedure for the synthesis of 3-(Carbamimidoyl)thymidine derivatives (7a-7g)
Compounds 6a 6g (40 mg) were added to a solution of TFA in dichloromethane (10 mL, 10% v/v). The reaction mixtures were stirred at room temperature for 4 h, the solvents were evaporated under reduced pressure, and the residues were purified by preparative HPLC. The percentage purity of the synthesized compounds were > 95%.
4.1.8.1. 3-(Propylcarbamimidoyl)thymidine (7a)
Retention time (analytical HPLC) = 4.8 min, yield = 9 mg, 40% (white amorphous solid); 1H NMR (400 MHz, CDOD3, δ ppm): 8.08 (s, 1H, H-6), 6.28 (t, J = 6.3 Hz, 1′H, 1H), 4.40-4.46 (m, 1H, 3′H), 3.96 (q, J = 3.0 Hz, 1H, 4′H), 3.82 (dd,J = 12.0 and 3.0 Hz, 1H, 5″H), 3.76 (dd, J = 12.0 and 3.3 Hz, 1H, 5′H), 3.42 (t, J = 6.3 Hz, 2H, NH-CH2), 2.20-2.35 (m, 2H, 2″H and 2′H), 1.94 (s, 3H, 5-CH3), 1.69-1.79 (m, 2H, NH-CH2-CH2), 1.06 (t, J = 7.4 Hz, 2H, proypl-CH3). 133 C NMR (CDOD3, 100 MHz, δ ppm): 162.66 (C-4 C=O), 154.02 (NH-C(=NH)-), 149.81 (C-2 C=O), 138.65 (C-6), 110.98 (C-5), 89.40 (C-1′), 87.29 (C-3′), 72.25 (C-4′), 62.73 (C-5′), 46.11 (C-2′), 41.43 (NH-CH2), 21.95 (NH-CH2-CH2), 12.59 (5-CH3), 11.44 (propyl-CH3). HR-MS (ESI-MS) for C14H23N4O5. Calcd: 327.1668. Found: m/z 327.1654 (M+H)+.
4.1.8.2. 3-(Butylcarbamimidoyl)thymidine (7b)
Retention time (analytical HPLC) = 4.4 min, yield = 11 mg, 46% (white amorphous solid); 1H NMR (400 MHz, CDCl3, δ ppm): 8.07 (s, 1H, H-6), 6.28 (t,J = 6.6 Hz, 1H, 1′H), 4.40-4.60 (m, 1H, 3′H), 3.90-4.00 (m, 1H, 4′H), 3.83 (dd, J = 3.0, 12.0 Hz, 1H, 5″H), 3.76 (dd, J = 3.3, 12.0 Hz, 1H, 5′H), 3.46 (t, J = 6.9 Hz, 2H, NH-CH2-), 2.18-2.38 (m, 2H, 2″H and 2′H), 1.94 (s, 3H, 5-CH3 1.65-1.75 (m, 2H, NH-CH2-CH2-), 1.46-155 (m, 2H, butyl-CH2-CH3), 0.99 (t, J = 7.3 Hz, 3H, butyl-CH3).13C NMR (CDCl3, 100 MHz, δ ppm): 162.69 (C-4 C=O), 153.85 (NH-C(=NH)-), 149.77 (C-2 C=O), 138.58 (C-6), 110.95 (C-5), 89.26 (C-1′), 87.26 (C-3′), 72.15 (C-4′), 62.67 (C-5′), 44.20 (NH-CH2), 41.34 (C-2′), 30.44 (NH-CH2-CH2), 20.79 (butyl-CH2-CH3), 13.87 (5-CH3), 12.60 (butyl-CH3). HR-MS (ESI-MS) for C15H25N4O5. Calcd: 341.1825. Found: m/z 341.1853 (M+H)+.
4.1.8.3. 3-(Phenylcarbamimidoyl)thymidine (7c)
Retention time (analytical HPLC) = 8.9 min, yield = 18 mg, 70% (white amorphous solid); 1H NMR (400 MHz, CD3OD, δ ppm): 8.11 (s, 1H, H-6), 7.35-7.63 (m, 5H, phenyl H), 6.27-6.40 (m, 1′H, 1H), 4.35-4.50 (m, 1H, 3′H), 3.90-4.00 (m, 1H, 4′H), 3.84 (d, J = 11.1 Hz, 1H, 5″H), 3.77 (d, J = 11.4 Hz, 1H, 5′H), 2.20-2.40 (m, 2H, 2′H and 2″H), 1.90-2.05 (s, 3H, 5-CH3). 13C NMR (CD3OD, 100 MHz, δ ppm): 162.84 (C-4 C=O), 153.50 (=N-C-NH), 149.96 (C-2 C=O), 138.68 (C-6), 135.96, 131.59, 130.20, 135.53 (Ar-C), 111.04 (C-5), 89.38 (C-1′), 87.33 (C-4′), 72.20 (C-3′), 62.71 (C-5′), 41.47 (C-2′), 12.63 (5-CH3); HR-MS (ESI-MS) for C17H21N4O5. Calcd: 361.1512. Found: m/z 361.1532 (M+H)+.
4.1.8.4. 3-(Benzylcarbamimidoyl)thymidine (7d)
Retention time (analytical HPLC) = 9.6 min, yield = 20 mg, 80% (white amorphous solid); 1H NMR (400 MHz, CD3OD, δ ppm): 8.08 (s, 1H, H-6), 7.34-7.50 (m, 5H, phenyl H), 6.29 (t, 6.6 Hz, 1H, 1′H), 4.71 (s, 2H,NH-CH2-), 4.40-4.47 (m, 1H, 3′H), 3.96 (dd, J = 2.9 and 5.9 Hz, 1H, 4′H), 3.83 (dd, J = 2.9 and 12.0 Hz, 1H, 5″H), 3.76 (dd, J = 3.3 and 12.0 Hz, 1H, 5′H), 2.21-2.37 (m, 2H, 2′H and 2″H), 1.96 (s, 3H, 5-CH3). 13C NMR (CD3OD, 100 MHz, δ ppm): 162.28 (C-4 C=O), 154.27 (=N-C-NH), 149.87 (C-2 C=O), 138.69 (C-6), 134.78, 130.13, 129.63, 128.86 (Aromatic-C), 111.00 (C-5), 89.35 (C-1′), 87.33 (C-4′), 72.18 (C-3′), 62.69 (C-5′), 47.97(NH-CH2-), 41.42 (C-2′), 12.60 (5-CH3); HR-MS (ESI-MS) for C18H23N4O5. Calcd: 375.1668. Found: 375.1673 (M+H)+.
4.1.8.5. 3-[2-(closo-1,7-Carboranyl)ethylcarbamimidoyl]thymidine (7e)
Retention time (analytical HPLC) = 15.0 min, yield = 18 mg, 69% (white wax); 1H NMR (400 MHz, CD3OD, δ ppm): 8.08 (s, 1H, H-6), 6.27 (t, J = 6.6 Hz, 1′H, 1H), 4.45-4.50 (m, 1H, 3′H), 3.93-4.00 (m, 1H, 4′H), 3.82 (dd, J = 2.8 and 12.0 Hz, 1H, 5″H), 3.75 (dd, J = 3.0 and 12.0 Hz, 1H, 5′H), 3.62 (Carboranyl-CH), 3.51 (t, J = 7.6 Hz, 2H, NH-CH2), 2.40 (t, J = 7.6 Hz, 2H, Carborane-CH2), 2.2-2.35 (m, 2H, 2′H and 2″H), 1.94 (s, 3H, 5-CH3). 13C NMR (CD3OD, 100 MHz, δ ppm): 162.67 (C-4 C=O), 154.29 (=N-C-NH), 149.81 (C-2 C=O), 138.84 (C-6), 111.07 (C-5), 89.52 (C-1′), 87.42 (C-4′), 73.84 (Carboranyl-C), 72.34 (C-3′), 62.81 (C-5′), 57.57 (Carboranyl-CH), 43.82 (NH-CH2), 41.51 (C-2′), 34.99 (Carborane-CH2), 12.65 (5-CH3); HR-MS (ESI-MS) for C15H31B10N4O5. Calcd: 455.3298. Found: m/z 455.3270 (M+H)+.
4.1.8.6. 3-[3-(closo-1,7-Carboranyl)propylcarbamimidoyl]thymidine (7f)
Retention time (analytical HPLC) = 20.2 min, yield = 22 mg, 29% (white wax); 1H NMR (400 MHz, CD3OD, δ ppm): 8.08 (s, 1H, H-6), 6.29 (t, J = 6.7 Hz, 1′H, 1H), 4.40-4.45 (m, 1H, 3′H), 3.90-3.95 (m, 1H, 4′H), 3.82 (dd, J = 2.9 and 12.0 Hz, 1H, 5″H), 3.75 (dd, J = 3.2 and 12.0 Hz, 1H, 5′H), 3.54 (s, 1H, Carboranyl-CH), 3.41 (t, J = 6.5 Hz, 2H, NH-CH2), 2.10-2.32 (m, 4H, 2′H and 2″H, Carborane-CH2), 1.95 (s, 3H, 5-CH3), 1.74-1.83 (m, 2H, NH-CH2-CH2). 13C NMR (CD3OD, 100 MHz, δ ppm): 162.66 (C-4 C=O), 153.98 (=N-C-NH), 149.85 (C-2 C=O), 138.65 (C-6), 111.02 (C-5), 89.43 (C-1′), 87.30 (C-4′), 76.67 (Carboranyl-C), 72.30 (C-3′), 62.76 (C-5′), 57.11 (Carboranyl-CH), 43.54 (NH-CH2), 41.42 (C-2′), 34.68 (Carborane-CH2), 29.10 (Carborane-CH2-CH2), 12.61 (5-CH3); HR-MS (ESI-MS) for C16H33B10N4O5. Calcd: 469.3454. Found: m/z 469.3382 (M+H)+.
4.1.8.7. 3-[4-(closo-1,7-Carboranyl)butylcarbamimidoyl]thymidine (7g)
Retention time (analytical HPLC) = 21.2 min, yield = 21 mg, 80% (white wax); 1H NMR (400 MHz, CD3OD, δ ppm): 8.08 (s, 1H, H-6), 6.29 (t, J = 6.7 Hz, 1′H, 1H), 4.40-4.44 (m, 1H, 3′H), 3.92-4.00 (m, 1H, 4′H), 3.83 (dd,J = 2.7 and 12.0 Hz, 1H, 5″H), 3.76 (dd, J = 2.9 and 12.0 Hz, 1H, 5′H), 3.51 (s, 1H, Carboranyl-CH), 3.44 (t, J = 6.4 Hz, 2H, NH-CH2), 2.10- 2.32 (m, 2H, 2′H and 2″H), 2.05 (t J = 8.2 Hz, 2H, Carborane-CH2), 1.95 (s, 3H, 5-CH3), 1.40-1.70 (m, 4H, NH-CH2CH2CH2). 13C NMR (CD3OD, 100 MHz, δ ppm): 162.66 (C-4 C=O), 159.45 (=N-C-NH), 154.17 (C-2 C=O), 138.74 (C-6), 111.06 (C-5), 89.49 (C-1′), 87.40 (C-4′), 77.58 (Carboranyl-C), 72.34 (C-3′), 62.82 (C-5′), 57.03 (Carboranyl-CH), 44.06 (NH-CH2-), 41.51 (C-2′), 37.59 (Carborane-CH2), 28.25 and 28.13 (NH-CH2-CH2), 12.66 (5-CH3); HR-MS (ESI-MS) for C17H35B10N4O5. Calcd: 483.3611. Found: m/z 483.3558 (M+H)+.
4.1.9. 3-(Tetrazol-5-ylmethyl)thymidine (8)
A mixture of 2 (500 mg, 1.75 mmol), sodium azide (350 mg, 5.4 mmol), and zinc bromide (350 mg, 1.5 mmol) were dissolved in water/isopropanol (15 mL, 2/1, v/v) and heated in a sealed tube at 90 °C for three days [23]. The reaction mixture was passed through a 0.2 μm filter (Fisher Scientific, USA), the filtrate evaporated under reduced pressure, and the residue purified by preparative HPLC. Retention time (analytical HPLC) = 15.5 min, Yield = 575 mg, 90% (white amorphous solid); 1H NMR (400 MHz, CD3OD, δ ppm): 7.92 (s, 1H, H-6), 6.31 (t, J = 6.6 Hz, 1H, 1′H), 5.44 (s, 2H, N3-CH2), 4.38-4.48 (m, 1H, 3′H), 3.90-4.00 (m, 1H, 4′H), 3.83 (dd, J = 3.0 and 12.0 Hz, 1H, 5″H), 3.76 (dd, J = 3.6 and 12.0 Hz, 1H, 5′H), 2.20-2.38 (m, 2H, 2′H and 2″H), 1.92 (s, 3H, 5-CH3). 13C NMR (CD3OD, 100 MHz, δ ppm): 164.72 (C-4 C=O), 154.93 (=N-C=N-), 152.00 (C-2 C=O), 137.09 (C-6), 110.68 (C-5), 88.82 (C-1′), 87.23 (C-4′), 72.00 (C-3′), 62.70 (C-5′), 41.23 (C-2′), 35.62 (N3-CH2), 13.13 (5-CH3); HR-MS (ESI-MS) for C12H17N6O5. Calcd: 325.1260. Found: m/z 325.1275 (M+H)+, for C12H16N6Na12O5. Calcd: 347.1080. Found: m/z 347.1108 (M+Na)+ and for C24H33N12O10. Calcd: 649.2443. Found: m/z 649.2494 (2M+H)+.
4.1.10. 3-(Tetrazol-5-yl)thymidine (10)
A mixture of 2 (100 mg, 0.36 mmol), sodium azide (70 mg, 1.1 mmol), and zinc bromide (70 mg, 0.3 mmol) were dissolved in a mixture of DMF, water, and isopropanol (6 mL, 3/2/1, v/v/v). The reaction mixture was stirred for 5 h at 0 °C, then suspended in water (200 mL), and extracted with ethyl acetate (100 mL × 3) [23]. The organic layer was evaporated, the residue dissolved in TFA in dichloromethane (10 mL, 10% v/v), and the resulting mixture was stirred for 4 h at room temperature. The solvents were evaporated and the residue was purified by preparative HPLC. Retention time (analytical HPLC) = 13.2 min, Yield = 25 mg, 44% (white amorphous solid); 1H NMR (400 MHz, CD3OD, δ ppm): 8.05 (s, 1H, H-6), 6.28 (t, J = 6.5 Hz, 1H, 1′H), 4.43 (dd, J = 4.8 and 8.2 Hz, 1H, 3′H), 3.94 (dd, J = 3.2 and 6.5 Hz, 1H, 4′H), 3.84 (dd, J = 3.0 and 12.1 Hz, 1H, 5″H), 3.76 (dd, J = 3.5 and 12.0 Hz, 1H, 5′H), 2.30 (t, J = 5.4 Hz, 2H, 2′H and 2″H), 1.96 (m, 3H, 5-CH3). 13C NMR (CD3OD, 100 MHz, δ ppm): 165.06 (N-C=N), 164.67 (C-4 C=O), 151.51 (C-2 C=O), 138.43 (C-6), 111.12 (C-5), 89.23 (C-1′), 87.55 (C-4′), 72.11 (C-3′), 62.80 (C-5′), 41.54 (C-2′), 13.03 (5-CH3); HR-MS (ESI-MS) for C11H14N6NaO5. Calcd: 333.0923. Found: m/z 333.0939 (M+Na)+.
4.1.11. General procedure for the synthesis of N-alkylated tetrazolyl-type thymidine derivatives (9a, 9b1/2-9d1/2 and 11a, 11b1/2-11d1/2)
Compound 8 or 10 (0.046 mmol), iodoalkyl derivative (0.09 mmol), and potassium carbonate (20 mg, 0.15 mmol) were dissolved in DMF, and acetone (2 mL, 1/1, v/v) and stirred at 50 °C for 24 h. The residues were purified as two isomers (Nb- and Nc-subsituted tetrazolyl dThd analogues) by preparative HPLC. The percentage purities of the synthesized compounds were > 95%.
4.1.11.1. 3-[1(2)-Propyltetrazol-5-ylmethyl]thymidine (9a)
Retention time (analytical HPLC) = 22 min, yield = 10 mg, 59% (white amorphous solid). 1H NMR (400 MHz, CD3OD, δ ppm): 7.93 (s, 1H, H-6), 6.30 (t, J = 6.6 Hz, 1H, 1′H), 5.36 (s, 2H, N3-CH2), 4.57 (t, J = 6.9 Hz, 2H, Nc-CH2), 4.35-4.45 (m, 1H, 3′H), 3.87-3.95 (m, 1H, 4′H), 3.81 (dd, J = 2.8 and 12.1 Hz, 1H, 5″H), 3.76 (dd, J = 3.5 and 12.1 Hz, 1H, 5′H), 2.18-2.38 (m, 2H, 2′H and 2″H), 2.00 (hexaplet, J = 7.2 Hz, 2H, Nc-CH2-CH2), 1.93 (s, 3H, 5-CH3), 0.91 (t, J = 7.4 Hz, 2H, propyl-CH3). 13C NMR (CD3OD, 100 MHz, δ ppm): 165.05 (C-4 C=O), 163.88 (=N-C=N-), 152.26 (C-2 C=O), 137.14 (C-6), 110.83 (C-5), 89.04 (C-1′), 87.40 (C-4′), 72.17 (C-3′), 62.88 (C-5′), 56.02 (N-CH2), 41.23 (C-2′), 37.41 (N3-CH2), 23.88 (N-CH2-CH2), 13.35 (5-CH3), 11.33 (propyl-CH3); HR-MS (ESI-MS) for C15H22N6NaO5. Calcd: 389.1549. Found: m/z 389.1536 (M+Na)+.
4.1.11.2. 3-[1(2)-closo-1,7-Carboranylethyltetrazol-5-ylmethyl]thymidine (9b1/2)
Retention time (analytical HPLC) = 16.1 min, yield = 14 mg, 64% (white wax); isomeric ratio 9b1:9b2 = 20:80. 1H NMR (400 MHz, CD3OD, δ ppm): 7.93 (s, 1H, H-6), 6.29 (t, J = 6.6 Hz, 1H, 1′H), 5.35 (s, 2H, N3-CH2), 4.65 (t, J = 7.5 Hz, 1.7 H, Nc-CH2), 4.52 (t, J = 8.4 Hz, 0.3 H, Nb-CH2), 4.35-4.45 (m, 1H, 3′H), 3.87-3.95 (m, 1H, 4′H), 3.81 (dd, J = 2.9 and 12.0 Hz, 1H, 5″H), 3.74 (dd, J = 3.5 and 12.0 Hz, 1H, 5′H), 3.55 (s, 1H, Carboranyl H), 2.75 (t, J = 7.5 Hz, 2H, Carborane-CH2), 2.19-2.33 (m., 2H, 2′H and 2″H), 1.93 (s, 3H, 5-CH3). 13C NMR (CD33OD, 100 MHz, δ ppm): 164.96 (C-4 C=O), 164.07 (=N-C=N-), 152.19 (C-2 C=O), 137.12 (C-6), 110.80 (C-5), 89.06 (C-1′), 87.35 (C-4′), 73.64 (Carboranyl C), 72.17 (C-3′), 62.84 (C-5′), 57.54 (CarboranylCH), 52.99 (N-CH2), 41.53 (C-2′), 37.28 (Carborane-CH2), 36.07 (N3-CH23); HR-MS for C16H31B10N6O5. Calcd: 495.3359. Found: m/z 495.3372 (M+H)+ and for C16H30B10N6NaO5. Calcd: 517.3179. Found: m/z 517.3160 (M+Na)+.
1H-1H NOESY (800 MHz, CD3OD): Cross-peak observed between the triplet at δ 4.52 and the singlet at 5.35 ppm. T1 relaxation rates were measured using an inversion recovery experiment at 250 MHz and were calculated using TopSpin 3.1 (Bruker, USA). A 1H-1H NOESY with presaturation on the methanol-OH at δ 4.892 ppm was collected at 800 MHz in CD3OD with a NOESY mixing time of 0.69 s based on the average T1 relaxation time of the protons of interest, a spectral width of 1.7 ppm in both dimensions centered at 4.874 ppm, 256 points in the indirect dimension, 1024 points in the direct dimension, and a 3 s recycle delay. The NOESY data were processed using TopSpin 3.1. The data were zero filled to 2048 points in the direct dimension and 1024 points in the indirect dimension (see Supplementary material).
4.1.11.3. 3-[1(2)-closo-1,7-Carboranylpropyltetrazol-5-ylmethyl]thymidine (9c1/2)
Retention time (analytical HPLC) = 16.8 min, yield = 14 mg, 60% (white wax); isomeric ratio 9c1:9c2 = 15:85. 1H NMR (400 MHz, CD3OD, δ ppm): 7.95 (s, 1H, H-6), 6.31 (t, J = 6.6 Hz, 1H, 1′H), 5.36 (s, 2H, N3-CH2), 4.57 (t, J = 6.1 Hz, 1.7 H, Nc-CH2), 4.46 (t, J = 6.5 Hz, 0.3 H, Nb-CH2), 4.36-4.43 (m, 1H, 3′H), 3.86-3.97 (m, 1H, 4′H), 3.82 (dd, J = 3.0 and 12.0 Hz, 1H, 5″H), 3.74 (dd, J = 3.5 and 12.0 Hz, 1H, 5′H), 3.51 (s, 1H, Carboranyl H), 2.18-2.33 (m, 2H, 2′H and 2″H), 1.96-2.15 (m, 4H, Carborane-CH2-CH2), 1.94 (s, 3H, 5-CH3). 13C NMR (CD3OD, 100 MHz, δ ppm): 165.02 (=N-C=N-), 164.13 (C-4 C=O), 152.26 (C-2 C=O), 137.15 (C-6), 110.83 (C-5), 89.10 (C-1′), 87.37 (C-4′), 76.66 (CarboranylC), 72.20 (C-3′), 62.87 (C-5′), 57.18 (Carboranyl CH), 53.31 (N-CH2), 41.58 (C-2′), 37.36 (Carborane-CH2), 34.60 (N3-CH2), 30.44 (Carborane-CH2-CH2), 13.30 (5-CH3); HR-MS (ESI-MS) for C17H33B10N6O5. Calcd: 509.3516. Found: m/z 509.3509 (M+H)+ and for C17H32B10N6NaO5. Calcd: 531.3335. Found: m/z 531.3341 (M+Na)+.
4.1.11.4. 3-[1(2)-closo-1,7-Carboranylbutyltetrazol-5-ylmethyl]thymidine (9d1/2)
Retention time (analytical HPLC) = 17.2 min, yield = 16 mg, 62% (white wax); isomeric ratio 9d1:9d2 = 30:70. 1H NMR (400 MHz, CD3OD, δ ppm): 7.94 (s, 1H, H-6), 6.30 (t, J = 6.4 Hz, 1H, 1′H), 5.36 (s, 2H, N3-CH ), 4.60 (t, J = 6.7 Hz, 1.5 H, Nc-CH2), 4.51 (t, J = 7.1 Hz, 0.5 H, Nb-CH2), 4.35-4.45 (m, 1H, 3′H), 3.87-3.95 (m, 1H, 4′H), 3.81 (dd, J = 2.9 and 12.0 Hz, 1H, 5″H), 3.73 (dd, J = 3.3 and 12.0 Hz, 1H, 5′H), 3.48 (s, 1H, Carboranyl H), 2.20-2.35 (m, 2H, 2′H and 2″H), 2.06 (t, J = 8.5 Hz, 0.5 H, Carborane-CH2), 1.99 (t, J = 8.5 Hz, 1.5 H, Carborane-CH2), 1.82-1.96 (m, 7H, Carborane-CH2-CH2, 5-CH3), 1.83 (t, J = 7.4 Hz, 2H, Carborane-CH2-CH2). 13C NMR (CD3CN, 100 MHz, δ ppm): 163.70 (C-4 C=O), 163.58 (=N-C=N-), 151.62 (C-2 C=O), 136.37 and 136.12 (C-6), 110.14 (C-5), 88.26 and 88.21 (C-1′), 86.52 and 86.40 (C-4′), 77.32 (Carboranyl C), 71.59 and 71.49 (C-3′), 62.42 and 62.35 (C-5′), 56.73 (Carboranyl CH), 53.19 (N-CH2), 40.70 (C-2′), 36.81 (Carborane-CH2), 36.41 (N3-CH2), 29.07 (N-CH2-CH2), 27.29 (Carborane-CH2-CH2), 13.21 (5-CH3); HR-MS (ESI-MS) for C18H35B10N6O5. Calcd: 523.3672. Found: m/z 523.3672 (M+H)+ and for C18H34B10N6NaO5. Calcd: 545.3492. Found: m/z 545.3411 (M+Na)+.
4.1.11.5. 3-[1(2)-Propyltetrazol-5-yl]thymidine (11a)
Retention time (analytical HPLC) = 19.5 min, yield = 12 mg, 71% (white amorphous solid). 1H NMR (400 MHz, CD3OD, δ ppm): 8.05 (s, 1H, H-6), 6.28 (t, J = 6.6 Hz, 1H, 1′H), 4.70 (t, J = 6.9 Hz, 2H, Nc-CH2), 4.43 (dd, J = 4.7 and 8.2 Hz, 1H, 3′H), 3.90-4.00 (m, 1H, 4′H), 3.83 (dd, J = 3.0 and 12.1 Hz, 1H, 5″H), 3.75 (dd, J = 3.5 and 12.1 Hz, 1H, 5′H), 2.30 (dd, J = 5.2 and 6.3 Hz, 2H, 2′H and 2″H), 2.07 (hexaplet, J = 7.2 Hz, 2H, N-CH2-CH2), 1.95 (m, 3H, 5-CH3), 0.98 (t, J = 7.4 Hz, 2H, propyl-CH3). 13C NMR (CD3OD, 100 MHz, δ ppm): 164.63 (C-4 C=O), 157.85 (=N-C=N), 151.56 (C-2 C=O), 138.56 (C-6), 111.06 (C-5), 89.26 (C-1′), 87.50 (C-4′), 72.18 (C-3′), 62.83 (C-5′), 56.88 (N-CH2), 41.45 (C-2′), 23.80 (N-CH2-CH2), 12.98 (5-CH3), 11.22 (propyl-CH3); HR-MS (ESI-MS) for C14H20N6NaO5. Calcd: 375.1393. Found: m/z 375.1409 (M+Na)+.
4.1.11.6. 3-[1(2)-closo-1,7-Carboranylethyltetrazol-5-yl]thymidine (11b1/2)
Retention time (analytical HPLC) = 16.6 min, yield = 13 mg, 56% (white wax); isomeric ratio 11b1:11b2 = 5:95. 1H NMR (400 MHz, CD3OD, δ ppm): 8.05 (s, 1H, H-6), 6.27 (t, J = 6.6 Hz, 1H, 1′H), 4.78 (t, J = 7.6 Hz, 1.9 H, Nc-CH b2), 4.55-4.65 (m, 0.1 H, Nb-CH2), 4.43 (dd, J = 4.6 and 8.1 Hz, 1H, 3′H), 3.91-3.94 (m, 1H, 4′H), 3.83 (dd, J = 2.9 and 12.0 Hz, 1H, 5″H), 3.75 (dd, J = 3.5 and 12.0 Hz, 1H, 5′H), 3.59 (s, 1H, Carboranyl H), 2.84 (t, J = 7.6 Hz, 2H, Carborane-CH2), 2.30 (t, J = 5.6 Hz, 2H, 2′H and 2″H), 1.96 (m, 3H, 5-CH3). 13C NMR (CD3OD, 100 MHz, δ ppm): 164.49 (C-4 C=O), 157.87 (N-C=N), 151.43 (C-2 C=O), 138.53 (C-6), 110.97 (C-5), 89.19 (C-1′), 87.43 (C-4′), 73.44 (CarboranylC), 72.09 (C-3′), 62.73 (C-5′), 57.59 (CarboranylCH), 53.79 (N-CH2), 41.36 (C-2′), 35.92 (Carborane-CH2), 12.87 (5-CH3); HR-MS (ESI-MS) for C15H28B10N6NaO5. Calcd: 503.3022. Found: m/z 503.3067 (M+Na)+.
4.1.11.7. 3-[1(2)-closo-1,7-Carboranylpropyltetrazol-5-yl]thymidine (11c1/2)
Retention time (analytical HPLC) = 16.6 min, yield = 10 mg, 42% (white wax); isomeric ratio 11c1:11c2 = 5:95. 1H NMR (400 MHz, CD3OD, δ ppm): 8.05 (s, 1H, H-6), 6.28 (t, J = 6.6 Hz, 1H, 1′H), 4.70 (t, J = 6.8 Hz, 2H, Nc-CH2), 4.35-4.48 (m, 1H, 3′H), 3.90-4.00 (m, 1H, 4′H), 3.84 (dd, J = 2.7 and 12.0 Hz, 1H, 5″H), 3.76 (dd, J = 3.3 and 12.0 Hz, 1H, 5′H), 3.51 (s, 1H, Carboranyl H), 2.31 (t, J = 6.6 Hz, 2H, 2′H and 2″H), 2.04-2.19 (m, 9H, Carborane-CH2-CH2), 1.96 (s, 3H, 5-CH3). NMR (CD3OD, 100 MHz, δ ppm): 164.63 (C-4 C=O), 157.90 (N-C=N), 151.47 (C-2 C=O), 138.52 (C-6), 110.99 (C-5), 89.19 (C-1′), 87.43 (C-4′), 76.55 (Carboranyl C), 72.10 (C-3′), 62.74 (C-5′), 57.08 (Carboranyl CH), 54.18 (N-CH2), 41.35 (C-2′), 34.44 (N-CH2-CH2), 30.34 (Carborane-CH2), 12.88 (5-CH3); HR-MS (ESI-MS) for C16H30B10N6NaO5. Calcd: 517.3179. Found: m/z 517.3128 (M+Na)+.
4.1.11.8. 3-[1(2)-closo-1,7-Carboranylbutyltetrazol-5-yl]thymidine (11d1/2)
Retention time (analytical HPLC) = 17.8 min, yield = 11 mg, 45% (white wax); isomeric ratio 11d1:11d2 = 10:90. 1H NMR (400 MHz, CD3OD, δ ppm): 8.05 (s, 1H, H-6), 6.28 (t, J 6.6 Hz, 1H, 1′H), 4.73 (t, J = 6.8 Hz, 1.6 H, Nc-CH2), 4.56 (t, J = 6.9 Hz, 0.4 H, Nb-CH2), 4.43 (dd, J = 4.6 and 8.2 Hz, 1H, 3′H), 3.94 (dd, J = 3.0 and 6.2 Hz, 1H, 4′H), 3.84 (dd, J = 2.9 and 12.0 Hz, 1H, 5″H), 3.76 (dd, J = 3.5 and 12.0 Hz, 1H, 5′H), 3.47 (s, 1H, Carboranyl H), 2.30 (t, J = 5.7 Hz, 2H, 2′H and 2″H), 1.84-2.10 (m, 9 H, Carborane-CH2-CH2,-CH2 5-CH3). 13C NMR (CD3OD, 100 MHz, δ ppm): 164.59 (C-4 C=O), 157.93 (N-C=N), 151.53 (C-2 C=O), 138.57 (C-6), 111.05 (C-5), 89.28 (C-1′), 87.52 (C-4′), 77.58 (CarboranylC), 72.20 (C-3′), 62.84 (C-5′), 57.01 (Carboranyl CH), 54.67 (N-CH2), 41.46 (C-2′), 37.23 (N-CH2-CH2), 29.66 (Carborane-CH2), 27.89 (Carborane-CH2-CH2), 12.99 (5-CH3); HR-MS (ESI-MS) for C17H33B10N6O5. Calcd: 509.3516. Found: m/z 509.3457 (M+H)+, for C17H32B10N6NaO5. Calcd: 531.3335. Found: m/z 531.3365 (M+Na)+ 5 and for C17H32B10N6KO5. Calcd: 547.3074. Found: m/z 5 547.3081 (M+K)+.
4.2. Enzymatic studies
4.2.1. Phosphorylation Transfer Assays (PTAs)
Recombinant hTK1 was expressed and purified using the bacterial expression system [BL21(DE3) pLysS transformed with the vector hTK1-pET-14b], as described previously [29,30]. The activity of the enzyme with 100 μM dThd was determined to be 441 nmol dThd monophosphate formed per minute and per mg of hTK1 protein.
For PTA studies, dThd and the N3-substituted dThd derivatives were dissolved in DMSO (100 mM concentration) and further diluted with water to produce stock solutions of concentrations (0.4 mM). The phosphorylation transfer assays were carried out as described previously with minor modifications [29-31]. The reaction mixtures contained 100 μM nucleoside and 100 μM ATP (with a small fraction of 0.13 μM [γ-32P]ATP (10 μCi/μL), 25 mM Tris-HCl (pH 7.6), 5 mM MgCl2, 125 mM KCl, 10 mM DTT, and 0.5 mg/mL bovine serum albumin (BSA) in a total volume of 20 μL. In all reactions, the final concentration of DMSO was set to < 1%. The reaction mixtures were incubated at 37 °C for 20 min in the presence of 135 ng of enzyme. Following the incubation period, the enzyme was inactivated by heating for 5 min at 99 °C. The reaction mixtures were centrifuged and 2 μL sample portions were spotted on PEI–cellulose TLC plates (EMD Chemicals Inc.). The TLC plates were developed in a solvent system containing isobutyric acid:ammonium hydroxide:water (66:1:33) over a period of 6 h. The radiolabeled spots were visualized by placing the TLC plates on a BioMax XAR film (Kodak) overnight and developing the films using a X-ray film developer (Tiba M6B, Series VI B Rapid processor; Commonwealth X-Ray Inc.). The spot intensities of the phosphorylated compounds were calculated with Adobe Photoshop (Adobe Systems Incorporated, USA).
4.2.2. Enzyme Kinetic Studies (Km and kcat values)
The enzyme kinetics studies were carried out using the protocol described above for PTAs with varying substrate concentrations. The reaction mixtures contained 0.1-300 μM nucleoside and 100 μATP (with a small fraction of 0.325 μM [γ-32P]ATP (Perkin Elmer), 25 mM Tris-HCl (pH 7.6), 5 mM MgCl2, 125mM KCl, 10 mM DTT, and 0.5 mg/mL bovine serum albumin (BSA). The kinetic parameters were calculated with the enzyme kinetic module of SigmaPlot 12 (Systat Software Inc, USA) using the Michaelis-Menton equation.
4.2.3. Phosphorylation Transfer Assays for in vitro Metabolite Determination
Compound 7f was dissolved in DMSO (10 mM concentration) and further diluted with water to a concentration of 1 mM. The phosphorylation transfer assay was carried out as described in section 4.2.1 with minor modifications. The reaction mixture contained 250 μM 7f and 1 mM 31P-ATP, 25 mM Tris-HCl (pH 7.6), 5 mM MgCl2, 125 mM KCl, 10 mM DTT, and 0.5 mg/mL bovine serum albumin (BSA) in a total volume of 2 mL. The reaction mixture was incubated at 37 °C for 24 h in the presence of 1.5 μg of enzyme. Following the incubation period, the enzyme was inactivated by heating for 5 min at 99 °C. The reaction mixture was centrifuged and the supernatant were analyzed by LR-ESI-MS to detect the presence of dTMP and 7f-monophosphate.
4.3. Solubility Studies
The protocol for the solubility studies was generously provided by Dr. Hardeep Singh Saluja (College of Pharmacy, The Southwestern Oklahoma State University). Calibration curves for known compound concentrations were generated using area under the curves (AUCs) from analytical HPLC data. These calibration curves were used to determine the unknown quantities of compounds dissolved in PBS buffer (pH 7.4). The quantity of ~ 5 mg of N5-2OH, 3e, 3f, 9b1/2-9d1/2, or 11b1/2-11d1/2 were added to micro centrifuge tubes containing PBS (0.2-1 mL, pH 7.4). The micro centrifuge tubes were placed in a shaker overnight and centrifuged at 3300 rpm for 5 min. The solutions were passed through 0.2 μm filters and analyzed by HPLC to obtain the AUCs corresponding to amount of the drug soluble in PBS buffer.
Supplementary Material
Highlights.
Four libraries of N3-carboranyl thymidine analogues (3CTAs) were synthesized.
3CTAs were evaluated for improved binding to human thymidine kinase 1.
Selected 3CTAs were subjected to in-depth enzyme kinetics studies (rkcat/Km
Solubility studies of 3CTAs were conducted in PBS at pH 7.4.
Table 2.
Enzymatic properties of selected N3-substituted dThd analogues.
| Compounds | Km (^M) ± SD1 | kcat (S-1) ± SD1 | rkcat/Km (%) |
|---|---|---|---|
| dThd | 4.9 ± 1.2 | 0.46 ± 0.03 | 100.0 |
| N5-2OH | 41.5 ± 10.0 | 0.42 ± 0.03 | 10.8 |
| 9a | 37.3 ± 7.0 | 0.45 ± 0.03 | 12.9 |
| 9b1/2 | 31.8 ± 8.4 | 0.44 ± 0.03 | 14.9 |
| 9c1/2 | 55.2 ± 15.8 | 0.39 ± 0.04 | 7.6 |
| 9d1/2 | 56.3 ± 13.2 | 0.47 ± 0.04 | 8.9 |
| 11a | 14.0 ± 2.4 | 0.34 ± 0.02 | 26.0 |
| 11b1/2 | 35.6 ± 10.8 | 0.35 ± 0.03 | S 10.7 |
| 11c1/2 | 79.9 ± 36.4 | 0.37 ± 0.06 | 5.0 |
| 11d1/2 | 17.4 ± 5.4 | 0.32 ± 0.02 | 19.7 |
Data represent the means of three replicates ± SD
Acknowledgment
The present study was supported by funds from The Ohio State University College of Pharmacy, NIH grant 5R01 CA127935, and the National Center for Research Resources award UL1RR025755. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Center for Research Resources or the National Institutes of Health. The authors thank Drs. Robert A. Baiocchi, John Byrd, and Fengting Yang from the Division of Hematology and Oncology, Department of Internal Medicine, at The Ohio State University for generously availing their laboratory facilities and assistance for work related to hTK1 expression and purification as well as some of the phosphoryl transfer assays. The authors also thank Dr. Hardeep Singh Saluja from College of Pharmacy, The Southwestern Oklahoma State University, for providing the protocol for the solubility studies and Dr. Robert W. Curley, Jr., for his help with the 1H-1H NOESY NMR studies.
Abbreviations
- ATP
Adenosine triphosphate
- 3CTAs
3-Carboranyl Thymidine Analogues
- BNCT
Boron Neutron Capture Therapy
- PBS
phosphate buffered saline
- 10B
boron-10
- hTK1
human thymidine kinase 1
- KMT
Kinase Mediated Trapping
- DMSO
dimethyl sulfoxide
- dThd
thymidine
- DMF
dimethyl formamide
- TBDMS
tert.-butyldimethylsilyl
- TBAF
tetrabutylammonium fluoride
- HR-MS
High Resolution Mass Spectra
- LR-MS
Low Resolution Mass Spectra
- ESI-MS
Electrospray Ionization Mass Spectra
- AUCs
areas under the curves
- PTA
Phosphorylation Transfer Assay
- BSA
Bovine serum albumin
- dTMP
thymidine monophosphate
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Appendix A. Supplementary material Supplementary data related to this article can be found online, at ……………… Data include original MS-, 1H-NMR-, and 13C-NMR spectra, HPLC chromatograms, 1H-1H NOESY NMR data, HPLC methods, as well as methods for hTK1 purification and the determination of hTK1 concentrations.
References
- [1].Tjarks W, Tiwari R, Byun Y, Narayanasamy S, Barth RF. Carboranyl thymidine analouges for neutron capture therapy. Chem. Commun. 2007:4978–4991. doi: 10.1039/b707257k. [DOI] [PubMed] [Google Scholar]
- [2].Soloway AH, Tjarks W, Barnum BA, Rong F–G, Barth RF, Codogni IM, Wilson JG. The chemistry of neutron capture therapy. Chem. Rev. 1998;98:1515–1562. doi: 10.1021/cr941195u. [DOI] [PubMed] [Google Scholar]
- [3].Sherley JL, Kelly TJ. Human cytosolic thymidine kinase. Purification and physical characterization of the enzyme from HeLa cells. J. Biol. Chem. 1988;263:375–382. [PubMed] [Google Scholar]
- [4].Shields AF, Grierson JR, Dohmen BM, Machulla HJ, Stayanoff JC, Crews J. M. Lawhorn, Obradovich JE, Muzik1 O, Mangner TJ. Imaging proliferation in vivo with [F-18]FLT and positron emission tomography. Nat. Med. 1998;4:1334–1336. doi: 10.1038/3337. [DOI] [PubMed] [Google Scholar]
- [5].Byun Y, Thirumamagal BTS, Yang W, Eriksson S, Barth RF, Tjarks W. Preparation and biological evaluationof 10B-Enriched 3-[5-{2-(2,3-dihydroxyprop-1-yl)-o-carboran-1-yl}pentan-1-yl]thymidine (N5-2OH) J. Med. Chem. 2006;49:5513–5523. doi: 10.1021/jm060413w. [DOI] [PubMed] [Google Scholar]
- [6].Barth RF, Yang W, Madhoun A.S. Al, Johnsamuel J, Byun Y, Chandra S, Smith DR, Tjarks W, Eriksson S. Boron-containing nucleosides as potential delivery agents for neutron capture therapy of brain tumors. Cancer Res. 2004;64:6287–6295. doi: 10.1158/0008-5472.CAN-04-0437. [DOI] [PubMed] [Google Scholar]
- [7].Madhoun A. S. Al, Johnsamuel J, Yan J, Ji W, Wang J, Zhuo JC, Lunato AJ, Woollard JE, Hawk AE, Cosquer GY, Blue TE, Eriksson S, Tjarks W. Synthesis of a small library of 3-(carboranylalkyl)thymidines and their biological evaluation as substrates for human thymidine kinases 1 and 2. J. Med. Chem. 2002;45:4018–4028. doi: 10.1021/jm020047q. [DOI] [PubMed] [Google Scholar]
- [8].Barth RF, Yang W, Wu G, Swindall M, Byun Y, Narayanasamy S, Tjarks W, Tordoff K, Moeschberger ML, Eriksson S, Binns PJ, Riley KJ. Thymidine kinase 1 as a molecular target for boron neutron capture therapy of brain tumors. Proc. Natl. Acad. Sci. USA. 2008;105:17493–17497. doi: 10.1073/pnas.0809569105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Madhoun A. S. Al, Johnsamuel J, Barth RF, Tjarks W, Eriksson S. Evaluation of human thymidine kinase 1 substrates as new candidates for boron neutron capture therapy. Cancer Res. 2004;64:6280–6286. doi: 10.1158/0008-5472.CAN-04-0197. [DOI] [PubMed] [Google Scholar]
- [10].Welin M, Kosinska U, Mikkelsen NE, Carnrot C, Zhu C, Wang L, Eriksson S, Pertersen B. Munch, Eklund H. Structures of thymidine kinase 1 of human and mycoplasmic origin. Proc. Natl. Acad. Sci. U. S. A. 2004;101:17970–17975. doi: 10.1073/pnas.0406332102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Kosinska U, Carnrot C, Eriksson S, Wang L, Eklund H. Structure of the substrate complex of thymidine kinase from Ureaplasma urealyticum and investigations of possible drug targets for the enzyme. FEBS J. 2005;272:6365–6372. doi: 10.1111/j.1742-4658.2005.05030.x. [DOI] [PubMed] [Google Scholar]
- [12].Peña D. Segura, Lutz S, Monnerjahn C, Konrad M, Lavie A. Binding of ATP to TK1-like enzymes is associated with a conformational change in the quaternary structure. J. Mol. Biol. 2007;369:129–141. doi: 10.1016/j.jmb.2007.02.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Birringer MS, Claus MT, Folkers G, Kloer DP, Schulz GE, Scapozza L. Structure of a type II thymidine kinase with bound dTTP. FEBS Lett. 2005;579:1376–1382. doi: 10.1016/j.febslet.2005.01.034. [DOI] [PubMed] [Google Scholar]
- [14].Peña1 D. Segura, Lichter J, Trani M, Konrad M, Lavie A, Lutz S. Quaternary structure change as a mechanism for the regulation of thymidine kinase 1-like enzymes. Structure. 2007;15:1555–1566. doi: 10.1016/j.str.2007.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Kosinska U, Carnrot C, Sandrini MPB, Clausen AR, Wang L, Piskur J, Eriksson S, Eklund H. Structural studies of thymidine kinases from Bacillus anthracis and Bacillus cereus provide insights into quaternary structure and conformational changes upon substrate binding. FEBS J. 2007;274:727–737. doi: 10.1111/j.1742-4658.2006.05617.x. [DOI] [PubMed] [Google Scholar]
- [16].Omari KE, Solaroli N, Karlsson A, Balzarini J, Stammers DK. Structure of vaccinia virus thymidine kinase in complex with dTTP: Insights for drug design. BMC Struct. Biol. 2006;6:22. doi: 10.1186/1472-6807-6-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Wojtczak BA, Andrysiak A, Grüner B, Lesnikowski ZJ. “Chemical Ligation”: A versatile method for nucleoside modification with boron clusters. Chem. Eur. J. 2008;14:10675–10682. doi: 10.1002/chem.200801053. [DOI] [PubMed] [Google Scholar]
- [18].Semioshkin A, Laskova J, Wojtczak B, Andrysiak A, Godovikov I, Bregadze V, Lesnikowski ZJ. Synthesis of closo-dodecaborate based nucleoside conjugates. J. Organomet. Chem. 2009;694:1375–1379. [Google Scholar]
- [19].Agarwal HK, Ricks KG, Buszek B, Tjarks W. Synthesis of closo-1,7-carboranyl alkyl amines. Tetrahedron Lett. 2011;52:5664–5667. doi: 10.1016/j.tetlet.2011.08.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Litosh VA, Wu W, Stupi BP, Wang J, Morris SE, Hersh MN, Metzker ML. Improved nucleotide selectivity and termination of 3′-OH unblocked reversible terminators by molecular tuning of 2-nitrobenzyle alkylated HOMedU triphosphates. Nucleic Acids Res. 2011;39:e39. doi: 10.1093/nar/gkq1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Kaupp G, Schmeyers J, Boy J. Quantitative gas-solid reactions with ClCN and BrCN: Synthesis of cynamides, cynates, thiocynates and their derivatives. Chem. Eur. J. 1998;4:2467–2474. [Google Scholar]
- [22].Snider BB, O′Hare SM. Synthesis of the hindered N,N,N’-trisubstituted guanidine moiety of martinelline and martinellic acid. Tetrahedron Lett. 2001;42:2455–2458. [Google Scholar]
- [23].Himo F, Demko ZP, Noodleman L, Sharpless KB. Why is tetrazole formation by addition of azide to organic nitriles catalyzed by Zinc(II) salts? J. Am. Chem. Soc. 2003;125:9983–9987. doi: 10.1021/ja030204q. [DOI] [PubMed] [Google Scholar]
- [24].Sveshnikov NN, Nelson JH. Discrimination of structural isomers of N-methylated and N-tert-butylated tetrazoles by 13C and 15N NMR. Long-Range 15N, 1H coupling constants. Magn. Reson. Chem. 1997;35:209–212. [Google Scholar]
- [25].Purchase CF, White AD. Alkylation of tetrazoles using Mitsunobu conditions. Synthetic Commun. 1996;26:2687–2694. [Google Scholar]
- [26].Narayanasamy S, Thirumamagal BTS, Johnsamuel J, Byun Y, Al-Madhoun AS, Usova E, Cosquer GY, Yan J, Bandyopadhyaya AK, Tiwari R, Eriksson S, Tjarks W. Hydrophilically enhanced 3-carboranyl thymidine analogues (3CTAs) for boron neutron capture therapy (BNCT) of cancer. Bioorg. Med. Chem. 2006;14:6886–6899. doi: 10.1016/j.bmc.2006.06.039. [DOI] [PubMed] [Google Scholar]
- [27].Dai W, Dove C. Pollock, Dong LC, Li S. Advanced screening assays to rapidly identify solubility-enhancing formulations: High-throughput. Adv. Drug Deliv. Rev. 2008;60:657–672. doi: 10.1016/j.addr.2007.10.017. [DOI] [PubMed] [Google Scholar]
- [28].Hasabelnaby S, Goudah A, Agarwal HK, Abd alla MM, Tjarks W. Synthesis, chemical and enzymatic hydrolysis, and aqueous solubility of amino acid ester prodrugs of 3-carboranyl thymidine analogs for boron neutron capture therapy of brain tumors. Eur. J. Med. Chem. 2012;55:325–334. doi: 10.1016/j.ejmech.2012.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Lunato AJ, Wang J, Woollard JE, Anisuzzaman AKM, Ji W, Rong FG, Ikeda S, Soloway AH, Eriksson S, Ives DH, Blue TE, Tjarks W. Synthesis of 5-(carboranylalkylmercapto)-2-deoxyuridines and 3-(carboranylalkyl)thymidines and their evaluation as substrates for human thymidine kinases 1 and 2. J. Med. Chem. 1999;42:3373–3389. doi: 10.1021/jm990125i. [DOI] [PubMed] [Google Scholar]
- [30].Wang L, Petersen B. Munch, Sjöberg AH, Hellman U, Bergman T, Jörnvall H, Eriksson S. Human thymidine kinase 2: Molecular cloning and charactersation of the enzyme activity with antiviral and cytostatic nucleoside substrates. FEBS Lett. 1999;443:170–174. doi: 10.1016/s0014-5793(98)01711-6. [DOI] [PubMed] [Google Scholar]
- [31].Tiwari R, Toppino A, Agarwal HK, Hua T, Byun Y, Gallucci J, Hasabelnaby S, Goudah A, Darby MV, Barth RV, Tjarks W. Synthesis, biological evaluation, and radioiodination of halogenated closo-carboranylthymidine analogues. Inorg. Chem. 2012;51:629–639. doi: 10.1021/ic202150b. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.