

Abstract
Background
We initiated this preclinical study in order to analyze the impact of sorafenib single treatment versus combination treatment in human colorectal cancer.
Methods
The effect of increasing sorafenib doses on proliferation, apoptosis, migration, and activation of signal cascades was analyzed in vitro. The effect of sorafenib single treatment versus 5-fluorouracil (5-FU) single treatment and combination therapy on in vivo proliferation and target cytokine receptor/ligand expression was analyzed in a human colon cancer xenograft mouse model using HT29 tumor cells.
Results
In vitro, SW480 and HT29 cell lines were sensitive to sorafenib, as compared to Caco2 and SW620 cell lines, independent of the mutation status of K-ras, Raf, PTEN, or PI3K. The effect on migration was marginal, but distinct differences in caspases activation were seen. Combination strategies were beneficial in some settings (sorafenib + 5-FU; irinotecan) and disadvantageous in others (sorafenib + oxaliplatin), depending on the chemotherapeutic drug and cell line chosen. Sensitive cell lines revealed a downregulation of AKT and had a weak expression level of GADD45β. In resistant cell lines, pp53 and GADD45β levels decreased upon sorafenib exposure. In vivo, the combination treatment of sorafenib and 5-FU was equally effective as the respective monotherapy concerning tumor proliferation. Interestingly, treatment with either sorafenib or 5-FU resulted in a significant decrease of VEGFR1 and PDGFRβ expression intensity.
Conclusions
In colorectal cancer, a sensitivity towards sorafenib exists, which seems similarly effective as a 5-FU monotherapy. A combination therapy, in contrast, does not show any additional effect.
Keywords: Colorectal cancer, TKI, Sorafenib
Introduction
Colorectal cancer ranges among the three most frequent malignancies in Western countries [1, 2]. Survival is determined by local recurrence, lymphatic, and hematogenous dissemination [3]. Due to improved therapeutic strategies, the overall survival in stage IV colorectal cancer has increased from 8 months to more than 2 years during the last decade [4–6].
Besides new chemotherapeutic drugs, such as platinum derivates (oxaliplatin) and topoisomerase II inhibitors (irinotecan), the introduction of biologicals targeting tumor neovascularization or growth signaling significantly has improved response and prognosis [4–6].
Specific mutations in tumor-suppressor genes (APC, DCC, p53) and oncogenes (K-ras) occur during the adenoma–carcinoma sequence of colorectal cancer [7–9]. The K-ras mutation status was the first key to personalized therapy in colorectal cancer, as anti-EGFR strategies were shown to be effective in K-ras wild types only [10].
Receptor tyrosine kinases (RTKs) are transmembrane receptors containing extracellular ligand-binding domains connected to intracellular catalytic domains [11]. The growth factors VEGF/PDGF/EGF and their receptors VEGFR1-3, PDGFRα/β, and EGFR are critical in the process of (lymphatic) neo-angiogenesis and dissemination in human cancer [12–16]. Inhibition of RTKs with sorafenib has been successful in renal and hepatocellular cancers [17, 18]. Two phase I studies revealed a disease stabilization in pretreated colorectal cancer patients receiving sorafenib in combination with either irinotecan or oxaliplatin [19, 20]. However, recent phase II/III studies testing other multi-tyrosine kinase inhibitors in human colorectal cancer have failed to show any benefit [21]. So far only one randomized Phase III study with Regorafenib improved survival times after failure of all approved standard therapies [22]. Therefore, the impact of combinational therapies (sorafenib + chemotherapy) remains controversial. Preclinical data as well as experimental data explaining interaction mechanisms are widely missing. Thus, we initiated this study to examine sorafenib targeted RTK expression and to analyze the in vivo effect of sorafenib alone or in combination with the classical chemotherapeutic backbone 5-fluorouracil (5-FU).
Material and methods
Cell culture
The human colorectal cancer cell lines SW480 [K-ras mt, B-Raf wt, PI3K wt, p53 mt], SW620 [K-ras mt, B-raf wt, PI3K wt, p53 mt], and HT29 [K-ras wt, B-raf mt, PI3K wt, p53 mt] were cultured in RPMI 1640 (Invitrogen, Germany) supplemented with 10 % FCS, 100 U/ml penicillin, 100 μg/ml streptomycin (Cambrex, Germany), and 1 mM l-glutamine (Invitrogen, Germany). The human colorectal cancer cell line Caco2 [K-ras wt, B-Raf wt, PI3K wt, p53 mt] was cultured in DMEM (Invitrogen, Germany) supplemented with 10 % FCS, 100 U/ml penicillin, 100 μg/ml streptomycin (Cambrex, Germany), and 1 mM l-glutamine (Invitrogen, Germany).
Proliferation assays and chemosensitvity
For proliferation assays, 5 × 103 SW480, SW620, Caco2, or HT-29 cells were plated in 96-well plates and cultured as described above. Twelve hours after plating sorafenib (0, 5, and 10 μg/ml), 5-FU (0.5 mg/ml) ± sorafenib (5 μg/ml), irinotecan (1 mg/ml) ± sorafenib (5 μg/ml), or oxaliplatin (0.5 mg/ml) ± sorafenib (5 μg/ml) were added to the medium. The amount of cells per well was determined by luminescence assay (CellTiter-Glo Cell Viability assay, Promega, USA). Each condition was performed in quadruplicates.
For apoptosis analyses, 2 × 105 cells were seeded per 6 wells, respectively. Twelve hours after plating, cells were treated for 24 h as mentioned above. Suspended cells were collected, and adherent cells were trypsinized prior to fixation with 100 % ethanol, staining with propidium iodide and analyzation by FACS without gating. Each condition was performed in quadruplicates.
Migration assay
SW480, SW620, Caco2, or HT29 cells (2 × 106 ) were seeded per 6 wells, cultured for 24 h, serum-starved (2 % FCS only) for 12 h, and exposed to sorafenib at different concentrations (0, 5, or 10 μg/ml) for 6 h. Migration was assayed with 24-well HTS FluoroBlock Inserts in triplet approaches (8 μM pore size; Becton Dickinson, USA).
In brief, 4×104 cells were resuspended in RPMI1640/DMEM medium containing 2 % FCS and 10 ng/ml CXCL12 and added to the upper chamber. Subsequently, RPMI1640/DMEM medium with 20 % FCS and 100 ng/ml CXCL12 (Sigma, Germany) was added to the lower chamber. Chambers were incubated for 24 h at 37 °C in a humid atmosphere of 5 % CO2. After incubation, the amount of migrated cells in the lower chamber was determined by luminescence assay (CellTiter-Glo, Cell Viability assay, Promega, USA). Each condition was performed in triplicate.
Caspase assay
Cells were treated with placebo or sorafenib (5 and 10 μg/ml, respectively). After incubation for 16 h, cells were lysed in buffer containing 20 mM Tris/HCl pH 8.0, 5 mM EDTA, 0.5 % Triton X-100, and onefold complete protease inhibitor cocktail (Roche, Germany). Protein concentration was determined by Bradford assay (Sigma, Germany). Sixty micrograms of protein was incubated in reaction buffer (25 mM HEPES pH 7.5, 50 mM NaCl, 10 % glycerol, 0.05 % CHAPS, and 5 mM DTT) in the presence of 50 μM fluorogenic substrate (Biomol, Germany), which was specific for caspase 3 (DEVD-AMC); caspase 6, 8, and 10 (Ac-IETD-AFC); or caspase 9 (Ac-LEHD-AFC). Analyses were performed in triplicates.
Assays were performed in black micro-titer plates (Nunc, Germany), and after 1 h incubation at 37 °C, the generation of free AMC or AFC was measured using a fluorometer plate reader (Appliscan, Thermo Fisher, Germany) at an excitation wavelength of 380 nm (AMC and AFC) and an emission wavelength of 460 nm (AMC) or 505 nm (AFC).
Western blot analysis
SW480, SW620, Caco2, or HT29 cells (2 × 106) were harvested after a 12-h-long exposition to placebo or sorafenib (5 and 10 μg/ml, respectively). Cells were washed twice with phosphate-buffered saline (PBS; 1×) and lysed in 2× RIPA solution. For Western blot analysis, 100 μg of protein was loaded on 8–12 % SDS-PAGE gels, respectively. After separation, the gel was transferred to a PVDF membrane (Roth, Karlsruhe, Germany). Proteins (AKT/pAKT, MEK/pMEK, PI3K/pPI3K, mTOR/pmTOR, P53/pp53, FoxO3a/pFoxO3a, GADD45β, and alpha tubulin) were detected with specific primary antibodies (Table 1; 4 °C, overnight). The specific secondary antibodies were exposed for 1 h at room temperature (Table 1). For visualisation, the Roti Lumin systems 1 and 2 were applied (P79 and P80, Roth, Karlsruhe, Germany). Each condition was performed in duplicates.
Table 1.
Antibodies used for Western blotting
| Antibody | Manufacturer | Order number | Secondary antibody | Size (kDa) | Dilution |
|---|---|---|---|---|---|
| Rabbit-anti-human pPI3K | Cell Signaling | 4228 | Goat-anti-rabbit IgG | 85/60 | 1:1,000 |
| Rabbit-anti-human PI3K | Cell Signaling | 4257 | Goat-anti-rabbit IgG | 85 | 1:1,000 |
| Rabbit-anti-human pAKT | Cell Signaling | 9267 | Goat-anti-rabbit IgG | 60 | 1:1,000 |
| Rabbit-anti-human AKT | Cell Signaling | 4685 | Goat-anti-rabbit IgG | 60 | 1:1,000 |
| Rabbit-anti-human pmTOR | Cell Signaling | 2971 | Goat-anti-rabbit IgG | 289 | 1:1,000 |
| Rabbit-anti-human mTOR | Cell Signaling | 2983 | Goat-anti-rabbit IgG | 289 | 1:1,000 |
| Rabbit-anti-human pMEK | Cell Signaling | 9121 | Goat-anti-rabbit IgG | 45 | 1:1,000 |
| Rabbit-anti-human MEK | Cell Signaling | 9122 | Goat-anti-rabbit IgG | 45 | 1:1,000 |
| Goat-anti-human GADD45β | Santa Cruz Biotechnology | sc-8776 | Donkey-anti-goat IgG | 18 | 1:500 |
| Mouse-anti-human α-Tubulin | Sigma Aldrich | t5168 | Goat-anti-mouse IgG | 48,5 | 1:2,000 |
| Goat-anti-mouse IgG | Santa Cruz Biotechnology | sc-2031 | – | – | 1:10,000 |
| Goat-anti-rabbit IgG | Santa Cruz Biotechnology | sc-2030 | – | – | 1:10,000 |
| Donkey-anti-goat IgG | Santa Cruz Biotechnology | sc-2033 | – | – | 1:10,000 |
Subcutaneous xenograft tumor system
HT29 tumor cells (1 × 107) were suspended in 0.2 ml pure RPMI1640 medium and 1× PBS (1:1) and applied by subcutaneous injection into the left flank of 7–8-week-old female nod-SCID mice. Nod-SCID mice were irradiated with 1.8 Gy 1 day prior to s.c. injection of tumor cells. As soon as the tumors reached a size of 10 mm, animals received i.p. injections of placebo (group 1; 200 μl, 5 days/week; 25 % cremophor in NaCl 0,9 %), sorafenib (group 2; 200 μl; 5 days per week; 0.12 mg/dose solved in 25 % cremophor; 30 mg/kg/week), 5-FU (group 3; 200 μl; three times a week; 0.18 mg/dose solved in 25 % cremophor; 25 mg/kg/week) or sorafenib + 5-FU (group 4; 200 μl; combination of group 2 and 3). The size of tumors was measured manually twice weekly. Tumors grew for 4 weeks. Thereafter, tumor nodules were excised and measured manually with a vernier micrometer.
Immunohistochemistry
Excised tumors obtained from the experimental animals were paraffin-embedded. After obtaining adequate slides, the tissue samples were screened for Ki-67, PDGFA, VEGFA, VEGFR1, VEGFR2, PDFGRα, and PDGFRβ protein expression by immunohistochemistry. To that purpose, the tissues were deparaffinized, rehydrated, and subsequently incubated with the respective primary antibodies [anti-PDGFRα (sc-338); 1:200, 2 h, Santa Cruz Biotechnology, CA, USA; anti-PDGFRβ (3169), 1:40, 2 h, Cell Signaling Technology, MA, USA; PDGFA (NBP1-19781), 1:100, 2 h, Novus Biologicals, Cambridge, UK; VEGFA (ab46154), 1:200, 2 h, Abcam plc, Cambridge UK; VEGFR1 (RB-9049-R7), 1:50, 2 h, Thermo Fisher Scientific GmbH Neomarkers, Germany; VEGFR2 (RB-9239-R7), 1:50, 2 h, Thermo Fisher Scientific GmbH Neomarkers, Germany; VEGFR3 (sc-321), 1:200, 2 h, Santa Cruz Biotechnology, Germany; Ki-67 (mib1), 1:100, 2 h, Dako, Germany; Envision flex plusTM, Autostainer, Dako, Germany]. The secondary antibody (anti-rabbit-mouse-goat antibody) was incubated for 15 min at room temperature, followed by incubation with streptavidin-POD (Dako, Germany) for 15 min. Antibody binding was visualized using AEC solution (Dako, Germany). Afterwards, the tissues were counterstained by haemalaun solution (Dako, Germany). The expression of the respective tyrosine kinase was evaluated using a scoring system. Expression strength of PDGFA, VEGFA, VEGFR1, VEGFR2, VEGFR3, PDFGRα, and PDGFRβ was classified as negative (0), low (1), medium, (2) and high (3). All slides were independently evaluated by three investigators. The Ki-67 expression was measured as percentage of Ki-67 expressing cells.
Statistics
In order to assess dependence of growth factor and Ki-67 expression on treatment with 5-FU and sorafenib, the minimum, the maximum, the median, and the quartiles in subgroups were calculated. For Ki-67 analyses, the mean and standard deviations were calculated and displayed in box plots. Ki-67 was measured three times for each specimen; averages were analyzed using two-way analysis of variance. To compare growth factor expression between treatment groups the Kruskal–Wallis tests was used, followed by pairwise Wilcoxon test if the Kruskal–Wallis test gave a p value ≤0.05.
All tests were performed with exploratory intention, associations with p values ≤0.05 might warrant further consideration. Statistical analysis was performed using SAS 9.3 2002–2010 by SAS Institute Inc., Cary, NC, USA.
Results
Proliferation assay
Inhibition of tumor growth through low dose sorafenib was seen in all cell lines, except for Caco2 (Table 2; Fig. 1a). High-dose sorafenib eradicated SW480 and HT29 cells significantly and SW620 cell less effectively, whereas Caco2 cells revealed a decelerated tumor cell growth only. Mutation status of K-ras, B-Raf, PI3K, or p53 did not correlate with resistance.
Table 2.
Functional effects of sorafenib in distinct colorectal cancer cell lines
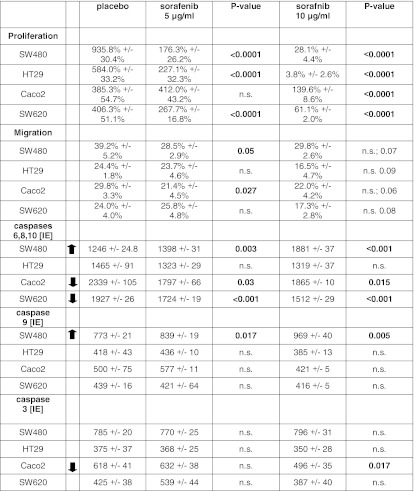
Fig. 1.

Effect of sorafenib monotherapy and combining sorafenib with standard chemotherapeutics 5-FU, oxalipaltin, or irinotecan. a While SW480 and HT29 cells were almost eradicated by high dose sorafenib treatment, low doses resulted in a significantly reduced growth as compared to placebo treatment. In contrast, Caco2 and SW620 cells were resistant to low-dose sorafenib while high-dose sorafenib stabilized tumor cell load of SW620 and Caco2. b 5-Fluorouracil. Combining 5-FU and sorafenib revealed additive effects (proliferation and apoptosis) in SW480 and HT 29 cells but not in SW620 or Caco2 cells. In contrast, the combination even increased proliferation of Caco2 cells. c Irinotecan. Combining 5-FU and irinotecan demonstrated clear additive effects in SW480, HT29 (proliferation and apoptosis), and SW620 (proliferation only) cells and even slightly increased apoptosis of Caco2 cells. However, no effect was seen in SW620 cells. d Oxaliplatin. Combining oxalipaltin and sorafenib revealed an additive effect (proliferation and apoptosis) in HT29 cells only but not in SW480, SW620, or Caco2 cells. In contrast, the combination increased proliferation of SW480, Caco2, and SW620 cells and inhibited apoptosis of SW480 and SW620 cells
Migration assay
The treatment with low-dose sorafenib (5 μg/ml) significantly inhibited migration only in SW480 and Caco2 cells (Table 2). High-dose (10 μg/ml) sorafenib nonsignificantly inhibited migration in HT29 and SW620 cells.
Caspases assays
Caspases 6, 8, and 10
Treatment with sorafenib significantly induced caspases 6, 8, and 10 activity in SW480 but not in HT29 cells (Table 2). In contrast, exposure to sorafenib significantly decreased caspases 6, 8, and 10 activity in Caco2 and SW620 cells
Caspase 9
Treatment with sorafenib significantly induced caspase 9 activity in SW480 but did not impact on HT29, Caco2, or SW620 cells.
Caspase 3
Treatment with sorafenib did not modify caspase 3 activity in SW480, HT29, or SW620 cells. However, caspase 3 activity was significantly decreased in Caco2 cells.
Chemosensitivity assay (apoptosis and proliferation)
5-Fluorouracil
Combining 5-FU and sorafenib revealed additive effects (proliferation and apoptosis) in SW480 and HT 29 cells but not in SW620 or Caco2 cells (Fig. 1b; Table 3). In contrast, the combination even increased proliferation of Caco2 cells.
Table 3.
Effect of combining sorafenib with classical chemotherapeutic drugs
| SW480 (%) | p value | HT29 (%) | p value | Caco2 (%) | p value | SW620 (%) | p value | ||
|---|---|---|---|---|---|---|---|---|---|
| Proliferation | |||||||||
| 5-FU + | Placebo | 150.3 ± 16.0 | 0.061 | 262.3 ± 25.9 | 0.004 | 337.2 ± 25.3 | <0.001 | 97.2 ± 8.5 | 0.186 |
| Sorafenib | 127.7 ± 11.9 | 218.5 ± 20.2 | 468.2 ± 29.1 | 104.1 ± 8.5 | |||||
| Irinotecan + | Placebo | 59.0 ± 7.0 | 0.002 | 217.7 ± 23.8 | <0.001 | 585.1 ± 51.9 | 0.66 | 120.6 ± 9.7 | <0.001 |
| Sorafenib | 39.1 ± 2.6 | 116.2 ± 13.1 | 570.4 ± 78.6 | 75.1 ± 8.1 | |||||
| Oxaliplatin + | Placebo | 56.6 ± 3.5 | 0.1 | 204.7 ± 36.1 | <0.001 | 308.4 ± 33.2 | <0.001 | 10.2 ± 0.8 | <0.001 |
| Sorafenib | 61.7 ± 6.6 | 114.8 ± 28.6 | 450.1 ± 20.0 | 13.7 ± 1.7 | |||||
| Apoptosis | |||||||||
| 5−FU | 17.23 ± 2.67 | 0.002 | 5.28 ± 0.07 | 0.047 | 14.38 ± 1.15 | 0.32 | 5.84 ± 2.45 | 0.15 | |
| 5−FU + sorafenib | 42.27 ± 0.74 | 20.55 ± 1.62 | 16.98 ± 3.38 | 10.98 ± 1.73 | |||||
| Sorafenib | 16.43 ± 1.45 | <0.001 | 11.78 ± 1.56 | 0.031 | 17.12 ± 0.37 | 0.95 | 9.07 ± 4.65 | 0.66 | |
| Irinotecan | 41.62 ± 4.12 | 0.004 | 24.08 ± 2.82 | <0.001 | 12.38 ± 2.79 | 0.041 | 39.07 ± 3.01 | 0.238 | |
| Irinotecan + sorafenib | 71.57 ± 1.25 | 56.23 ± 1.1 | 19.74 ± 3.2 | 26.44 ± 7.96 | |||||
| Sorafenib | 16.43 ± 1.45 | <0.001 | 11.78 ± 1.56 | 0.002 | 17.12 ± 0.37 | 0.29 | 9.07 ± 4.65 | 0.146 | |
| Oxaliplatin | 39.26 ± 1.23 | <0.001 | 24.93 ± 1.87 | <0.001 | 38.52 ± 3.12 | 0.6 | 40.71 ± 1.31 | 0.058 | |
| Oxaliplatin + sorafenib | 31.47 ± 1.15 | 46.92 ± 1.21 | 36.84 ± 2.13 | 30.62 ± 0.04 | |||||
| Sorafenib | 16.43 ± 1.45 | <0.001 | 11.78 ± 1.56 % | <0.001 | 17.12 ± 0.37 | <0.001 | 9.07 ± 4.65 | <0.001 | |
Irinotecan
Combining 5-FU and irinotecan demonstrated clear additive effects in SW480, HT29 (proliferation and apoptosis), and SW620 (proliferation only) cells and even slightly increased apoptosis of Caco2 cells (Fig. 1c). However, no effect was seen in SW620 cells.
Oxaliplatin
Combining oxalipaltin and sorafenib revealed an additive effect (proliferation and apoptosis) in HT29 cells only but not in SW480, SW620, or Caco2 cells (Fig. 1d). In contrast, the combination increased proliferation of SW480, Caco2, and SW620 cells and inhibited apoptosis of SW480 and SW620 cells.
Signal cascade inhibition
In order to investigate the relevance of sorafenib in the inhibition of signal cascades, we analyzed diverse pathways (Fig. 2a). Upon exposure with increasing sorafenib doses, we observed an inhibition of the Ras–Raf pathway (pMEK) in SW620 but an induction in Caco2 cell lines. This pathway remained unchanged in SW480 and HT29 cells.
Fig. 2.

a Upon exposure with augmenting sorafenib doses, we observed an inhibition of the Ras–Raf pathway (pMEK) in Sw620 cell lines, only. In contrast, this pathway was activated in Caco2 cells. The AKT pathway was particularly altered in sensitive cell lines. SW480 and HT29 cells revealed only a hint–absent pAKT expression, but AKT expression was clearly suppressed upon exposure to increasing sorafenib doses. In contrast, the resistant cell line Caco2 did not show such AKT inhibitory behavior. An inhibition of pPI3K and pAKT was seen in Caco2 but not in SW620. b Analyses of the signaling pathways showed that sorafenib-sensitive cell lines reveal almost absent pAKT, absent–weak GADD45β, and medium–strong FoxO3a expression levels. In contrast, resistant cell lines showed a medium pAKT, very strong GADD45β, and weak–medium FoxO3a expression levels. GADD45β expression levels discriminated best between sensitive and resistant cell lines. When analyzing the impact of sorafenib, we observed that the sensitive cell line SW480 revealed a pp53 and a GADD45ß upregulation upon exposure to increasing sorafenib doses. In contrast, resistant cell lines showed initially high (Caco2, SW620) and, upon drug exposure, decreasing GADD45β (Caco2) and decreasing pp53 (Caco2, SW620) expression levels
The AKT pathway was specifically altered in sensitive cell lines: SW480 and HT29 cells revealed only weak–absent pAKT, but AKT expression was significantly suppressed upon exposure with increasing sorafenib doses. In contrast, the resistant cell line Caco2 did not show any AKT suppressive behavior. An inhibition of pPI3K and pAKT was seen in Caco2 but not in SW620.
These results raise the question of whether a suppression of AKT expression correlates with responsiveness to sorafenib. As it has been previously reported that GADD45ß takes control when AKT is absent, we analyzed GADD 45-associated genes.
Potential resistance indicators
We observed that sorafenib-sensitive cell lines revealed almost absent pAKT, weak GADD45β, and medium–strong FoxO3a expression levels (Fig. 2b). In contrast, resistant cell lines showed medium pAKT, intensive GADD45β, and weak–medium FoxO3a expression levels. GADD45β expression levels discriminated best between sensitive and resistant cell lines.
Analyzing the impact of sorafenib on protein expression, we observed that the sensitive cell line SW480 reacts with a pp53 and a GADD45β upregulation upon increasing sorafenib doses. In contrast, resistant cell lines revealed primarily intense (Caco2, SW620) and, upon sorafenib exposure, decreasing (Caco2) GADD45β expression levels. Similarly, both resistant cell lines decreased pp53 levels upon exposure to increasing sorafenib doses.
In vivo xenograft model
Ex vivo analyses of tumor size indicated that—compared to placebo control—only a sorafenib monotherapy significantly decreased tumor size (220 ± 3.06 % versus 95.8 ± 4.34 %; P = <0.0001), while a 5-FU monotherapy inhibited tumor growth nonsignificantly (220 ± 3.06 % versus 124 ± 20.8 %; P = 0.097) (Fig. 3). The 5-FU plus sorafenib combination therapy was equipotent to 5-FU monotherapy (220 ± 3.06 % versus 146 ± 24.89 %; P = 0.085), but inferior compared to sorafenib monotherapy (P = 0.068), in this explorative analysis.
Fig. 3.

In vivo, only sorafenib monotherapy inhibited tumor growth significantly as compred to the control group. 5-FU treatment or the combination demonstrated only a nonsignificant inhibition. When all treatment groups were compared, no significant differences were observed
Therapeutic effect on Ki-67 proliferation index
Analysis of the proliferation index showed that sorafenib monotherapy and 5-FU monotherapy were equally effective in reducing the proliferation index as compared to placebo. However, the combination therapy of sorafenib and 5-FU did not result in further reduction of proliferation rates.
For Ki-67, analysis of variance demonstrated a significant effect for sorafenib (p = 0.0101), and for 5-FU + sorafenib interaction (p = 0.0049), the 5-FU main effect was borderline significant (p = 0.0537) as compared to the control group. On average, treatment with 5-FU lowered Ki-67 expression by 4.9 %, treatment with sorafenib on average lowered Ki-67 by 6.8 %. However, the effect was not additive: animals treated with both substances had higher Ki-67 values than animals treated with only one substance. The means for each treatment group are given in Fig. 4.
Fig. 4.

a Analysis of Ki-67 proliferation index in three different treated animal groups (sorafenib monotherapy versus 5-FU monotherapy versus combination therapy) show a significant reduction of proliferation in all treatment groups but no superior therapeutic effect of the combination therapy group. b Interaction plot for Ki-67 depicts no additive effect of combination therapy with 5-FU and sorafenib. Analysis of the expression intensity of PDGFA, PDGFRβ, and VEGFR1 in the control group and under treatment with sorafenib monotherapy, 5-FU monotherapy, and combination therapy. PDGFRβ and VEGFR1 expression intensities were significantly reduced by 5-FU and sorafenib, respectively
Therapeutic effect on cytokine expression
Specimens (N = 21) resected from 20 animals were included in the analysis. Each treatment group consisted of five animals, with six specimens available in the group treated with combined 5-FU and sorafenib (due to two tumors in one animal). The distribution of growth factor expression is given in Table 4. A summary of the Ki-67 measurements is given in Table 5.
Table 4.
Summary measures for growth factor expression in treatment groups
| Treated with 5-FU | Treated with sorafenib | Label | N | Minimum | Lower quartile | Median | Upper quartile | Maximum |
|---|---|---|---|---|---|---|---|---|
| No | No | VEGFR 1 | 5 | 2 | 2 | 2.5 | 2.5 | 2.5 |
| VEGFR 2 | 2 | 2 | 2 | 2 | 2 | 2 | ||
| VEGFR 3 | 5 | 1.5 | 2 | 2.5 | 3 | 3 | ||
| PDGFR alpha | 5 | 0 | 0.5 | 0.5 | 1 | 1.5 | ||
| PDGFR beta | 5 | 2 | 2 | 2 | 2 | 2 | ||
| VEGF A in cytoplasm | 5 | 0.5 | 2 | 2.5 | 3 | 3 | ||
| VEGF A in nucleus | 3 | 2 | 2 | 2 | 2 | 2 | ||
| PDGF A | 5 | 0 | 1 | 1.5 | 1.5 | 2 | ||
| Yes | VEGFR 1 | 5 | 0.5 | 0.5 | 1 | 1 | 1 | |
| VEGFR 2 | 5 | 0 | 0 | 0.5 | 1 | 1.5 | ||
| VEGFR 3 | 5 | 1.5 | 1.5 | 2 | 2.5 | 3 | ||
| PDGFR alpha | 5 | 0 | 0.5 | 0.5 | 0.5 | 1.5 | ||
| PDGFR beta | 5 | 0 | 0.5 | 0.5 | 0.5 | 1 | ||
| VEGF A in cytoplasm | 5 | 1 | 1 | 1 | 1 | 2 | ||
| VEGF A in nucleus | 5 | 0 | 0 | 0 | 1 | 2 | ||
| PDGF A | 5 | 0 | 0 | 0 | 1 | 1.5 | ||
| Yes | No | VEGFR 1 | 5 | 0.5 | 0.5 | 1 | 1 | 1.5 |
| VEGFR 2 | 5 | 0 | 0 | 0 | 0.5 | 1 | ||
| VEGFR 3 | 5 | 1 | 1.5 | 1.5 | 2 | 3 | ||
| PDGFR alpha | 5 | 0 | 0 | 0.5 | 1.5 | 1.5 | ||
| PDGFR beta | 5 | 0 | 0 | 0 | 1 | 1.5 | ||
| VEGF A in cytoplasm | 5 | 0.5 | 1 | 1 | 2 | 2.5 | ||
| VEGF A in nucleus | 5 | 0 | 0 | 1 | 2 | 3 | ||
| PDGF A | 5 | 0 | 0 | 0 | 0 | 0.5 | ||
| Yes | VEGFR 1 | 6 | 1 | 1 | 1.3 | 1.5 | 1.5 | |
| VEGFR 2 | 4 | 0 | 0 | 0.3 | 1 | 1.5 | ||
| VEGFR 3 | 6 | 1.5 | 2 | 2.3 | 2.5 | 3 | ||
| PDGFR alpha | 6 | 0.5 | 0.5 | 1 | 1 | 1 | ||
| PDGFR beta | 6 | 0 | 0 | 0.5 | 0.8 | 1 | ||
| VEGF A in cytoplasm | 6 | 1 | 1 | 1 | 1 | 1.5 | ||
| VEGF A in nucleus | 6 | 0 | 0 | 1 | 2 | 2 | ||
| PDGF A | 6 | 0 | 0 | 0 | 1 | 1 |
Table 5.
Summary of Ki-67 measurements
| Treated with 5-FU | Treated with sorafenib | N | Mean | Standard deviation | Minimum | Lower quartile | Median | Upper quartile | Maximum |
|---|---|---|---|---|---|---|---|---|---|
| No | No | 5 | 48.9 | 3.3 | 44.3 | 47 | 49.7 | 51.7 | 52 |
| Yes | 5 | 34.5 | 4.8 | 28 | 33 | 33.7 | 37.3 | 40.7 | |
| Yes | No | 5 | 36.5 | 5.3 | 29.7 | 34.3 | 35 | 40 | 43.3 |
| Yes | 6 | 37.3 | 7 | 28.5 | 34 | 36.7 | 38 | 49.7 |
There were substantial differences in the expression of PDGFRβ (p = 0.002) and VEGFR1 (p = 0.0002) between the control group and the different treatment groups. There was also a tendency for different expression of PDGFA (p = 0.0799) between different groups (Fig. 5a). Representative tissue examples are shown in Fig. 5b.
Fig. 5.

a Analysis of the expression intensity of PDGFA, PDGFRβ, and VEGFR1 in the control group and under treatment with sorafenib monotherapy, 5-FU monotherapy, and combination therapy. PDGFRβ and VEGFR1 expression intensities were significantly reduced by 5-FU and sorafenib, respectively. b Immunostaining was evaluated by three authors independently. The immunohistochemical staining was analyzed according to a scoring method as previously validated and described by Laverdiere et al. The tumors were classified into four groups based on the homogeneous staining intensity: 0, no expression; 1, weak expression; 2, moderate expression; and 3, strong expression. In the case of heterogeneous staining within the same sample, the respective higher score was chosen if more than 50 % of cells revealed a higher staining intensity. If evaluations did not agree, specimens were re-evaluated and reclassified according to the assessment given most frequently by the observers
The highest expression levels of VEGFR1 were seen in the untreated control group and differed in relation to the group treated with 5-FU (p = 0.0079), sorafenib (p = 0.0079), and 5-FU + sorafenib (p = 0.0022). The difference among treatment groups remained within random variation.
A similar result was seen for PDGFRβ expression. The untreated control group showed the highest expression level differing from all treatment groups (p = 0.0079 when compared with 5-FU or sorafenib, p = 0.0043 when compared to 5-FU + sorafenib).
The tentative differences in PDGFA expression resulted from higher values in the untreated control group as compared to lower values in the treated groups.
The other growth factors did not show any differences beyond random variation(p > 0.1). The p values in the Kruskal–Wallis test were: PDGFRα, p = 0.8355; VEGFR2, p = 0.1020; VEGFR 3, p = 0.5058; cytoplasmic VEGF A, p = 0.2701; and nuclear VEGF A, p = 0.3397.
Discussion
The approach of inhibiting RTKs with sorafenib has been successful in renal and hepatocellular cancers [17, 18]. A phase I study revealed disease stabilization in pretreated colorectal cancer patients [20]. Except of one recent study with Regorafenib, recent phase II/III studies testing other multi-tyrosine kinase inhibitors in colorectal cancer failed to show any benefit [21, 22]. So far, no molecular markers have been identified which are helpful in stratifying the patients.
We performed defined functional in vitro analyses in order to identify sorafenib-sensitive and sorafenib-resistant cell lines. While HT29 and SW480 were found to be sorafenib sensible, Caco2 was resistant and SW620 showed features of resistance. However, the mutation status of K-ras, B-Raf, PI3K, or p53 did not correlate with resistance.
Combining sorafenib with chemotherapeutic drugs used in colorectal cancer revealed an additive effect in growth inhibition and apoptosis induction in SW480 (except for oxaliplatin) and HT29 cells, whereas in Caco2 cells, apoptosis was not increased and proliferation even stimulated (5-FU or oxaliplatin). These data are in line with previous reports describing a reduced cellular uptake of oxaliplatin and generation of DNA adducts in specific colorectal cancer cells through sorafenib [23]. Thus, combination with oxaliplatin seems disadvantageous in specific settings. The effect of sorafenib on migration was marginal and of no significant importance.
Induction of apoptosis might explain the different observations made upon sorafenib exposure: While activity of caspases 6, 8, and 10 was induced in sensitive SW480 cells, it was decreased in the resistant cell lines. Furthermore, SW480 reacted with an increased activity of caspase 9. In contrast, activity of caspase 3 was decreased in Caco2 cells upon exposure to sorafenib. An induction of caspase 3 activity, as seen in prostate cancer cells, was not observed in colorectal cancer cells [24]. Our data reveal that resistance to sorafenib is associated with inhibition of specific pro-apoptotic pathways. However, sorafenib is also known to induce caspase-independent apoptosis, mediated through nuclear translocation of AIF [25].
We observed an inhibition of the Ras–Raf pathway (pMEK) in SW620 cell lines only, matching sorafenib’s function as a Raf inhibitor [25]. While sensitive cell lines revealed only a weak–absent pAKT expression, AKT expression was clearly suppressed upon exposure with increasing sorafenib doses. In contrast, the resistant cell line Caco2 did not show such AKT suppressive behavior. These observations match a previous report that a constitutively active AKT protects cells against sorafenib/bortezomib-induced apoptosis [26, 27].
Sorafenib-sensitive cells lines were defined by almost absent pAKT, medium–strong FoxO3a, and hint GADD45β levels. The tumor suppressor FoxO3 belongs to a subclass of the forkhead transcription factors, being inhibited by activation of the PI3K pathway. Downregulation of FoxO3 is thus considered a consequence of pAKT activity.
In contrast, resistant cell lines showed medium pAKT, weak FoxO3a, and very intense GADD45β levels. GADD45β expression levels discriminated best between sensitive and resistant cell lines. GADD45 is a stress sensor modulating the response of cells to genotoxic or oxidative stress [28–30]. In specific colon cancer cells, GADD45β over-expression was linked to protection from platin induced death, matching our observations [31]. Being an apoptosis modulator, activation of GADD45β prevents the propagation of damaged cells, causing an arrest in cell growth and apoptosis after exposure to toxins [32]. This regulation seems intact in SW480 cells but reversed in resistant cells; GADD45ß was downregulated in Caco2 upon sorafenib treatment, going along with a sorafenib-mediated inhibition of caspases 6, 8, and 10. As a downstream effector of p53, GADD45β was confirmed to be specifically downregulated in HCC, which was associated to the extent of p53 mutation [33]. We observed a pp53 and a GADD45β upregulation in some sensitive cell line (SW480) upon exposure to sorafenib. In contrast, resistant cell lines showed primarily high (Caco2, SW620) and, upon sorafenib exposure, decreasing (Caco2) GADD45β and pp53 levels. These data are in line with observations in HCC, in which GADD45β induction by sorafenib occurred only in sensitive hepatocellular carcinoma cell lines, independent of the Raf/MEK/ERK signaling pathway [34].These findings confirm our definition of sensitive cell lines, in which sorafenib induces apoptosis and inhibits proliferation.
In vivo, Wilhelm and colleagues described a potent growth inhibition of HT29 xenografts at sorafenib doses of 7.5 mg/kg. We studied four different groups in vivo: placebo, 5-FU, sorafenib, and 5-FU + sorafenib. 5-FU was chosen, being the backbone of most chemotherapeutic protocols in colorectal cancer. Sorafenib was applied at 5 mg/kg, matching 400 mg/day as used in combination therapies [19, 20].
Interestingly, we observed that a sorafenib monotherapy was at least equally effective as the 5-FU monotherapy or as the combination therapy and even tended to inhibit in vivo tumor growth somewhat better than the combination therapy.
The proliferation index was significantly reduced in all treatment groups as compared to the control group but displayed similar results for mono-agent therapy and the combination therapy. Since only small numbers were analyzed, a possibility exists that larger treatment groups might demonstrate even more distinct differences. However, we clearly demonstrate that combination of sorafenib and chemotherapy did not result in any additive effects. In contrast, it seems that treatment effects are partially cancelled when 5-FU and sorafenib are applied simultaneously.
Expression rates of receptor tyrosine kinases VEGFR1 and PDGFRβ as well as of the ligand PDGFA were decreased by all treatment regimens used. However, no significant differences were detected between treatment groups.
Inhibition of receptor tyrosine kinases through sorafenib could potentially lead to a selection of low target expressing tumor cells. Combination regimens of sorafenib and 5-FU might reduce sorafenib target expression leading to a similar proliferation effect as under 5-FU monotherapy. However, the adverse events in humans might rather be additive. Our results indicate that there is no additive effect in combination of these two treatment mechanisms and that combination might only add adverse events. Therefore, in future studies preferentially sorafenib monotherapy versus sequential treatment regimens (inductiontherapy via chemotherapy–maintenance via sorafenib) should be explored.
Conclusion
Diverse tyrosine kinase inhibitors have failed in colorectal cancer. However, sorafenib still seems promising in distinct settings, if applied as monotherapy. In our human colon cancer xenograft model, it seems that treatment effects are partially cancelled when 5-FU and sorafenib are applied simultaneously. However, monotherapy with sorafenib seems to be sufficient for tumor control in our human colon cancer xenograft model, especially considering the application advantage and toxicity profile.
Acknowledgments
This study was financially supported by the Bayer Healthcare AG.
Disclaimer
Carl Schimanski received research funding; Markus Moehler, speech honoraria; and Peter R. Galle, consulting and speech honoraria.
Open Access
This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
Contributor Information
Thomas C. Wehler, Email: thomas.wehler@unimedizin-mainz.de
Markus Moehler, Phone: +49-6131-176076, FAX: +49-6131-176472, Email: moehler@mail.uni-mainz.de.
References
- 1.Greenlee RT, Murray T, Bolden S, Wingo PA, et al. Cancer statistics, 2000. CA Cancer J Clin. 2000;50:7–33. doi: 10.3322/canjclin.50.1.7. [DOI] [PubMed] [Google Scholar]
- 2.Weir HK, Thun MJ, Hankey BF, Ries LA, Howe HL, Wingo PA, Jemal A, Ward E, Anderson RN, Edwards BK. Annual report to the nation on the status of cancer, 1975–2000, featuring the uses of surveillance data for cancer prevention and control. J Natl Cancer Inst. 2003;95:1276–1299. doi: 10.1093/jnci/djg040. [DOI] [PubMed] [Google Scholar]
- 3.August DA, Ottow RT, Sugarbaker PH. Clinical perspective of human colorectal cancer metastasis. Cancer Metastasis Rev. 1984;3:303–324. doi: 10.1007/BF00051457. [DOI] [PubMed] [Google Scholar]
- 4.Douillard JY, Siena S, Cassidy J, Tabernero J, Burkes R, Barugel M, Humblet Y, Bodoky G, Cunningham D, Jassem J, Rivera F, Kocakova I, et al. Randomized, phase III trial of panitumumab with infusional fluorouracil, leucovorin, and oxaliplatin (FOLFOX4) versus FOLFOX4 alone as first-line treatment in patients with previously untreated metastatic colorectal cancer: the PRIME study. J Clin Oncol. 2010;28(31):4697–7705. doi: 10.1200/JCO.2009.27.4860. [DOI] [PubMed] [Google Scholar]
- 5.Sobrero A, Ackland S, Clarke S, Perez-Carrion R, Chiara S, Gapski J, Mainwaring P, Langer B, Young S. Phase IV study of bevacizumab in combination with infusional fluorouracil, leucovorin and irinotecan (FOLFIRI) in first-line metastatic colorectal cancer. Oncology. 2009;77:113–119. doi: 10.1159/000229787. [DOI] [PubMed] [Google Scholar]
- 6.Welch S, Spithoff K, Rumble RB, Maroun J. Bevacizumab combined with chemotherapy for patients with advanced colorectal cancer: a systematic review. Ann Oncol. 2010;21(6):1152–1162. doi: 10.1093/annonc/mdp533. [DOI] [PubMed] [Google Scholar]
- 7.Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61:759–767. doi: 10.1016/0092-8674(90)90186-I. [DOI] [PubMed] [Google Scholar]
- 8.Cho KR, Vogelstein B. Genetic alterations in the adenoma–carcinoma sequence. Cancer. 1992;70:1727–1731. doi: 10.1002/1097-0142(19920915)70:4+<1727::AID-CNCR2820701613>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 9.Vogelstein B, Kinzler KW. The multistep nature of cancer. Trends Genet. 1993;9:138–141. doi: 10.1016/0168-9525(93)90209-Z. [DOI] [PubMed] [Google Scholar]
- 10.Khambata-Ford S, Garrett CR, Meropol NJ, Basik M, Harbison CT, Wu S, Wong TW, Huang X, Takimoto CH, Godwin AK, Tan BR, Krishnamurthi SS, et al. Expression of epiregulin and amphiregulin and K-ras mutation status predict disease control in metastatic colorectal cancer patients treated with cetuximab. J Clin Oncol. 2007;25:3230–3237. doi: 10.1200/JCO.2006.10.5437. [DOI] [PubMed] [Google Scholar]
- 11.Li E, Hristova K. Role of receptor tyrosine kinase transmembrane domains in cell signaling and human pathologies. Biochemistry. 2006;45:6241–6251. doi: 10.1021/bi060609y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mobius C, Stein HJ, Becker I, Feith M, Theisen J, Gais P, Jutting U, Siewert JR. The ‘angiogenic switch’ in the progression from Barrett’s metaplasia to esophageal adenocarcinoma. Eur J Surg Oncol. 2003;29:890–894. doi: 10.1016/j.ejso.2003.07.002. [DOI] [PubMed] [Google Scholar]
- 13.Liu XE, Sun XD, Wu JM. Expression and significance of VEGF-C and FLT-4 in gastric cancer. World J Gastroenterol. 2004;10:352–355. doi: 10.3748/wjg.v10.i3.352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yonemura Y, Endo Y, Tabata K, Kawamura T, Yun HY, Bandou E, Sasaki T, Miura M. Role of VEGF-C and VEGF-D in lymphangiogenesis in gastric cancer. Int J Clin Oncol. 2005;10:318–327. doi: 10.1007/s10147-005-0508-7. [DOI] [PubMed] [Google Scholar]
- 15.Takahashi Y, Cleary KR, Mai M, Kitadai Y, Bucana CD, Ellis LM. Significance of vessel count and vascular endothelial growth factor and its receptor (KDR) in intestinal-type gastric cancer. Clin Cancer Res. 1996;2:1679–1684. [PubMed] [Google Scholar]
- 16.Wang KL, Wu TT, Choi IS, Wang H, Reseetkova E, Correa AM, Hofstetter WL, Swisher SG, Ajani JA, Rashid A, Albarracin CT. Expression of epidermal growth factor receptor in esophageal and esophagogastric junction adenocarcinomas: association with poor outcome. Cancer. 2007;109:658–667. doi: 10.1002/cncr.22445. [DOI] [PubMed] [Google Scholar]
- 17.Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc JF, de Oliveira AC, Santoro A, Raoul JL, Forner A, Schwartz M, Porta C, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359:378–390. doi: 10.1056/NEJMoa0708857. [DOI] [PubMed] [Google Scholar]
- 18.Escudier B, Eisen T, Stadler WM, Szczylik C, Oudard S, Staehler M, Negrier S, Chevreau C, Desai AA, Rolland F, Demkow T, Hutson TE, et al. Sorafenib for treatment of renal cell carcinoma: final efficacy and safety results of the phase III treatment approaches in renal cancer global evaluation trial. J Clin Oncol. 2009;27:3312–3318. doi: 10.1200/JCO.2008.19.5511. [DOI] [PubMed] [Google Scholar]
- 19.Mross K, Steinbild S, Baas F, Gmehling D, Radtke M, Voliotis D, Brendel E, Christensen O, Unger C. Results from an in vitro and a clinical/pharmacological phase I study with the combination irinotecan and sorafenib. Eur J Cancer. 2007;43:55–63. doi: 10.1016/j.ejca.2006.08.032. [DOI] [PubMed] [Google Scholar]
- 20.Kupsch P, Henning BF, Passarge K, Richly H, Wiesemann K, Hilger RA, Scheulen ME, Christensen O, Brendel E, Schwartz B, Hofstra E, Voigtmann R, et al. Results of a phase I trial of sorafenib (BAY 43-9006) in combination with oxaliplatin in patients with refractory solid tumors, including colorectal cancer. Clin Colorectal Cancer. 2005;5:188–196. doi: 10.3816/CCC.2005.n.030. [DOI] [PubMed] [Google Scholar]
- 21.Saltz LB, Rosen LS, Marshall JL, Belt RJ, Hurwitz HI, Eckhardt SG, Bergsland EK, Haller DG, Lockhart AC, Rocha Lima CM, Huang X, DePrimo SE, et al. Phase II trial of sunitinib in patients with metastatic colorectal cancer after failure of standard therapy. J Clin Oncol. 2007;25:4793–4799. doi: 10.1200/JCO.2007.12.8637. [DOI] [PubMed] [Google Scholar]
- 22.Cuteem EV, Sobrero AF, Siena S, Falcone A, Ychou M, Humblet Y, Bouche O, Mineur L, Barone C, Adenis A, Tabernero J, Yoshino T et al (2012) Phase III CORRECT trial of regorafenib in metastatic colorectal cancer (mCRC). J Clin Oncol 30(suppl; abstr 3502)
- 23.Heim M, Scharifi M, Zisowsky J, Jaehde U, Voliotis D, Seeber S, Strumberg D. The Raf kinase inhibitor BAY 43-9006 reduces cellular uptake of platinum compounds and cytotoxicity in human colorectal carcinoma cell lines. Anticancer Drugs. 2005;16:129–136. doi: 10.1097/00001813-200502000-00003. [DOI] [PubMed] [Google Scholar]
- 24.Huang R, Chen XQ, Huang Y, Chen N, Zeng H. The multikinase inhibitor sorafenib induces caspase-dependent apoptosis in PC-3 prostate cancer cells. Asian J Androl. 2010;12:527–534. doi: 10.1038/aja.2010.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Panka DJ, Wang W, Atkins MB, Mier JW. The Raf inhibitor BAY 43-9006 (Sorafenib) induces caspase-independent apoptosis in melanoma cells. Cancer Res. 2006;66:1611–1619. doi: 10.1158/0008-5472.CAN-05-0808. [DOI] [PubMed] [Google Scholar]
- 26.Yu C, Friday BB, Lai JP, Yang L, Sarkaria J, Kay NE, Carter CA, Roberts LR, Kaufmann SH, Adjei AA. Cytotoxic synergy between the multikinase inhibitor sorafenib and the proteasome inhibitor bortezomib in vitro: induction of apoptosis through Akt and c-Jun NH2-terminal kinase pathways. Mol Cancer Ther. 2006;5:2378–2387. doi: 10.1158/1535-7163.MCT-06-0235. [DOI] [PubMed] [Google Scholar]
- 27.Chen KF, Yu HC, Liu TH, Lee SS, Chen PJ, Cheng AL. Synergistic interactions between sorafenib and bortezomib in hepatocellular carcinoma involve PP2A-dependent Akt inactivation. J Hepatol. 2010;52:88–95. doi: 10.1016/j.jhep.2009.10.011. [DOI] [PubMed] [Google Scholar]
- 28.Kastan MB, Zhan Q, el-Deiry WS, Carrier F, Jacks T, Walsh WV, Plunkett BS, Vogelstein B, Fornace AJ., Jr A mammalian cell cycle checkpoint pathway utilizing p53 and GADD45 is defective in ataxia-telangiectasia. Cell. 1992;71:587–597. doi: 10.1016/0092-8674(92)90593-2. [DOI] [PubMed] [Google Scholar]
- 29.Liebermann DA, Hoffman B. Myeloid differentiation (MyD)/growth arrest DNA damage (GADD) genes in tumor suppression, immunity and inflammation. Leukemia. 2002;16:527–541. doi: 10.1038/sj.leu.2402477. [DOI] [PubMed] [Google Scholar]
- 30.Nemoto S, Finkel T. Redox regulation of forkhead proteins through a p66shc-dependent signaling pathway. Science. 2002;295:2450–2452. doi: 10.1126/science.1069004. [DOI] [PubMed] [Google Scholar]
- 31.Smith ML, Kontny HU, Bortnick R, Fornace AJ., Jr The p53-regulated cyclin G gene promotes cell growth: p53 downstream effectors cyclin G and Gadd45 exert different effects on cisplatin chemosensitivity. Exp Cell Res. 1997;230:61–68. doi: 10.1006/excr.1996.3402. [DOI] [PubMed] [Google Scholar]
- 32.Zhang W, Bae I, Krishnaraju K, Azam N, Fan W, Smith K, Hoffman B, Liebermann DA. CR6: A third member in the MyD118 and Gadd45 gene family which functions in negative growth control. Oncogene. 1999;18:4899–4907. doi: 10.1038/sj.onc.1202885. [DOI] [PubMed] [Google Scholar]
- 33.Qiu W, David D, Zhou B, Chu PG, Zhang B, Wu M, Xiao J, Han T, Zhu Z, Wang T, Liu X, Lopez R, et al. Down-regulation of growth arrest DNA damage-inducible gene 45beta expression is associated with human hepatocellular carcinoma. Am J Pathol. 2003;162:1961–1974. doi: 10.1016/S0002-9440(10)64329-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ou DL, Shen YC, Yu SL, Chen KF, Yeh PY, Fan HH, Feng WC, Wang CT, Lin LI, Hsu C, Cheng AL. Induction of DNA damage-inducible gene GADD45{beta} contributes to sorafenib-induced apoptosis in hepatocellular carcinoma cells. Cancer Res. 2010;70:9309–9318. doi: 10.1158/0008-5472.CAN-10-1033. [DOI] [PubMed] [Google Scholar]