

SUMMARY
Little is known about the mechanisms governing neonatal growth and maturation of organs. Here we demonstrate that calcineurin/Nuclear Factor of Activated T cells (Cn/NFAT) signaling regulates neonatal pancreatic development in mouse and human islets. Inactivation of calcineurin b1 (Cnb1) in mouse islets impaired dense core granule biogenesis, decreased insulin secretion, and reduced cell proliferation and mass, culminating in lethal diabetes. Pancreatic β cells lacking Cnb1 failed to express genes revealed to be direct NFAT targets required for replication, insulin storage, and secretion. In contrast, glucokinase activation stimulated Cn-dependent expression of these genes. Calcineurin inhibitors, such as tacrolimus, used for human immunosuppression, induce diabetes. Tacrolimus exposure reduced Cn/NFAT-dependent expression of factors essential for insulin dense core granule formation and secretion and neonatal β cell proliferation, consistent with our genetic studies. Discovery of conserved pathways regulating β cell maturation and proliferation suggests new strategies for controlling β cell growth or replacement in human islet diseases.
INTRODUCTION
Defects in β cell function and number underlie many human diseases, most notably diabetes mellitus. Emerging strategies to achieve replacement or regeneration of pancreatic β cells rely on knowledge about β cell development and growth. β cells form in the embryonic pancreas (Seymour and Sander, 2011), and after birth, normal β cell development culminates in two crucial milestones. First, enhancement of glucose sensing, insulin production per cell, and increase of insulin-containing dense core secretory granules, result in the maturation of β cell stimulus-secretion coupling (Bruin et al., 2008; Kim et al., 2006). Second, proliferation in neonatal mice and human islets leads to expansion and establishment of appropriate β cell mass (Georgia and Bhushan, 2004; Meier et al., 2008; Teta et al., 2005). Defective β cell maturation or growth promotes pathogenesis of diabetes and other diseases (McKnight et al., 2010). Despite the importance of β cell functional maturation and expansion to human health, little is known about the mechanisms controlling and coordinating these crucial steps of β cell development.
To achieve effective glucose sensing and insulin secretion, β cells enhance expression of genes encoding hallmark factors, including Preproinsulin, glucose transporters (like Glut2 and Glucokinase), and the transcription factor Pdx1, during the first 3 postnatal weeks in mice (Aguayo-Mazzucato et al., 2011; Jermendy et al., 2011). In β cells, processed insulin is stored in secretory granules that have an electron-dense core in ultra-structural analysis (Kim et al., 2006). These intracellular dense core granules (DCGs) harbor several principal protein components, including the hormones insulin and islet amyloid polypeptide (IAPP), granins encoded by chromogranin A (ChgA) and chromogranin B (ChgB), and transmembrane proteins like IA2 (also called ICA152) (Kim et al., 2006; Suckale and Solimena, 2010). Prior studies suggest that IA2 is an important regulator of DCG formation and insulin secretion via linked transcriptional and posttranscriptional mechanisms (Harashima et al., 2005; Mziaut et al., 2006; Saeki et al., 2002). For example, studies of immortalized β cell lines suggest that depolarization stimulates Ca2+-dependent cleavage of IA2 to induce transcription of genes encoding Insulin, Prohormone convertase 1/3, and IA2 itself (Mziaut et al., 2006; Trajkovski et al., 2004). Other studies suggest that IA2 is necessary and sufficient for regulating DCG number in the mouse MIN6 β cell line (Harashima et al., 2005). These in vitro studies suggest how activity-dependent regulation maintains DCGs in adult β cells, but it remains unclear how Ca2+-dependent β cell pathways might regulate transcription of hallmark DCG components like granins and IAPP or how β cell DCG formation is regulated in vivo.
In concert with their maturation, β cells replicate, and this β cell expansion is postulated to modulate diabetes susceptibility (Butler et al., 2007). Studies have identified regulators required for neonatal β cell replication and establishment of β cell mass, including cyclin dependent kinase 4 (Cdk4) and D type cyclins (Georgia and Bhushan, 2004; Kushner et al., 2005; Rane et al., 1999), the transcription factor FoxM1 (Zhang et al., 2006), and other factors. Islet CyclinD2 (encoded by CcnD2) and FoxM1 protein levels are highest in neonatal mice, then decline in adults, indicating that transcription of CcnD2 and FoxM1 may regulate and limit β cell proliferation, but this possibility has not been previously explored. Moreover, it is unknown if these or other factors regulate neonatal β cell expansion in humans (Davis et al., 2010; Heit et al., 2006b).
Glucose signaling is a physiological regulator of β cell functional maturation and proliferation. Glucokinase is a crucial regulator of β cell glucose metabolism, and prior studies demonstrate that glucokinase activation stimulates Ca2+ transients and depolarization, which in turn enhance β cell production of insulin (Lawrence et al., 2001), insulin secretion (Grimsby et al., 2003), and proliferation (Pechhold et al., 2009; Porat et al., 2011; Salpeter et al., 2011). Glucokinase mRNA and activity increase during the period of postnatal β cell growth and maturation (Aguayo-Mazzucato et al., 2011; Rozzo et al., 2009; Taniguchi et al., 2000); thus, glucokinase regulated depolarization and Ca2+ signaling may be physiological regulators of pathways governing β cell proliferation and functional specialization. However, the identity of these pathways remains unclear.
The calcineurin/Nuclear Factor of Activated T cells (Cn/NFAT) pathway regulates gene transcription to coordinate proliferation, survival, and differentiation of diverse cell types, including lymphocytes and neurons (Wu et al., 2007). Calcineurin is a Ca2+-activated serine/threonine phosphatase required for activation of the NFATc family of transcription factors (NFATc1-c4). With sustained rises in intracellular Ca2+, calcineurin activation leads to dephosphorylation of NFATc proteins and other substrates (Crabtree and Olson, 2002), a step permitting NFATc nuclear translocation and regulation of gene transcription. A role for Cn/NFAT in human β cell function has been indirectly inferred from the striking observation that 10%–30% of patients requiring immunosuppression with calcineurin inhibitors, like tacrolimus (FK506), develop diabetes mellitus (reviewed in Heit, 2007). We previously reported (Heit et al., 2006a) a role for Cn/NFAT signaling in adult mouse pancreatic β cells. Conditional genetic disruption of Cn/NFAT signaling in that study, however, resulted in a nonlethal adult phenotype, where β cell development was not investigated. β cell proliferation and mass from birth through 8 weeks of age was indistinguishable from littermate controls, and by 10 weeks, these mice developed mild hyperglycemia accompanied by a reduced β cell mass. However, a role for Cn/NFAT in insulin secretion was not established. Here, we used conditional genetics to inactivate Cnb1 in neonatal islets, revealing a requirement for Cn/NFAT signaling in neonatal β cell development, including DCG biogenesis, functional maturation, and mass establishment. Additionally, studies of islets from young human subjects show that Cn/NFAT-regulated mechanisms governing DCG formation and β cell replication are conserved in humans. Changes of gene expression in human islets exposed to FK506 described here also unveil molecular and cellular rationales for the long-standing clinical observation that calcineurin inhibitors promote diabetes mellitus.
RESULTS
Lethal Postnatal Diabetes from Loss of Pancreatic Islet Cn/NFAT Signaling
To investigate Cn/NFAT regulation of postnatal β cell development, we intercrossed mice (Figure S1A available online) to produce progeny harboring a Cnb1 null allele (Cnb1Δ), a Cre recombinase-sensitive conditional allele (Cnb1lox) (Winslow et al., 2006), and Ngn3-Cre, which produces Cre in pancreatic endocrine progenitors (Schonhoff et al., 2004). On postnatal day 1 (P1), Ngn3-Cre; Cnb1Δ/lox mice (hereafter nCnb1KO) had normal β cell mass (Figures 1A and 1B), were born at expected mendelian frequency (λ2 = 1.452, p = 0.2282), and had normal hormone+ islet cell composition (Figure S1B). Consistent with these findings, serum glucose levels in nCnb1KO mice were indistinguishable from littermate controls before P20 (Figure 1C and data not shown). Thus, islet development was not detectably disrupted in embryonic and newborn nCnb1KO mice.
Figure 1. nCnb1KO Mice Develop Severe Postnatal Diabetes, Hypoinsulinemia, and Early Onset Lethality.
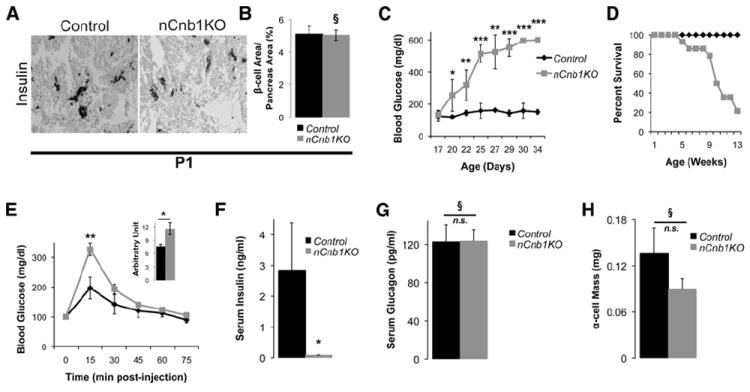
(A and B) Representative insulin immunostaining and quantification of total β cell area/Pancreatic area (in percentage) in postnatal day 1 (P1) control (black bar) and nCnb1KO (gray bar) pancreas (n = 3 per genotype).
(C) Blood glucose levels of postnatal nCnb1KO mice (gray lines) and littermate controls (black lines) during ad libitum feeding (n = 4 per genotype minimum per time point).
(D) Percent survival of aging mice (n = 31, controls, black lines; n = 14, nCnb1KO, gray lines).
(E) Glucose tolerance test performed on P19, normoglycemic mice (n = 5, controls, black; n = 4, nCnb1KO, gray). Inset, area under the curve calculated for indicated genotypes.
(F and G) Serum insulin (F) and serum glucagon (G) levels from fasted P26 mice.
(H) α cell mass in P26 mice. All data are from both female and male mice and represented as means ± SEM. *p < 0.05, **p < 0.025, ***p < 0.002. §, not significant (n.s.).
See also Figure S1.
Cnb1 mRNA levels were significantly reduced in postnatal nCnb1KO islets (Figure S1C) accompanied by reduced nuclear localization and levels of NFATc1 in nCnb1KO β cells (Figure S1D), consistent with evidence of transcriptional autoregulation by Nfatc1 (Serfling et al., 2006). By weaning at P20–P21, nCnb1KO mice were overtly diabetic, with severe hyperglycemia (Figure 1C) and significant weight loss (Figure S1E). By 12 weeks of age only 20% of nCnb1KO mice were viable (Figure 1D). Thus, Cnb1 inactivation in islet progenitors produced lethal diabetes in young mice. Insulin challenge suggested systemic insulin sensitivity was unaffected in nCnb1KO mice (Figure S1F). However, intraperitoneal glucose challenge revealed impaired glucose tolerance in nCnb1KO mice (Figure 1E). Serum insulin levels during ad libitum feeding were markedly reduced in nCnb1KO mice by P26 (Figure 1F), suggesting severe β cell defects. By contrast, neither serum glucagon levels nor α cell mass were detectably affected in nCnb1KO mice (Figures 1G and 1H). We did not detect behavioral defects like those previously described in mice with brain-specific calcineurin inactivation or inhibition (Malleret et al., 2001). Development of other pancreatic islet cells, including delta (somatostatin), pancreatic polypeptide (PP), and epsilon (ghrelin) cells also appeared unchanged in nCnb1KO mice compared to controls (Figures S1B and S1H). Additionally, Pdx1-Cre; Cnb1Δ/lox (pCnb1KO) mice, in which Cre is expressed from cis-regulatory elements of the pancreatic duodenal homeobox 1 (Pdx1) promoter (Gu et al., 2002), phenocopied nCnb1KO mice. pCnb1KO mice had no reduction in β cell mass at birth but developed lethal diabetes in the early postnatal period (Figures S1I and S1J). Thus, disrupted neonatal β cell development contributed to the lethal diabetes in nCnb1KO mice, and we investigated nCnb1KO β cell function and growth.
Neonatal β Cell Development Requires Cnb1
We postulated that β cell Cnb1 deficiency might compromise β cell function or growth. Prior to diabetes onset, insulin content was reduced by 50% in nCnb1KO islets compared to controls (Figure 2A). In addition, nCnb1KO β cells had severely impaired insulin secretion. Cultured islets from prediabetic nCnb1KO P20 mice showed no increase in insulin secretion on glucose challenge (Figure 2B). Likewise, insulin secretion stimulated by arginine was significantly blunted in nCnb1KO islets (Figure 2B). Thus, insulin content and secretion were reduced in nCnb1KO islets. During neonatal β cell maturation, expression of insulin 2 (Ins2), Pdx1, Glut2, and glucokinase (Gck) increases (Aguayo-Mazzucato et al., 2011; Jermendy et al., 2011). Levels of mRNAs encoding all of these factors were reduced in islets from prediabetic nCnb1KO mice at P20 (Figure 2C). Chromatin immunoprecipitation (ChIP) studies of wild-type (WT) P20 islets revealed association of NFATc1 at cis-regulatory elements in the Ins2 and Gck genes (Figures S2A and S2B), suggesting that NFATc1 directly regulates neonatal islet expression of these genes. Immunohistology supported these findings, revealing reductions of Insulin, Glut2, and Pdx1 protein in P20 nCnb1KO islets (Figures 2D, S2C, and S2D). However, expression of Hif1a, which encodes another β cell metabolic regulator (Cheng et al., 2010), was unaltered in nCnb1KO islets (Figure S1C). Thus, Cn/NFAT signaling is required during β cell maturation for expression of Insulin, Pdx1, Glut2, and Gck.
Figure 2. Decreased Insulin Production and Secretion in nCnb1KO Islets.
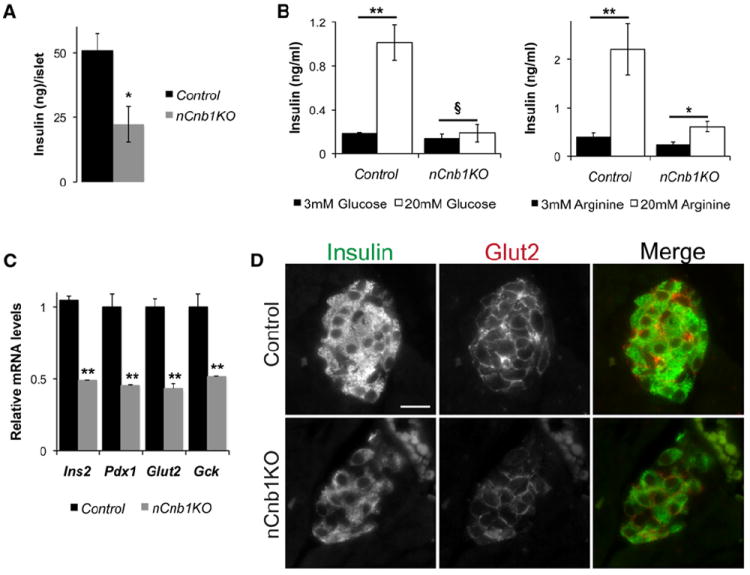
(A) Whole islet insulin content by insulin EIA in size-matched control (black bars) and nCnb1KO (gray bars) islets assessed on postnatal day 20 (P20).
(B) Glucose-stimulated (left) and arginine-stimulated (right) insulin secretion in static culture assays of islets from P20, normoglycemic nCnb1KO and control mice.
(C) Quantitative real-time PCR (qRT-PCR) of β cell factors involved in insulin production and secretion, including Insulin 2 (Ins2), pancreatic and duodenal homeobox 1 (Pdx1), glucose transporter 2 (Glut2), and glucokinase (Gck) in P20 nCnb1KO islets as compared to size-matched islets from littermate controls (n = 4 per genotype). All data presented as means ± SEM. *p < 0.05, **p < 0.025, ***p < 0.002. §, not significant (n.s.).
(D) Immunohistochemical detection of β cell factors Insulin and Glut2 in P20 nCnb1KO and control pancreatic islets. Scale bar = 10 μM.
See also Figure S2.
Cn/NFAT Signaling Regulates β Cell Dense Core Granule Formation
To determine if Cn/NFAT signaling regulates DCG formation, we investigated DCGs in nCnb1KO mutant and control islets. We used transmission electron microscopy criteria (Pictet et al., 1972) to quantify DCG subsets, including mature, immature, crystal-containing, and empty DCGs (Figures 3A and 3B). Compared to control islets, β cells in nCnb1KO islets from prediabetic P20 mice had a 40% decrease in average DCG number (Figure 3C). Levels of mature DCGs were also decreased in nCnb1KO β cells, matched by an increase of immature DCGs (Figure 3D). These results suggest that Cn/NFAT signaling is required for formation and maturation of DCGs.
Figure 3. Dense Core Granule Biogenesis and Maturation in Mouse β Cells Requires Cn/NFAT Signaling.
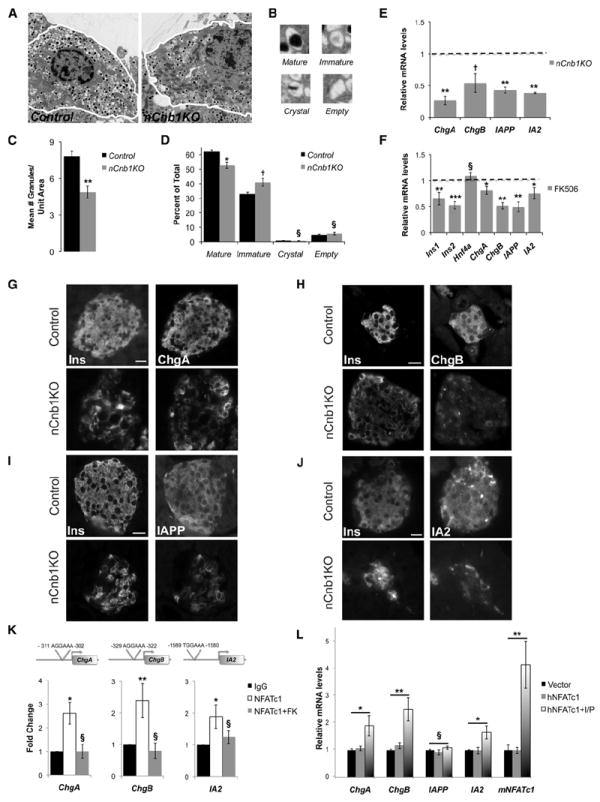
(A) Transmission electron micrographs of postnatal day 20 (P20) control and nCnb1KO β cells.
(B) Representative pictures of the four insulin granule types: (1) mature, (2) immature, (3) crystal-containing, and (4) empty.
(C and D) Quantification and morphometric analysis of dense core granules (DCGs) from WT (black bars) and nCnb1KO (gray bars) β cells showing (C) number of granules per unit area and (D) abundance of the different granule subtypes (as a percentage of the total number of granules).
(E) qRT-PCR of DCG components in P20 nCnb1KO islets as compared to size-matched islets from littermate controls (n = 4 per genotype). Dashed line represents control levels normalized to 1.0.
(F) qRT-PCR of β cell factors and DCG components in P20 islets from wild-type (WT), C57BL/6 male mice treated with FK506 (10 μM) or vehicle (EtOH) for 72 hr (n = 5).
(G–J) Immunohistochemical detection of DCG components ChgA (G), ChgB (H), IAPP (I), and IA2 (J) in P26 nCnb1KO and control islets. Scale bar = 10 μM.
(K) Chromatin immunoprecipitation (ChIP) of NFATc1 at indicated loci in islets isolated and fixed from P20 WT, C57BL/6 mice. Islets were treated for 24 hr with either vehicle (EtOH) or FK506 (10 μM) (n = 4 per condition). ChIP data are presented as fold change of signal relative to IgG background with comparisons to leftmost data bar (black).
(L) Relative mRNA levels of indicated genes after in vitro transfection of MIN6 cells with human NFATc1 expression construct (hNFATc1) or empty expression vector (Vector) and treated with either vehicle DMSO or a combination of Ionomycin and PMA (I/P). All data presented as means ± SEM. *p < 0.05, **p < 0.025, ***p < 0.002. §, not significant (n.s.).
Genes encoding DCGs proteins (reviewed in Suckale and Solimena, 2010) include Insulin, chromogranin A (ChgA), chromogranin B (ChgB), islet amyloid polypeptide (IAPP), and the protein tyrosine phosphatase receptor IA2. mRNA and protein levels of ChgA, ChgB, IAPP, and IA2 were all decreased in nCnb1KO islets (Figures 3E and 3G–3J). To validate these findings, we exposed islets isolated from WT P20 mice to FK506. Compared to islets exposed to vehicle, islets exposed to FK506 had significantly reduced levels of Ins1, Ins2, ChgA, ChgB, IAPP, and IA2 mRNA. By contrast, levels of mRNA encoding a regulator of insulin secretion, HNF4a, remained unchanged (Figure 3F). Thus, genetic and pharmacological studies provide evidence that Cnb1 is required for expression of hallmark components of β cell DCGs. We next used ChIP to investigate links between Cnb1 and NFAT transcriptional regulation of these genes. We found consensus NFAT-binding sites in the promoter regions of ChgA, ChgB, and IA2, but not IAPP (Figure 3K; see Experimental Procedures). ChIP studies of neonatal WT islets revealed significant association of NFATc1 at a subset of sites in ChgA, ChgB, and IA2 compared to IgG controls (Figure 3K). In WT islets exposed to FK506, NFATc1 binding at ChgA, ChgB, and IA2 promoters was reduced to levels comparable to IgG controls (Figure 3K). These results suggest that NFATc1 directly regulates Cn-dependent expression of genes encoding hallmark β cell DCG components.
To determine whether NFATc1 was sufficient to induce levels of DCG components, we expressed human NFATc1 (hNFATc1) (Beals et al., 1997) in the murine β cell line MIN6. Transfected or control cells were exposed to ionomycin and phorbol 12-myristate 13-acetate (PMA), factors that stimulate NFAT nuclear localization and activity (Beals et al., 1997), or to vehicle. Consistent with studies revealing NFATc1 autoregulation (Serfling et al., 2006), we observed that murine NFATc1 (mNFATc1) mRNA increased in MIN6 cells transfected with hNFAT and induced with ionomycin/PMA (Figure 3L). By contrast mNFATc1 levels were not elevated in MIN6 cells expressing hNFATc1 exposed to vehicle (Figure 3L). Levels of mRNAs encoding the NFAT targets ChgA, ChgB, and IA2 also increased in hNFATc-transfected MIN6 cells exposed to ionomycin/PMA (Figure 3L). By contrast, IAPP mRNA levels were not increased, consistent with the lack of NFATc1 binding sites discussed previously. Thus, Cn/NFAT signaling is sufficient to stimulate expression of genes encoding β cell DCG components.
To investigate if Cn/NFAT regulation of DCG components was conserved, we studied cultured human islets exposed to FK506 or vehicle control. Compared to control islets, we found reduction of mRNAs encoding INS, CHGA, CHGB, IAPP, and IA2 in islets exposed to FK506 (Figure 4A). By contrast, islet levels of mRNA encoding HNF4a were unaffected by FK506 (Figure 4A). Thus, similar to findings from mouse islets (Figures 3E and 3F), expression of genes encoding the principal DCG components in human islets was reduced by calcineurin inhibition. We next used ChIP to investigate if NFAT associated with cis-regulatory elements in CHGA, CHGB, IAPP, and IA2. Based on discovery of candidate NFAT-binding sites within the promoter regions of CHGA, CHGB, IAPP, and IA2 (Figure 4B; see Experimental Procedures), ChIP revealed significant association of NFATc1 at a subset of sites in all these loci, compared to IgG controls (Figures 4B and 4C). In human islets exposed to FK506, NFATc1 binding at these targets was consistently reduced (Figure 4B). Thus, Cn/NFAT signaling regulates expression of hallmark β cell DCG components in human islets.
Figure 4. Cn/NFAT Signaling Regulates Expression of DCG Components in Human Islets.
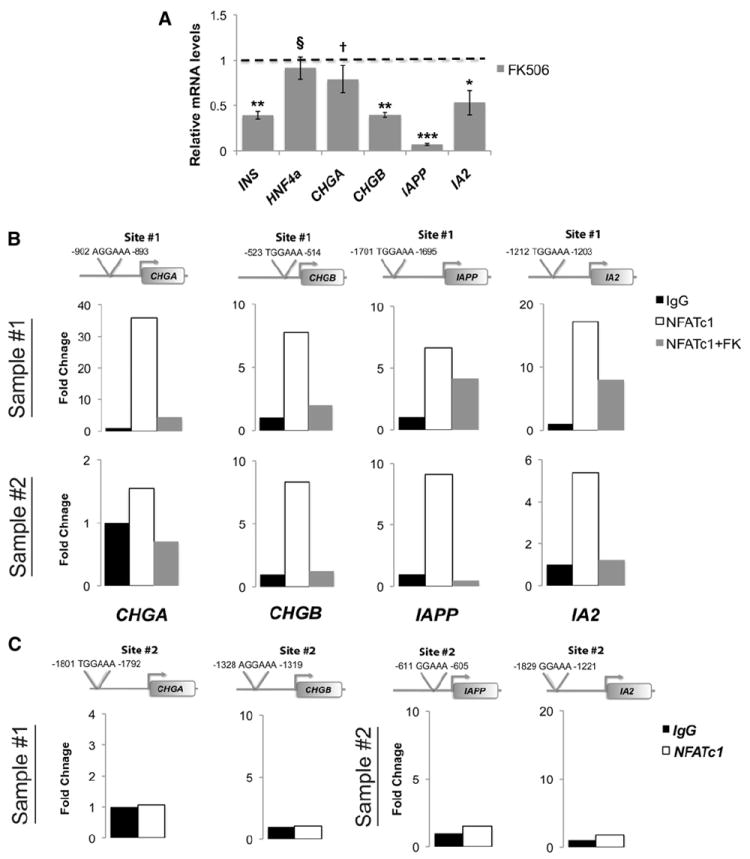
(A) Relative quantification of mRNAs encoding indicated DCG components in isolated human islets (n = 3) treated with FK506 (10 μM) or vehicle (EtOH) for 72 hr. Dashed line represents vehicle-treated control levels normalized to 1.0.
(B) ChIP of NFATc1 on isolated human islets (sample #1: 5 years old, sample #2: 13 years old). Each human sample was divided and treated for 24 hr with either vehicle (EtOH) or FK506 (10 μM).
(C) Additional adjacent NFAT consensus sites (“Site #2”) within the indicated gene promoter regions did not bind NFATc1 (see also Table S2). ChIP data are presented as fold change of NFATc1 signal (white bar) or NFATc1+FK506 (gray bar) relative to IgG (black bar) control signal. All data presented as means ± SEM. *p < 0.05, **p < 0.025, ***p < 0.002. †p < 0.15. §, not significant (n.s.)
Proliferation to Establish Neonatal β Cell Mass Requires Cn/NFAT Signaling
To investigate whether Cn/NFAT signaling regulates postnatal β cell proliferation, we examined nCnb1KO pancreata. Compared to littermate controls, nCnb1KO mice at P26 exhibited a 7-fold decrease in β cell mass (Figures 5A and 5B). In nCnb1KO mice at P26 we observed a 3-fold reduction in β cells expressing the proliferation marker Ki67, indicating impaired β cell proliferation (Figure 5C). By contrast, the percentage of β cells producing activated caspase 3, a marker of apoptosis, was not significantly increased (Figure S1G). Thus, Cnb1 is required for neonatal β cell proliferation and expansion. To identify the basis for impaired expansion of juvenile β cells lacking Cnb1, we measured expression of known regulators of neonatal β cell proliferation, including cyclin D1 (CcnD1), cyclin D2 (CcnD2), cyclin-dependent kinase 4 (Cdk4), and the Forkhead box (Fox) factor FoxM1 (Kushner et al., 2005; Rane et al., 1999; Zhang et al., 2006). In islets from prediabetic P20 nCnb1KO mice, CcnD2 and FoxM1 mRNAs were significantly reduced compared to levels in size- and age-matched control islets. Likewise, nCnb1KO islet mRNA levels of Cyclin A2 (CcnA2), another regulator of the G1-to-S phase transition, were also reduced (Figure 5D). By contrast, CcnD1 and Cdk4 mRNAs were not detectably reduced in nCnb1KO islets (Figure 5D). To validate these findings, we exposed neonatal, WT islets to FK506. Like in nCnb1KO islets, levels of CcnA2, CcnD2, and FoxM1 mRNAs were significantly decreased (Figure 5E). Immunohistology revealed accompanying reductions of CcnD2 and FoxM1 protein in nCnb1KO β cell nuclei (Figures 5F and 5G). These results reveal a requirement for Cn/NFAT signaling during a crucial neonatal stage of β cell proliferation and expansion.
Figure 5. Mouse Neonatal β Cell Proliferation and Mass Regulated by Cn/NFAT Signaling.
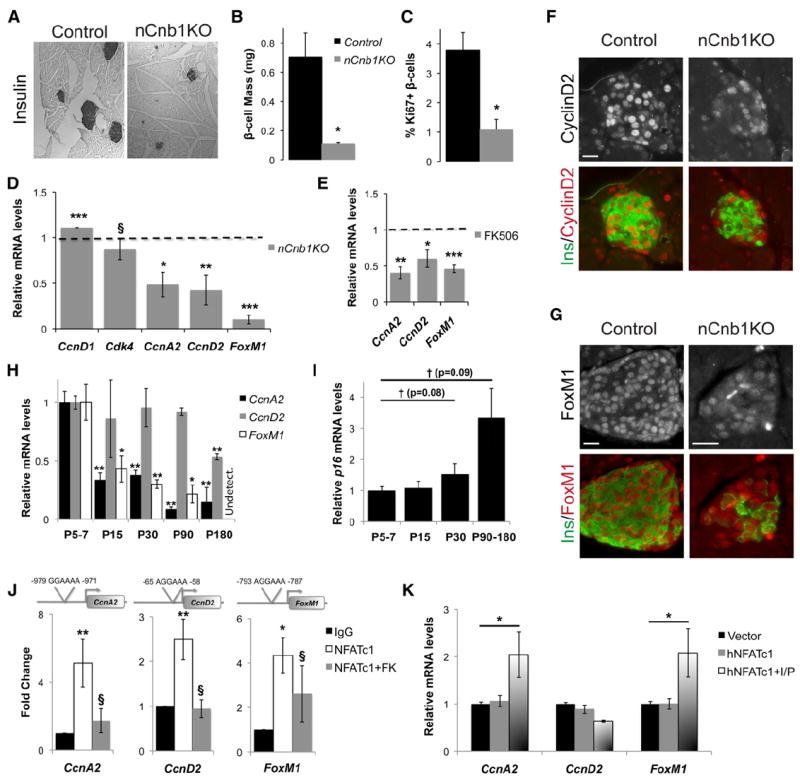
(A) Representative insulin stains of Control and nCnb1KO pancreatic tissue at postnatal day 26 (P26).
(B) Quantification of β cell mass by morphometry in control (black bar) and nCnb1KO (gray bar) mice.
(C) Quantification of β cell proliferation by scoring the percentage of Ki67+ β cells in control (black bar) and nCnb1KO (gray bar) pancreatic islets.
(D) Quantification of mRNAs encoding indicated cell cycle regulators in P20 nCnb1KO islets and size-matched control islets (n = 4 per genotype). Dashed line represents control levels normalized to 1.0.
(E) mRNA quantification of CcnA2, CcnD2, and FoxM1 in P20 islets from WT C57BL/6J male mice treated with FK506 (10 μM) or vehicle (EtOH) for 72 hr (n = 5). Dashed line represents control levels normalized to 1.0.
(F and G) Immunohistochemical detection of Cyclin D2 (F), gray, or red in merge), and FoxM1 (G), gray, or red in merge) in P26 nCnb1KO and control pancreatic islets. Insulin (Ins) in green. Scale bar = 10 μM.
(H and I) qRT-PCR time course of cell cycle regulators in FACS-isolated β cells from MIP-GFP mice at indicated ages (n = 4, 2, 3, 2, 2 at each time point, respectively).
(J) ChIP of NFATc1 on indicated loci from islets isolated and fixed from P20 wild-type, C57BL/6 mice. Islets were treated for 24 hr with either vehicle (EtOH) or FK506 (10 μM) (n = 4 per condition). ChIP data are presented as fold change of NFATc1 (white bar) or NFATc1+FK506 (gray bar) signal relative to IgG (black bar) background signal.
(K) Quantification of mRNA levels of indicated genes after in vitro transfection of MIN6 cells with human NFATc1 expression construct (hNFATc1) or empty expression vector (Vector) treated with either Ionomycin and PMA (I/P) or vehicle DMSO. All data presented as means ± SEM. *p < 0.05, **p < 0.025, ***p < 0.002. §, not significant (n.s.), with comparisons to leftmost data bar (black), unless otherwise noted.
CcnA2, CcnD2, and FoxM1 Regulation in Mouse and Human Islets by NFATc1
To investigate CcnD2, FoxM1, and CcnA2 regulation in islets, we measured mRNA levels encoding these factors in β cells purified by FACS from juvenile and adult MIP-GFP mouse islets (Sugiyama et al., 2007). Levels of mRNAs encoding CcnD2, FoxM1 (Ackermann Misfeldt et al., 2008), and CcnA2 were enriched in juvenile β cells, then declined in adult β cells (Figure 5H). By contrast, mRNA levels encoding the cyclin-dependent kinase inhibitor p16Ink4a increased during this interval (Figure 5I), confirming prior results (Chen et al., 2009). Age-dependent β cell expression of CcnD2 and FoxM1 mRNA reported here is consistent with prior studies of dynamic CyclinD2 and FoxM1 protein abundance in mouse β cells (Georgia and Bhushan, 2004; Zhang et al., 2006). We identified consensus promoter-proximal NFAT-binding sites in mouse CcnD2, FoxM1, and CcnA2 loci (Figure 5J). NFATc1 protein association at these sites in WT neonatal islets was revealed by ChIP and exposure of islets to FK506 consistently reduced NFATc1 binding to background levels (Figure 5J). Thus, NFATc1 directs Cn-dependent expression of known regulators of neonatal β cell proliferation.
To test whether NFATc1 was sufficient to induce the expression of these cell cycle regulators, we expressed human NFATc1 (hNFATc1) (Beals et al., 1997) in the MIN6 β cell line. hNFATc1-transfected cells were then exposed to ionomycin/PMA or to vehicle. hNFATc1 transfection followed by ionomycin/PMA treatment increased expression of CcnA2 and FoxM1, compared to controls (Figure 5K). Baseline levels of CcnD2 in MIN6 cells are known to be elevated (Cozar-Castellano et al., 2006); so, as expected, hNFATc1 transfection with ionomycin/PMA treatment did not further increase CcnD2 mRNA in MIN6 cells (Figure 5K). Thus, Cn/NFAT signaling regulates CcnA2, CcnD2, and FoxM1 expression and β cell proliferation in postnatal mouse islets.
Human β cell proliferation, assessed by Ki67 staining, is highest in children less than 5–10 years of age (Meier et al., 2008). To test the relevance of our findings to human β cell proliferation, we measured mRNA levels of CCNA2, CCND2, and FOXM1 in islets isolated from donors aged 1–5 years and control adult donors. mRNAs encoding CCNA2, CCND2, and FOXM1 were higher in islets purified from young donors than in islets from adults (Figures 6A–6C). By contrast, levels of mRNA encoding cyclin-dependent kinase 2 (CDK2) did not change significantly with human islet age (Figure 6D). Thus, our findings reveal elevated islet expression of CCNA2, CCND2, and FOXM1 during a period of established physiologic human β cell expansion. Based on similarities to our findings in mice, we tested whether Cn/NFAT signaling governs human islet CCNA2, CCND2, and FOXM1 expression. We identified consensus NFAT-binding sites in the promoter regions of human CCNA2, CCND2, and FOXM1 (Figure 6E), and ChIP revealed FK506-sensitive NFATc1 association at these sites in neonatal human islets (Figure 6E). Next, we exposed human islets to FK506 or vehicle control, and quantitative real-time PCR (qRT-PCR) revealed that CCNA2, CCND2, and FOXM1 mRNA levels were significantly decreased in FK506-exposed islets compared to control islets (Figure 6F). Consistent with these findings, exposure of cultured neonatal human islets to FK506 reduced β cell BrdU incoporation by nearly 3-fold, compared to islets exposed to vehicle control (Figures 6G and 6H). Collectively, these findings provide evidence that Cn/NFAT signaling regulates CCNA2, CCND2, and FOXM1 expression and β cell proliferation in human islets.
Figure 6. CCNA2, CCND2, and FOXM1 mRNA Levels Peak during the Neonatal Period in Human Islets, and Cn/NFAT Regulates Their Expression.

(A–D) qRT-PCR time course of CCNA2, CCND2, FOXM1, and CDK2 mRNA transcript levels in isolated islets from humans of increasing age (n = 2, except time point “39-56 y”, where n = 4).
(E) ChIP of NFATc1 at indiated loci from isolated human islets (sample #1: 5 years old, sample #2: 13 years old) treated for 24 hr with either vehicle (EtOH) or FK506 (10 μM). Note: Sample #1 was only treated with vehicle because of limited islet yield from donor sample. Additional adjacent NFAT consensus sites (“Site #2”) within the gene promoter region did not bind NFATc1 (see also Table S2). ChIP data are presented as fold change of NFATc1 signal (white bar) or NFATc1+FK506 (gray bar) relative to IgG (black bar) control signal.
(F) mRNA quantification of indicated genes in isolated human islets (n = 3) treated with FK506 (10 μM) or vehicle (EtOH) for 72 hr. Dashed line represents vehicle-treated control levels normalized to 1.0.
(G and H) Quantification of BrdU+ insulin+ cells as a percentage of all insulin+ cells in islets isolated from a 4-year-old human donor pancreas. Islets were divided and exposed to vehicle (DMSO) or FK506 (10 μM) (see Experimental Procedures). (H) Representative immunofluorescence staining of insulin+BrdU+ double-positive cells (arrowheads) from 4-year-old donor islets. Insulin (green) and BrdU (red). All data presented as means ± SEM. *p < 0.05, **p < 0.025, ***p < 0.002. §, not significant (n.s.).
Glucokinase Activator Induces NFAT Target Genes Governing β Cell Growth and Maturation
Our findings suggested that Cn/NFAT signaling is developmentally regulated in postnatal islets. If so, we predicted that expression of mRNAs encoding NFATc1 and other factors would be elevated in neonatal islets because NFATc1 activates expression of itself and other pathway components (Arron et al., 2006; Serfling et al., 2006). Consistent with this possibility, we observed that levels of mRNAs encoding NFATc1, NFATc2, and NFATc4 were higher in islets from P10 mice compared to those in P28 islets (Figure S3A). FACS purification confirmed that NFATc1 mRNA levels were approximately 50% greater in β cells from P5–P15 islets than those in β cells from adult mice (Figure S3B). Thus, expression and activity of Cn/NFAT signaling appears to be enhanced in neonatal islets when expression of NFAT targets governing β cell proliferation and maturation peaks.
Prior work suggests that glucokinase activation is a physiological mechanism for stimulating depolarization and Ca2+-dependent β cell proliferation and maturation (reviewed in Salpeter et al., 2011). Thus, we postulated that β cell Cn/NFAT signaling might be induced by glucokinase activators. Consistent with this possibility, we found that the glucokinase activator (GKA) R0-28-1675 (Matschinsky, 2009) increased NFATc1 mRNA by 50% in cultured P10 islets (Figure 7A), compared to islets exposed to vehicle control. This induction was blocked by simultaneous exposure of WT islets to FK506 and GKA (Figure 7A), a result supported by our finding that GKA-induced expression of NFATc1 was also blocked in nCnb1KO islets (Figure 7A). Ins2, ChgA, ChgB, IAPP, and IA2 were significantly increased in neonatal P10 islets exposed to GKA, an effect blunted or eliminated by FK506 (Figures 7B and 7C). Similar induction of mRNAs encoding β cell cycle regulators CcnA2, CcnD2, and FoxM1 were observed in P10 islets exposed to R0-28-1675; again, these effects were attenuated or blocked by simultaneous exposure to FK506 (Figure 7D). Together, these findings suggest that glucokinase activation of Cn/NFAT signaling in postnatal islets may regulate crucial regulators of β cell function and proliferation.
Figure 7. Glucokinase Activator Induces Transcription of NFATc1 and Its Targets in a Calcineurin-Dependent Manner.
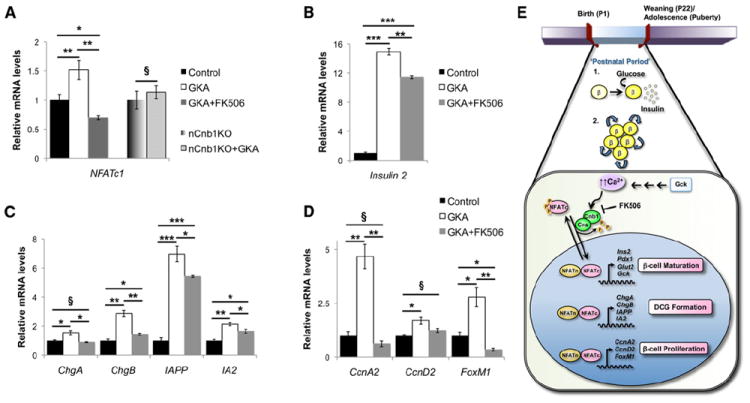
(A–D) Islets isolated from postnatal (P10) control C57Bl/6 or nCnb1KO mice treated with either vehicle, glucokinase activator (GKA) R0-28-1675 (10 μM) or GKA+FK506 (10 μM each) for 72 hr (n = 3 minimum per condition). qRT-PCR of (A) NFATc1, (B) Insulin 2, (C) indicated DCG components, and (D) indicated cell cycle regulators.
(E) Schematic summarizing a role for Cn/NFAT signaling in postnatal β cell (1) maturation and (2) proliferation via the direct transcriptional regulation of key β cell genes. Ins2 (insulin 2), Pdx1 (pancreatic duodenal homeobox 1), Glut2 (glucose transporter type 2), Gck (glucokinase), ChgA/B (chromogranins A and B), IAPP (islet amyloid polypeptide), IA2 (ICA512), CcnA2 (cyclinA2), CcnD2 (cyclinD2), and FoxM1 (forkhead homeobox factor M1). All data are from male mice and are represented as means ± SEM. *p < 0.05, **p < 0.025, ***p < 0.002. §, not significant (n.s.).
See also Figure S3.
DISCUSSION
Progress in creating replacement islets from renewable sources, like human stem cell lines, is limited by a lack of knowledge about physiological mechanisms promoting development of functional insulin-secreting β cells (McKnight et al., 2010). Likewise, the promise of advances in human islet transplantation is constrained by a demand for replacement β cells that exceeds supply (Meier et al., 2008). Thus, there is intense interest in understanding mechanisms regulating the in vivo maturation and proliferation of pancreatic islet β cells. Here we report that the Cn/NFAT pathway regulates maturation and expansion of functional β cells in both mouse and human islets (Figure 7E).
During physiological growth in neonatal mice and humans, islet β cells adapt by enhancing their hallmark functions, including glucose sensing, insulin production, DCG biogenesis, and stimulus-secretion coupling (Bruin et al., 2008; Kim et al., 2006; Suckale and Solimena, 2010). This functional maturation likely reflects the shift from intrauterine energy sources to the postnatal diet (Fowden and Hill, 2001). Exression of genes encoding the effectors of these functional adaptations, including Insulin2, Glut2, Glucokinase, Pdx1, ChromograninA, and IA2, increases in neonatal mice (Aguayo-Mazzucato et al., 2011; Jermendy et al., 2011; C. Benitez and S.K.K., unpublished data). Prior studies (Ahlgren et al., 1998; Gu et al., 2010; Mziaut et al., 2008; Zhang et al., 2005) suggest that subsets of these factors in adult islets or cell lines may be governed by transcriptional regulators, including MafA, Pdx1, NeuroD1, IA2, and Stat5. Other studies have previously demonstrated regulation of Insulin by NFATc1 in the β cell line MIN6 and in adult islets (Heit et al., 2006a; Lawrence et al., 2001). However, the basis for dynamic changes in neonatal expression of these β cell factors in mouse or human islets has not been established. The findings here demonstrate in vivo roles for Cn/NFAT in mouse islet maturation and in controlling expression of factors essential for hallmark β cell functions in neonatal human islets. We also present evidence that NFATc1 is sufficient to activate expression of target genes encoding β cell cycle regulators and dense core granule components. These findings do not rule out roles for other calcineurin-regulated factors in neonatal β cell development. In addition to NFATs, calcineurin has other important targets, including TORC2 and Erk1/2 (Arnette et al., 2003; Lawrence et al., 2005; Le Lay et al., 2009). Although there is evidence for Raf/Erk1/2 signaling in insulin secretion (Longuet et al., 2005), glucose regulation was not detectably perturbed in mice lacking TORC2 (Le Lay et al., 2009). Additional studies are needed to address the possibility that calcineurin-dependent Erk1/2 activation may regulate post-natal β cell development.
How are β cell growth and maturation coordinated in neonatal islets? Prior reports suggested that glucose metabolism by glucokinase may link β cell depolarization and Ca2+ influx to β cell proliferation (Pechhold et al., 2009; Porat et al., 2011; Salpeter et al., 2011) and function (Kassem et al., 2010; Terauchi et al., 1995, 2007; Vionnet et al., 1992). Moreover, neonatal β cell growth and maturation in rodents is accompanied by enhanced Ca2+ flux (Navarro-Tableros et al., 2007), requiring the voltage-gated calcium channel subunit α1D (Namkung et al., 2001). However, it remained unclear how Ca2+ signals were connected to genetic programs controlling β cell growth and maturation. Cn/NFAT signaling is regulated by Ca2+ transients (reviewed by Crabtree and Olson, 2002), and Ca2+-regulation of Cn/NFAT activity in β cells has been established (Lawrence et al., 2009). Our studies link glucokinase activation to Cn/NFAT signaling induction in mouse islet β cells. Thus, findings here suggest that Cn/NFAT signaling is a crucial pathway that links enhanced glucose metabolism and Ca2+ dynamics to transcriptional regulation that drives β cell proliferation and maturation in neonatal islets (Figure 7E). We speculate that further studies may reveal how Cn/NFAT signaling converts and integrates activity-dependent β cell Ca2+ transients into gene expression changes that orchestrate β cell developmental growth and functional maturation. Further studies are also required to establish if glucokinase activators can stimulate expression of β cell cycle regulators and hallmark dense core granule components in human islets in culture and in vivo. If so, discovery of Cn/NFAT activators might be used to stimulate proliferation and expansion of functional human β cells produced from expandable sources, including stem cell lines.
Based on frequency of iatrogenic diabetes mellitus from FK506 or cyclosporine A (Heit, 2007), we and others postulated that disrupted Cn/NFAT signaling might impair β cell function (Heit et al., 2006a; Redmon et al., 1996). Cultured human islets exposed to FK506 have impaired insulin secretion (Johnson et al., 2009) and reduced DCG numbers (Bugliani et al., 2009; Drachenberg et al., 1999), but the molecular basis for these findings remained unclear. Our results argue that impaired islet insulin secretion from exposure to calcineurin inhibitors reflects both disrupted expression of β cell secretion regulators, like Glut2 and Glucokinase, and impaired biogenesis or dynamics of β cell dense core granules. β cell depolarization may induce Ca2+-dependent processing of IA2 by the calpain protease, leading to Stat5 activation and increased expression of IA2 itself and other DCG components (Mziaut et al., 2006; Trajkovski et al., 2004). Recent studies have shown that calpain may also activate calcineurin activity in response to Ca2+ signaling (Chang et al., 2004), and we show here that Cn/NFAT signaling regulates islet expression of IA2 and Nfatc1. Thus, Ca2+-dependent signaling in β cells may lead both to autoactivation and cross-activation of transcriptional regulators that control expression and assembly of key DCG components, as well as essential glucose sensing factors, like glucose transporters and glucokinase. Studies of cultured cell lines and rodent islets provide evidence that signaling pathways regulated by GLP1, ERK/MAPK, and glucose may activate the Cn/NFAT pathway in β cells (Lawrence et al., 2001, 2005, 2009). Additional studies are needed to test if factors that modulate β cell Cn/NFAT activation may be useful for promoting maturation of replacement β cells produced from renewable sources.
In mice, CyclinD1, CyclinD2, Cdk4, and FoxM1 are required for β cell proliferation during physiological neonatal growth (Georgia and Bhushan, 2004; Kushner et al., 2005; Rane et al., 1999; Zhang et al., 2006). Work here reveals that Cn/NFAT signaling regulates in vivo mouse β cell expression of CcnD2 and FoxM1, as well as CcnA2, a known regulator of proliferation in smooth muscle and fibroblasts (Karpurapu et al., 2008; Tomono et al., 1998). Studies of neonatal mouse islets exposed to FK506 demonstrate Cn-dependent association of NFATc1 with these loci, supporting the view that NFATc1 regulates expression of these genes. Genetic studies allowing simultaneous inactivation of the eight alleles encoding NFATc1-c4 in β cells should test the requirement for these transcriptional regulators in neonatal β cell proliferation. Collectively, our results suggest that Cn/NFAT activity governs the expression of multiple cell cycle regulators essential for establishing β cell mass in juvenile mice. Here we show that levels of mRNA encoding CCND2, FOXM1, and CCNA2 are highest in islets from young human donors, then decline in adult islets, similar to their age-dependent reduction in mice. Moreover, molecular analysis reveals that Cn/NFAT signaling is required to sustain expression of these factors and BrdU incorporation by human neonatal β cells. Additional studies should also reveal whether age-dependent decline of human β cell proliferation is linked to attenuation of Cn/NFAT activity. Thus, our work unveils evolutionarily conserved mechanisms governing human β cell cycle regulation and suggests that impaired β cell replication may underlie iatrogenic diabetes in patients exposed to calcineurin inhibitors, like FK506. We speculate that development of novel Cn/NFAT pathway inhibitors with improved therapeutic index (Bernard et al., 2010) might prove useful to develop strategies for diseases reflecting increased β cell mass or function, including congenital or acquired hyperinsulinism, nesidioblastosis following bariatric surgery, insulinomas, and other neuroendocrine cancers.
EXPERIMENTAL PROCEDURES
Animals
Mice harboring the Cnb1f or Cnb1Δ alleles were described previously (Winslow et al., 2006). Transgenic Ngn3-Cre, Pdx1-Cre and MIP-EGFP mice were previously described (Gu et al., 2002; Schonhoff et al., 2004; Hara et al., 2003). C57/BL6 mice were from Charles River. Mice were used in accordance with the Institutional Animal Care and Use Committee of Stanford University. See Supplemental Experimental Procedures for details.
Molecular, Histological, and Electron Microscopy Methods
Standard histology and immunostaining protocols for various proteins on pancreas or islet sections were described previously (Chen et al., 2009; Heit et al., 2006a). β cell mass and proliferation were morphometrically quantified and details of these studies and mouse islet isolations, RT-PCRs, and ChIP analyses are provided in the Supplemental Experimental Procedures. For culture studies, islets were placed in standard media (RPMI 1640 with 4.5 mM glucose containing 10% FBS and 1% pen/strep) at 37°C and 5% CO2. In some experiments islets were maintained in media containing vehicle (DMSO), glucokinase activator (GKA) R0-28-1675 (10 μM; Axon Ligands) and/or FK506 (10 μM; LC Laboratories, Woburn, MA, USA) for 72 hr, with media changes every 24 hr. β cells from MIP-EGFP islets were isolated as previously described (Sugiyama et al., 2007). Analyses were performed in triplicate and islets from three to six mice of each genotype were independently tested.
For transmission EM experiments, 50 size-matched islets were isolated by collagenase perfusion from three prediabetic P20 nCnb1KO mice and three littermate controls as described above and in the Supplemental Experimental Procedures. Consecutive 75 to 90 nm sections were taken between, picked on formvar/carbon coated slot grids (EMS) or 100 mesh Cu grids (EMS). After contrast staining, sections were observed in a JEOL 1230 TEM at 80 kV and images captured with a digital camera. Three experiments were performed by independent researchers blinded to sample identity, with a minimum of 30 β cells scored per genotype per experiment.
Physiological and In Vitro Studies
Glucose and insulin tolerance tests were performed as previously described (Heit et al., 2006a). Serum insulin and glucagon levels were assessed in P26 mice fasted for 4 hr using the Mouse Insulin Ultrasensitive EIA and Glucagon ELISA kits (Alpco, Salem, NH, USA). Whole islet insulin content, GSIS and ASIS batch culture experiments were performed as previously described (Chen et al., 2009). Each condition was performed in quadruplicate using islets from at least three mice per genotype and levels of secreted insulin were normalized to islet DNA content.
MIN6 murine insulinoma cells (passage 26; Miyazaki et al., 1990) were transfected using lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) with 2 μg of control vector (pcDNA) or DNA permitting expression of human NFATc1 cDNA, as previously described (Beals et al., 1997). Cells grown for 48 hr were treated with vehicle (DMSO) or a combination of ionomycin (1 μM) and phorbol 12-myristate 13-acetate (PMA) (25 μM) to activate calcineurin/NFAT. Eight hours later cells were harvested for mRNA isolation.
Human Islet Studies
Institutional review board approval for research use of tissue was obtained from Stanford University School of Medicine. Human pancreata were obtained from previously healthy, nondiabetic organ donors by the National Diseases Resource Interchange. Islets were isolated by R. Bottino, (University of Pittsburgh) or at the University of Alabama (S. Bryant and T. Thompson). Seven independent human islet batches from donors aged 13, 19, and 23 months old or aged 4, 5, 19, and 20 years old, as well as five adult batches from donors of 28, 29, 49, 55, and 56 years old were used in this study. Within 24 hr of isolation, islets were transferred to fresh islet culture medium and handpicked after dithizone staining, detailed in the Supplemental Experimental Procedures. One thousand islet equivalents were concentrated and frozen for mRNA isolation, or prepared for ChIP as described above. BrdU analysis was performed following exposure to 50 μM BrdU for 24 hr. Islets were immunostained for insulin and BrdU and β cell proliferation rate was determined by quantifying the percentage of insulin+ and BrdU+ cells. We scored a minimum of 50 islets and over 2,000 β cells per condition.
Statistical Analysis
Results were expressed as the mean ± SEM. Statistical analysis was performed using the two-tailed or one-tailed, unpaired Student’s t test, with significance set at p < 0.05.
Supplementary Material
Acknowledgments
We thank Drs. I. Graef, W. Han, and M. Solimena for helpful discussions and advice, Drs. D. Melton, A. Leiter, and M. Hara for mice, the National Disease Research Interchange, A. Thompson and S. Bryant for human islet processing, J. Perrino and the Stanford Cell Sciences Imaging Facility for transmission electron microscopy and members of the Crabtree and Kim laboratories for comments on the manuscript. W.R.G. is a student in the Stanford Medical Scientist Training Program and was additionally supported by an American Diabetes Association (ADA) Medical Scholars grant. G.R.C. was supported by the National Institutes of Health (NIH; RO1HD55391). Work in the Kim laboratory was supported by a gift from the Snyder Foundation and Dewey Family Fund, grants from the Juvenile Diabetes Research Foundation and the NIH, and by the Howard Hughes Medical Institute (HHMI).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes three figures, two tables, and Supplemental Experimental Procedures and can be found with this article online at http://dx.doi.org/10.1016/j.devcel.2012.05.014.
References
- Ackermann Misfeldt A, Costa RH, Gannon M. Beta-cell proliferation, but not neogenesis, following 60% partial pancreatectomy is impaired in the absence of FoxM1. Diabetes. 2008;57:3069–3077. doi: 10.2337/db08-0878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguayo-Mazzucato C, Koh A, El Khattabi I, Li W-C, Toschi E, Jermendy A, Juhl K, Mao K, Weir GC, Sharma A, Bonner-Weir S. Mafa expression enhances glucose-responsive insulin secretion in neonatal rat beta cells. Diabetologia. 2011;54:583–593. doi: 10.1007/s00125-010-2026-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahlgren U, Jonsson J, Jonsson L, Simu K, Edlund H. beta-cell-specific inactivation of the mouse Ipf1/Pdx1 gene results in loss of the beta-cell phenotype and maturity onset diabetes. Genes Dev. 1998;12:1763–1768. doi: 10.1101/gad.12.12.1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnette D, Gibson TB, Lawrence MC, January B, Khoo S, McGlynn K, Vanderbilt CA, Cobb MH. Regulation of ERK1 and ERK2 by glucose and peptide hormones in pancreatic beta cells. J Biol Chem. 2003;278:32517–32525. doi: 10.1074/jbc.M301174200. [DOI] [PubMed] [Google Scholar]
- Arron JR, Winslow MM, Polleri A, Chang C-P, Neilson JR, Heit JJ, Kim SK, Francke U, Graef IA, Crabtree GR. NFAT dysregulation by increased dosage of DSCR1 and DYRK1a on chromosome 21. Nature. 2006;441:595–600. doi: 10.1038/nature04678. [DOI] [PubMed] [Google Scholar]
- Beals CR, Clipstone NA, Ho SN, Crabtree GR. Nuclear localization of NF-ATc by a calcineurin-dependent, cyclosporin-sensitive intramolecular interaction. Genes Dev. 1997;11:824–834. doi: 10.1101/gad.11.7.824. [DOI] [PubMed] [Google Scholar]
- Bernard B, Kline GA, Service FJ. Hypoglycaemia following upper gastrointestinal surgery: case report and review of the literature. BMC Gastroenterol. 2010;10:77. doi: 10.1186/1471-230X-10-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruin JE, Petre MA, Raha S, Morrison KM, Gerstein HC, Holloway AC. Fetal and neonatal nicotine exposure in Wistar rats causes progressive pancreatic mitochondrial damage and beta cell dysfunction. PLoS ONE. 2008;3:e3371. doi: 10.1371/journal.pone.0003371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bugliani M, Masini M, Liechti R, Marselli L, Xenarios I, Boggi U, Filipponi F, Masiello P, Marchetti P. The direct effects of tacrolimus and cyclosporin A on isolated human islets: A functional, survival and gene expression study. Islets. 2009;1:106–110. doi: 10.4161/isl.1.2.9142. [DOI] [PubMed] [Google Scholar]
- Butler PC, Meier JJ, Butler AE, Bhushan A. The replication of beta cells in normal physiology, in disease and for therapy. Nat Clin Pract Endocrinol Metab. 2007;3:758–768. doi: 10.1038/ncpendmet0647. [DOI] [PubMed] [Google Scholar]
- Chang I, Cho N, Kim S, Kim JY, Kim E, Woo J-E, Nam JH, Kim SJ, Lee M-S. Role of calcium in pancreatic islet cell death by IFN-gamma/TNF-alpha. J Immunol. 2004;172:7008–7014. doi: 10.4049/jimmunol.172.11.7008. [DOI] [PubMed] [Google Scholar]
- Chen H, Gu X, Su IH, Bottino R, Contreras JL, Tarakhovsky A, Kim SK. Polycomb protein Ezh2 regulates pancreatic beta-cell Ink4a/Arf expression and regeneration in diabetes mellitus. Genes Dev. 2009;23:975–985. doi: 10.1101/gad.1742509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng K, Ho K, Stokes R, Scott C, Lau SM, Hawthorne WJ, O’Connell PJ, Loudovaris T, Kay TW, Kulkarni RN, et al. Hypoxia-inducible factor-1alpha regulates beta cell function in mouse and human islets. J Clin Invest. 2010;120:2171–2183. doi: 10.1172/JCI35846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cozar-Castellano I, Fiaschi-Taesch N, Bigatel TA, Takane KK, Garcia-Ocaña A, Vasavada R, Stewart AF. Molecular control of cell cycle progression in the pancreatic beta-cell. Endocr Rev. 2006;27:356–370. doi: 10.1210/er.2006-0004. [DOI] [PubMed] [Google Scholar]
- Crabtree GR, Olson EN. NFAT signaling: choreographing the social lives of cells. Cell Suppl. 2002;109:S67–S79. doi: 10.1016/s0092-8674(02)00699-2. [DOI] [PubMed] [Google Scholar]
- Davis DB, Lavine JA, Suhonen JI, Krautkramer KA, Rabaglia ME, Sperger JM, Fernandez LA, Yandell BS, Keller MP, Wang IM, et al. FoxM1 is up-regulated by obesity and stimulates beta-cell proliferation. Mol Endocrinol. 2010;24:1822–1834. doi: 10.1210/me.2010-0082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drachenberg CB, Klassen DK, Weir MR, Wiland A, Fink JC, Bartlett ST, Cangro CB, Blahut S, Papadimitriou JC. Islet cell damage associated with tacrolimus and cyclosporine: morphological features in pancreas allograft biopsies and clinical correlation. Transplantation. 1999;68:396–402. doi: 10.1097/00007890-199908150-00012. [DOI] [PubMed] [Google Scholar]
- Fowden AL, Hill DJ. Intra-uterine programming of the endocrine pancreas. Br Med Bull. 2001;60:123–142. doi: 10.1093/bmb/60.1.123. [DOI] [PubMed] [Google Scholar]
- Georgia S, Bhushan A. Beta cell replication is the primary mechanism for maintaining postnatal beta cell mass. J Clin Invest. 2004;114:963–968. doi: 10.1172/JCI22098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimsby J, Sarabu R, Corbett WL, Haynes NE, Bizzarro FT, Coffey JW, Guertin KR, Hilliard DW, Kester RF, Mahaney PE, et al. Allosteric activators of glucokinase: potential role in diabetes therapy. Science. 2003;301:370–373. doi: 10.1126/science.1084073. [DOI] [PubMed] [Google Scholar]
- Gu G, Dubauskaite J, Melton DA. Direct evidence for the pancreatic lineage: NGN3+ cells are islet progenitors and are distinct from duct progenitors. Development. 2002;129:2447–2457. doi: 10.1242/dev.129.10.2447. [DOI] [PubMed] [Google Scholar]
- Gu C, Stein GH, Pan N, Goebbels S, Hörnberg H, Nave K-A, Herrera P, White P, Kaestner KH, Sussel L, Lee JE. Pancreatic beta cells require NeuroD to achieve and maintain functional maturity. Cell Metab. 2010;11:298–310. doi: 10.1016/j.cmet.2010.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara M, Wang X, Kawamura T, Bindokas VP, Dizon RF, Alcoser SY, Magnuson MA, Bell GI. Transgenic mice with green fluorescent protein-labeled pancreatic beta-cells. Am J Physiol Endocrinol Metab. 2003;284:E177–E183. doi: 10.1152/ajpendo.00321.2002. [DOI] [PubMed] [Google Scholar]
- Harashima S, Clark A, Christie MR, Notkins AL. The dense core transmembrane vesicle protein IA-2 is a regulator of vesicle number and insulin secretion. Proc Natl Acad Sci USA. 2005;102:8704–8709. doi: 10.1073/pnas.0408887102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heit JJ. Calcineurin/NFAT signaling in the beta-cell: From diabetes to new therapeutics. Bioessays. 2007;29:1011–1021. doi: 10.1002/bies.20644. [DOI] [PubMed] [Google Scholar]
- Heit JJ, Apelqvist AA, Gu X, Winslow MM, Neilson JR, Crabtree GR, Kim SK. Calcineurin/NFAT signalling regulates pancreatic beta-cell growth and function. Nature. 2006a;443:345–349. doi: 10.1038/nature05097. [DOI] [PubMed] [Google Scholar]
- Heit JJ, Karnik SK, Kim SK. Intrinsic regulators of pancreatic beta-cell proliferation. Annu Rev Cell Dev Biol. 2006b;22:311–338. doi: 10.1146/annurev.cellbio.22.010305.104425. [DOI] [PubMed] [Google Scholar]
- Jermendy A, Toschi E, Aye T, Koh A, Aguayo-Mazzucato C, Sharma A, Weir GC, Sgroi D, Bonner-Weir S. Rat neonatal beta cells lack the specialised metabolic phenotype of mature beta cells. Diabetologia. 2011;54:594–604. doi: 10.1007/s00125-010-2036-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson JD, Ao Z, Ao P, Li H, Dai L-J, He Z, Tee M, Potter KJ, Klimek AM, Meloche RM, et al. Different effects of FK506 rapamycin, and mycophenolate mofetil on glucose-stimulated insulin release and apoptosis in human islets. Cell Transplant. 2009;18:833–845. doi: 10.3727/096368909X471198. [DOI] [PubMed] [Google Scholar]
- Karpurapu M, Wang D, Singh NK, Li Q, Rao GN. NFATc1 targets cyclin A in the regulation of vascular smooth muscle cell multiplication during restenosis. J Biol Chem. 2008;283:26577–26590. doi: 10.1074/jbc.M800423200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kassem S, Bhandari S, Rodríguez-Bada P, Motaghedi R, Heyman M, García-Gimeno MA, Cobo-Vuilleumier N, Sanz P, Maclaren NK, Rahier J, et al. Large islets, beta-cell proliferation, and a glucokinase mutation. N Engl J Med. 2010;362:1348–1350. doi: 10.1056/NEJMc0909845. [DOI] [PubMed] [Google Scholar]
- Kim T, Gondré-Lewis MC, Arnaoutova I, Loh YP. Dense-core secretory granule biogenesis. Physiology (Bethesda) 2006;21:124–133. doi: 10.1152/physiol.00043.2005. [DOI] [PubMed] [Google Scholar]
- Kushner JA, Ciemerych MA, Sicinska E, Wartschow LM, Teta M, Long SY, Sicinski P, White MF. Cyclins D2 and D1 are essential for postnatal pancreatic beta-cell growth. Mol Cell Biol. 2005;25:3752–3762. doi: 10.1128/MCB.25.9.3752-3762.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence MC, Bhatt HS, Watterson JM, Easom RA. Regulation of insulin gene transcription by a Ca(2+)-responsive pathway involving calcineurin and nuclear factor of activated T cells. Mol Endocrinol. 2001;15:1758–1767. doi: 10.1210/mend.15.10.0702. [DOI] [PubMed] [Google Scholar]
- Lawrence MC, McGlynn K, Park B-H, Cobb MH. ERK1/2-dependent activation of transcription factors required for acute and chronic effects of glucose on the insulin gene promoter. J Biol Chem. 2005;280:26751–26759. doi: 10.1074/jbc.M503158200. [DOI] [PubMed] [Google Scholar]
- Lawrence MC, Shao C, McGlynn K, Naziruddin B, Levy MF, Cobb MH. Multiple chromatin-bound protein kinases assemble factors that regulate insulin gene transcription. Proc Natl Acad Sci USA. 2009;106:22181–22186. doi: 10.1073/pnas.0912596106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Lay J, Tuteja G, White P, Dhir R, Ahima R, Kaestner KH. CRTC2 (TORC2) contributes to the transcriptional response to fasting in the liver but is not required for the maintenance of glucose homeostasis. Cell Metab. 2009;10:55–62. doi: 10.1016/j.cmet.2009.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longuet C, Broca C, Costes S, Hani EH, Bataille D, Dalle S. Extracellularly regulated kinases 1/2 (p44/42 mitogen-activated protein kinases) phosphorylate synapsin I and regulate insulin secretion in the MIN6 beta-cell line and islets of Langerhans. Endocrinology. 2005;146:643–654. doi: 10.1210/en.2004-0841. [DOI] [PubMed] [Google Scholar]
- Malleret G, Haditsch U, Genoux D, Jones MW, Bliss TV, Vanhoose AM, Weitlauf C, Kandel ER, Winder DG, Mansuy IM. Inducible and reversible enhancement of learning, memory, and long-term potentiation by genetic inhibition of calcineurin. Cell. 2001;104:675–686. doi: 10.1016/s0092-8674(01)00264-1. [DOI] [PubMed] [Google Scholar]
- Matschinsky FM. Assessing the potential of glucokinase activators in diabetes therapy. Nat Rev Drug Discov. 2009;8:399–416. doi: 10.1038/nrd2850. [DOI] [PubMed] [Google Scholar]
- McKnight KD, Wang P, Kim SK. Deconstructing pancreas development to reconstruct human islets from pluripotent stem cells. Cell Stem Cell. 2010;6:300–308. doi: 10.1016/j.stem.2010.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier JJ, Butler AE, Saisho Y, Monchamp T, Galasso R, Bhushan A, Rizza RA, Butler PC. Beta-cell replication is the primary mechanism subserving the postnatal expansion of beta-cell mass in humans. Diabetes. 2008;57:1584–1594. doi: 10.2337/db07-1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazaki J, Araki K, Yamato E, Ikegami H, Asano T, Shibasaki Y, Oka Y, Yamamura K. Establishment of a pancreatic beta cell line that retains glucose-inducible insulin secretion: special reference to expression of glucose transporter isoforms. Endocrinology. 1990;127:126–132. doi: 10.1210/endo-127-1-126. [DOI] [PubMed] [Google Scholar]
- Mziaut H, Trajkovski M, Kersting S, Ehninger A, Altkrüger A, Lemaitre RP, Schmidt D, Saeger H-D, Lee M-S, Drechsel DN, et al. Synergy of glucose and growth hormone signalling in islet cells through ICA512 and STAT5. Nat Cell Biol. 2006;8:435–445. doi: 10.1038/ncb1395. [DOI] [PubMed] [Google Scholar]
- Mziaut H, Kersting S, Knoch K-P, Fan W-H, Trajkovski M, Erdmann K, Bergert H, Ehehalt F, Saeger H-D, Solimena M. ICA512 signaling enhances pancreatic beta-cell proliferation by regulating cyclins D through STATs. Proc Natl Acad Sci USA. 2008;105:674–679. doi: 10.1073/pnas.0710931105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namkung Y, Skrypnyk N, Jeong MJ, Lee T, Lee MS, Kim HL, Chin H, Suh PG, Kim SS, Shin HS. Requirement for the L-type Ca(2+) channel alpha(1D) subunit in postnatal pancreatic beta cell generation. J Clin Invest. 2001;108:1015–1022. doi: 10.1172/JCI13310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarro-Tableros V, Fiordelisio T, Hernández-Cruz A, Hiriart M. Physiological development of insulin secretion, calcium channels, and GLUT2 expression of pancreatic rat beta-cells. Am J Physiol Endocrinol Metab. 2007;292:E1018–E1029. doi: 10.1152/ajpendo.00457.2006. [DOI] [PubMed] [Google Scholar]
- Pechhold K, Koczwara K, Zhu X, Harrison VS, Walker G, Lee J, Harlan DM. Blood glucose levels regulate pancreatic beta-cell proliferation during experimentally-induced and spontaneous autoimmune diabetes in mice. PLoS ONE. 2009;4:e4827. doi: 10.1371/journal.pone.0004827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pictet RL, Clark WR, Williams RH, Rutter WJ. An ultrastructural analysis of the developing embryonic pancreas. Dev Biol. 1972;29:436–467. doi: 10.1016/0012-1606(72)90083-8. [DOI] [PubMed] [Google Scholar]
- Porat S, Weinberg-Corem N, Tornovsky-Babaey S, Schyr-Ben-Haroush R, Hija A, Stolovich-Rain M, Dadon D, Granot Z, Ben-Hur V, White P, et al. Control of pancreatic β cell regeneration by glucose metabolism. Cell Metab. 2011;13:440–449. doi: 10.1016/j.cmet.2011.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rane SG, Dubus P, Mettus RV, Galbreath EJ, Boden G, Reddy EP, Barbacid M. Loss of Cdk4 expression causes insulin-deficient diabetes and Cdk4 activation results in beta-islet cell hyperplasia. Nat Genet. 1999;22:44–52. doi: 10.1038/8751. [DOI] [PubMed] [Google Scholar]
- Redmon JB, Olson LK, Armstrong MB, Greene MJ, Robertson RP. Effects of tacrolimus (FK506) on human insulin gene expression, insulin mRNA levels, and insulin secretion in HIT-T15 cells. J Clin Invest. 1996;98:2786–2793. doi: 10.1172/JCI119105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozzo A, Meneghel-Rozzo T, Delakorda SL, Yang S-B, Rupnik M. Exocytosis of insulin: in vivo maturation of mouse endocrine pancreas. Ann N Y Acad Sci. 2009;1152:53–62. doi: 10.1111/j.1749-6632.2008.04003.x. [DOI] [PubMed] [Google Scholar]
- Saeki K, Zhu M, Kubosaki A, Xie J, Lan MS, Notkins AL. Targeted disruption of the protein tyrosine phosphatase-like molecule IA-2 results in alterations in glucose tolerance tests and insulin secretion. Diabetes. 2002;51:1842–1850. doi: 10.2337/diabetes.51.6.1842. [DOI] [PubMed] [Google Scholar]
- Salpeter SJ, Klochendler A, Weinberg-Corem N, Porat S, Granot Z, Shapiro AMJ, Magnuson MA, Eden A, Grimsby J, Glaser B, Dor Y. Glucose regulates cyclin D2 expression in quiescent and replicating pancreatic β-cells through glycolysis and calcium channels. Endocrinology. 2011;152:2589–2598. doi: 10.1210/en.2010-1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schonhoff SE, Giel-Moloney M, Leiter AB. Neurogenin 3-expressing progenitor cells in the gastrointestinal tract differentiate into both endocrine and non-endocrine cell types. Dev Biol. 2004;270:443–454. doi: 10.1016/j.ydbio.2004.03.013. [DOI] [PubMed] [Google Scholar]
- Serfling E, Chuvpilo S, Liu J, Höfer T, Palmetshofer A. NFATc1 autoregulation: a crucial step for cell-fate determination. Trends Immunol. 2006;27:461–469. doi: 10.1016/j.it.2006.08.005. [DOI] [PubMed] [Google Scholar]
- Seymour PA, Sander M. Historical perspective: beginnings of the beta-cell: current perspectives in beta-cell development. Diabetes. 2011;60:364–376. doi: 10.2337/db10-1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suckale J, Solimena M. The insulin secretory granule as a signaling hub. Trends Endocrinol Metab. 2010;21:599–609. doi: 10.1016/j.tem.2010.06.003. [DOI] [PubMed] [Google Scholar]
- Sugiyama T, Rodriguez RT, McLean GW, Kim SK. Conserved markers of fetal pancreatic epithelium permit prospective isolation of islet progenitor cells by FACS. Proc Natl Acad Sci USA. 2007;104:175–180. doi: 10.1073/pnas.0609490104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniguchi S, Tanigawa K, Miwa I. Immaturity of glucose-induced insulin secretion in fetal rat islets is due to low glucokinase activity. Horm Metab Res. 2000;32:97–102. doi: 10.1055/s-2007-978598. [DOI] [PubMed] [Google Scholar]
- Terauchi Y, Sakura H, Yasuda K, Iwamoto K, Takahashi N, Ito K, Kasai H, Suzuki H, Ueda O, Kamada N, et al. Pancreatic beta-cell-specific targeted disruption of glucokinase gene. Diabetes mellitus due to defective insulin secretion to glucose. J Biol Chem. 1995;270:30253–30256. doi: 10.1074/jbc.270.51.30253. [DOI] [PubMed] [Google Scholar]
- Terauchi Y, Takamoto I, Kubota N, Matsui J, Suzuki R, Komeda K, Hara A, Toyoda Y, Miwa I, Aizawa S, et al. Glucokinase and IRS-2 are required for compensatory beta cell hyperplasia in response to high-fat diet-induced insulin resistance. J Clin Invest. 2007;117:246–257. doi: 10.1172/JCI17645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teta M, Long SY, Wartschow LM, Rankin MM, Kushner JA. Very slow turnover of beta-cells in aged adult mice. Diabetes. 2005;54:2557–2567. doi: 10.2337/diabetes.54.9.2557. [DOI] [PubMed] [Google Scholar]
- Tomono M, Toyoshima K, Ito M, Amano H, Kiss Z. Inhibitors of calcineurin block expression of cyclins A and E induced by fibroblast growth factor in Swiss 3T3 fibroblasts. Arch Biochem Biophys. 1998;353:374–378. doi: 10.1006/abbi.1998.0667. [DOI] [PubMed] [Google Scholar]
- Trajkovski M, Mziaut H, Altkrüger A, Ouwendijk J, Knoch K-P, Müller S, Solimena M. Nuclear translocation of an ICA512 cytosolic fragment couples granule exocytosis and insulin expression in beta-cells. J Cell Biol. 2004;167:1063–1074. doi: 10.1083/jcb.200408172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vionnet N, Stoffel M, Takeda J, Yasuda K, Bell GI, Zouali H, Lesage S, Velho G, Iris F, Passa P, et al. Nonsense mutation in the glucokinase gene causes early-onset non-insulin-dependent diabetes mellitus. Nature. 1992;356:721–722. doi: 10.1038/356721a0. [DOI] [PubMed] [Google Scholar]
- Winslow MM, Gallo EM, Neilson JR, Crabtree GR. The calcineurin phosphatase complex modulates immunogenic B cell responses. Immunity. 2006;24:141–152. doi: 10.1016/j.immuni.2005.12.013. [DOI] [PubMed] [Google Scholar]
- Wu H, Peisley A, Graef IA, Crabtree GR. NFAT signaling and the invention of vertebrates. Trends Cell Biol. 2007;17:251–260. doi: 10.1016/j.tcb.2007.04.006. [DOI] [PubMed] [Google Scholar]
- Zhang C, Moriguchi T, Kajihara M, Esaki R, Harada A, Shimohata H, Oishi H, Hamada M, Morito N, Hasegawa K, et al. MafA is a key-regulator of glucose-stimulated insulin secretion. Mol Cell Biol. 2005;25:4969–4976. doi: 10.1128/MCB.25.12.4969-4976.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Ackermann AM, Gusarova GA, Lowe D, Feng X, Kopsombut UG, Costa RH, Gannon M. The FoxM1 transcription factor is required to maintain pancreatic beta-cell mass. Mol Endocrinol. 2006;20:1853–1866. doi: 10.1210/me.2006-0056. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.