

Abstract
Background & Aims
The role of trypsinogen activation in pathogenesis of acute pancreatitis (AP) has not been clearly established.
Methods
We generated and characterized mice with disruption in the gene that encodes trypsinogen7 (T7; T−/− mice), the mouse correlate of human cationic trypsinogen. The effects of pathologic activation of trypsinogen were studied in these mice, during induction of AP with caerulein. Acinar cell death, tissue damage, early intra-acinar activation of the transcription factor NF-κB, and local and systemic inflammation were compared between T−/− and wild-type mice with AP.
Results
Deletion of T7 reduced the total trypsinogen content by 60%, and resulted in total loss of cationic trypsinogen, but did not affect physiologic function. T−/− mice lacked pathologic activation of trypsinogen, which occurs within acinar cells early during AP progression. Absence of trypsinogen activation in T−/− mice led to near complete inhibition of acinar cell death in vitro and a 50% reduction in acinar necrosis during AP progression. However, T−/− mice had similar degrees of local and systemic inflammation during AP progression, as well as comparable intra-acinar levels of NF-κB activation—which was previously shown to occur concurrently with trypsinogen activation during early stages of pancreatitis.
Conclusions
T7 is activated during pathogenesis of AP in mice. Intra-acinar trypsinogen activation leads to acinar death during early stages of pancreatitis, which is responsible for 50% of the pancreatic damage in AP. However, progression of local and systemic inflammation in AP does not require trypsinogen activation. NF-κB is activated early in acinar cells, independently of trypsinogen activation, and might be responsible for progression of AP.
Keywords: PRSS1, pancreas, inflammatory response, regulation, protease, mouse model
Introduction
Acute pancreatitis (AP) is a necro-inflammatory disease of the pancreas with high mortality and significant morbidity due to lack of specific therapy (1, 2). The pathogenesis of this condition is not well understood. More than a century ago, Chiari observed activation of digestive enzymes in the pancreas in patients who succumbed to AP and suggested that pancreatitis is a disease of autodigestion by prematurely activated digestive enzymes (3). Consistent with this concept, research in the last few decades has shown intra-acinar trypsinogen activation in the early stages of pancreatitis in experimental models (4–10) as well as in clinical specimens (11, 12) and current research continues to focus on its mechanism (8, 13).
In multiple models of AP, colocalization of zymogens and lysosomes has been observed at early stages of pancreatic injury (6, 8, 9, 14–16). In these colocalization organelles, cathepsin B activates trypsinogen to trypsin (7, 17, 18). The intra-acinar trypsin is then believed to result in cell death eventually leading to AP. This “trypsin central” hypothesis is based on indirect evidence using chemical inhibitors (5, 18–20) or, more recently, using adenoviral mediated gene expression techniques (21–23), both of which have significant limitations. Further support comes from hereditary pancreatitis associated mutations in trypsinogen or trypsin inhibitors that lead to increased intra-acinar trypsin activity (24, 25). However, these are at best correlational and circumstantial evidence. Currently there is no direct and concrete experimental evidence showing that premature activation of trypsinogen is causally responsible for the pathogenesis of AP. Further, protease inhibitors have not proven effective in AP (26, 27). A group of investigators have suggested that intra-acinar trypsinogen activation may even be a protective response (28–31).
In this study, we report generation of a novel knockout mice lacking mouse trypsinogen isoform-7, a paralog of human cationic trypsinogen (PRSS1). These mice do not demonstrate pathologic trypsinogen activation and therefore are simple yet most rigorous tool to explore the role of intra-acinar trypsinogen activation in acute pancreatitis. Using these novel knock-out mice, we provide important data showing that while trypsinogen activation is important in causing cell injury early during pancreatitis, the progression of inflammation, both local and systemic, during acute pancreatitis does not require activation of trypsinogen.
Material and Methods
All experiments were performed according to protocols approved by the Institutional Animal Care and Use Committee of the University of Minnesota. AP was induced by caerulein i.p. injections at 50μg/kg/hour given 10 times. Animals were sacrificed 1 hour after the last injection, or 30 minutes after the first injection for early time point experiments. For further experimental and methodological details, see supplementary material and methods section.
Mouse trypsinogens
In mice, 20 genomic trypsinogen sequences located on chromosome 6 have been identified, numbered T1–20 (32–34). Of these, T 1, 3, 13, 14, 17, 18 and 19 are pseudogenes and T2 and 6 are relic genes (32, 34). Among the rest that are thought to be transcribed, only 4 isoforms are secreted at measurable levels by normal mouse pancreas (34). Similarly, in humans, of 9 known trypsinogen sequences, only 3 isoforms are secreted and the rest are pseudogenes (35). T7 isoform appeared as the best target for deletion based on careful review of available literature on mouse trypsinogens (32–34, 36–38).
Generation of trypsinogen isoform-7 knock-out mice
The entire 3844 nucleotide long T7 trypsinogen gene together with 440 nucleotide upstream and 386 nucleotide downstream flanking sequences was deleted by homologous recombination and replaced with an 1710 nucleotide sequence containing the neomycin resistance gene in embryonic stem [MK6(129S7)] cells. The targeting plasmid was constructed in the pKO Scrambler NTKV 1901 vector, which contains the neomycin resistance gene for positive selection and the thymidine kinase gene for negative selection. The 3510 nucleotide long 5′ homology arm was cloned between Kpn I and Xho I restriction sites and the 3491 nucleotide long 3′ homology arm was cloned between the EcoR I and Not I restriction sites. The plasmid was linearized with Xmn I digestion and electroporated into ES cells. Clones were screened by Southern blotting with probes annealing downstream and upstream of the 3′ and 5′ homology arms, respectively.
Three correctly targeted ES clones were identified by PCR and Southern analysis. These clones were used in separate C57Bl/6 blastocyst injection experiments to generate high-degree chimeric mice. Mating of the chimeric mice with C57BL/6N mice determined that one targeted clone (B59) gave rise to germ-line transmitting chimeric mice. Tail biopsy and Southern analysis of the subsequent agouti offspring identified F1 generation mice that were heterozygous for the T7 mutant allele. These mice were bred to C57BL/6N wildtype mice to establish a colony of heterozygous (T7 +/−) mice, and intercrossed to generate mice homozygous for the targeted allele (T7 −/−). These mice were further backcrossed with C57BL/6 strain for ten generations to obtain mice in C57BL/6 background.
Cathepsin B knock-out mice
These mice were obtained from Dr. G.J. Gores, Mayo Clinic, Rochester, MN and have been characterized previously (17, 39).
Statistical analysis
Results are expressed as means ± SEM. Unpaired Student’s t-test or ANOVA with Tukey-Kramer post-hoc test (> 2 groups) were used. α=0.05 and two-tailed p-values are reported.
Results
Phenotype characterization of T−/− mice
We successfully deleted trypsinogen isoform-7 (T7) gene in these novel knockout mice (T−/−). This was confirmed by absence of trypsinogen-7 gene transcript/mRNA (figure 1a). Probing with antibody against trypsinogen showed (figure 1b) two important results- 1) reduction of band intensity at 26 kD in T−/− mice (this band is known to correspond to trypsinogens based on previous studies on human and mouse (32, 40, 41)), and 2) absence of the peptide corresponding to 17 kD band in T−/− mice. Human cationic trypsinogen exists in two forms- single chain and double chain, and this phenomenon is specific to only cationic trypsinogen, not observed with other trypsinogen isoforms (40–42). Under reducing conditions, the two peptides of double chain trypsinogen (human form) separate and move to lower molecular weight segments during electrophoresis, one of these producing a prominent band at 17 kD (40, 41). Although this phenomenon has previously not been studied in mouse cationic trypsinogen, based on the properties of human cationic trypsinogen, the band observed at 17 kD in figure 1b is suspected to be T7 specific peptide of its double-chain form. The absence of this band in T−/− mice is strong evidence indicative of absence of cationic trypsinogen in T−/− mice.
Figure 1. Characterization of T−/− mice.

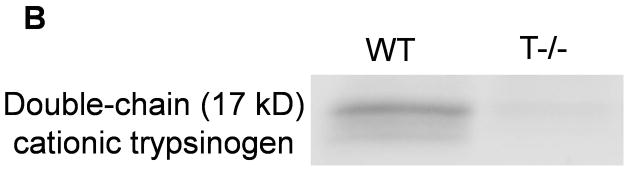
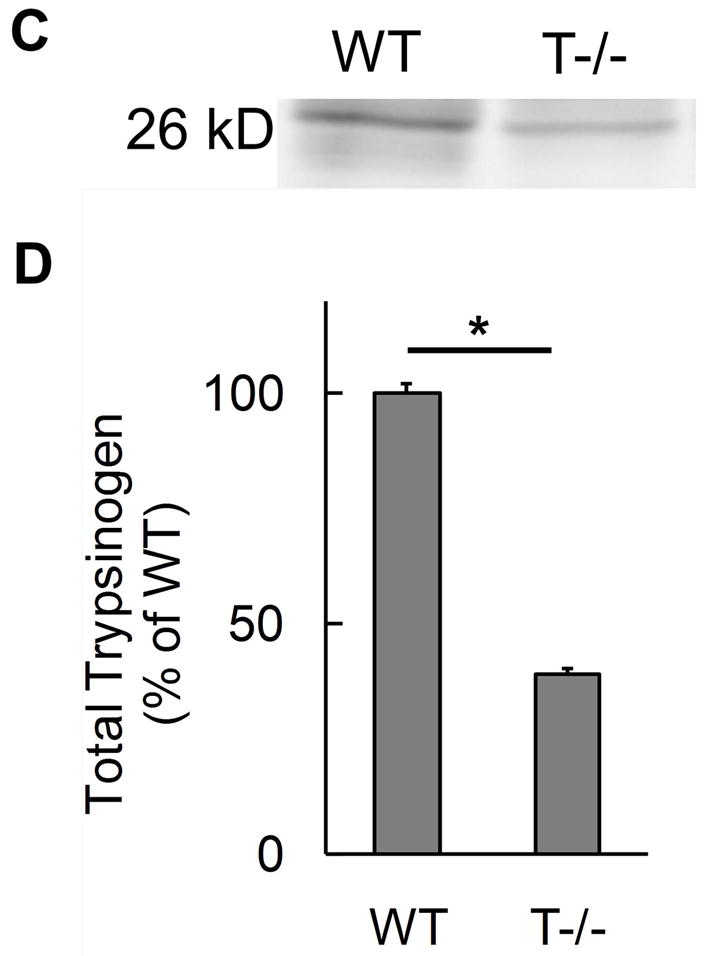

A) Absence of trypsinogen isoform-7 gene in T−/−mice: The mRNA transcript of trypsinogen isoform-7 gene is absent in T−/− mice while the expression of other isoforms of trypsinogen is not affected (n=8 each group). B and C) Reduction of total trypsinogen in T−/− mice: The 26kD band in T−/− lane is significantly reduced in intensity signifying reduction in total trypsinogen (B). On quantification (C), there was 60% reduction of total trypsingoen in T−/− mice as compared to WT (n≥3 for each group). The 17kD band (B) is suspected to be cationic trypsinogen/T7 specific peptide (from its double-chain form) based on studies on human cationic trypsinogen (40, 41), and its absence in T−/−mice strongly suggests the absence of cationic trypsinogen in T−/− mice. See text for further details. D) Dose response curve for amylase secretion in response to caerulein in-vitro. WT and T−/− acini demonstrate similar secretory response on stimulation with indicated concentrations of caerulein for 30 minutes. Data pooled from three experiments.
Cationic trypsinogen is the major trypsinogen isoform accounting for 60–65% of total trypsinogen in humans (40, 41, 43–46). Consistent with this fact, we found that the trypsinogen content in T−/− mice was only about 40% of that in the WT mice (figure 1c). Thus T7 isoform accounts for 60% of total secreted trypsinogen in mouse pancreas which is absent in T−/− mice. At the same time, expression of other trypsinogen isoforms is not altered in T−/− mice (figure 1a), and these account for 40% of total trypsinogen (figure 1c).
T−/− mice demonstrated a healthy phenotype. Weight gain, development and gross behavior were similar to WT mice suggesting lack of any physiological deficits. Similar secretory response to caerulein stimulation was seen in T−/− acini and WT acini as measured by amylase release in vitro (figure 1d) further confirming that physiological secretion is not altered in T−/− mice.
T−/− mice lack pathologic trypsinogen activation
Intra-acinar trypsinogen activation is known to peak around 30 minutes after pathologic stimulus in caerulein model (4). We measured trypsin activity in T−/− mice at 30 minutes when maximal activation is expected and at later time points (2 hr and 4 hours) to exclude any possibility of pathologic activation later. As demonstrated in figure 2a, T−/− did not demonstrate significant trypsinogen activation at 30 minutes while WT mice showed robust activation. At later time points, trypsinogen activation was not observed in any group. Similar results were obtained in-vitro demonstrating absence of trypsinogen activation in T−/− acini after supramaximal stimulation with caerulein for 30 minutes (2.3 fold activation in WT acini Vs no significant activation in T−/− acini compared to respective control group incubated without caerulein).
Figure 2. T−/− mice lack pathologic trypsinogen activation.


A) Trypsinogen activation was measured at indicated time points after caerulein (50μg/kg i.p. every hour). For controls and 30 minutes, n=12–15 per group, pooled from 3 independent experiments. N=6 per group for 2 hour and 4 hour points. p<0.0001 for WT controls Vs WT 30 minutes and for WT 30 minutes Vs T−/−30 minutes. p>0.90 for all other pair-wise comparisons. B) Chymotrypsinogen activation is dependent on trypsinogen activation and is absent in T−/− mice. Chymotrypsin activity was measured 30 minutes after caerulein (50μg/kg i.p.). N=10–13 per group, pooled from 2 independent experiments. p=0.007 for WT control Vs WT caerulein; p=0.02 for WT caerulein Vs T−/− caerulein.
Trypsinogen activation is the earliest event starting a cascade that eventually leads to activation of digestive enzymes present in the zymogen granules (47). To exclude the possibility that other components of the zymogen granules may be activated independent of trypsinogen activation, we explored the activation status of another enzyme component of zymogen, namely chymotrypsinogen. As demonstrated in figure 2b, there was no demonstrable chymotrypsinogen activation in T−/− mice while robust activation was observed in WT mice. It was confirmed that total chymotrypsinogen content was similar in WT and T−/− mice (WT: 409.95±28.41, T−/−: 382.46±45.43 (Units/mg protein), p=0.31, n=4).
Trypsinogen activation leads to acinar cell death in-vitro
Stimulation of acinar cells from WT mice with supramaximal doses of caerulein for 3 hours led to significant acinar necrosis demonstrated by LDH release (figure 3). Lack of trypsinogen activation in T−/− acini abrogated acinar necrosis providing clear evidence that intracellular trypsinogen activation is required for acinar cell death during pancreatitis in-vitro (figure 3).
Figure 3. Cell death in response to caerulein supramaximal stimulation is abrogated in acini from T−/− mice.

LDH activity was measured in the supernatant after 3 hours of incubation with or without 100nM caerulein and data were normalized to protein content in each well. The data are expressed as percentage of LDH release in the absence of caerulein. N=6 per group, each well in triplicate. p=0.003 WT caerulein Vs T−/− caerulein group.
Trypsinogen activation is responsible for half of pancreatic injury during acute pancreatitis
From figure 3, it is clear that intracellular trypsinogen activation leads to acinar cell death. To explore the contribution of this mode of acinar cell death in the pancreatic damage observed in acute pancreatitis, pancreatic necrosis during acute pancreatitis was compared in the presence (WT) and absence of trypsinogen activation (T−−). As shown in figure 4, absence of trypsinogen activation (T−/−) led to 50% reduction in necrosis suggesting that trypsinogen activation is responsible for half of local pancreatic damage during acute pancreatitis. Given that trypsinogen activation leads to acinar cell death at early time-points in-vitro (figure 3), these data (figure 4) together suggest that trypsinogen activation contributes to pancreatic injury during early stages of pancreatitis.
Figure 4. T−/− mice demonstrate about half of acinar necrosis Vs WT mice during acute pancreatitis.


Acute pancreatitis was induced by caerulein (50μg/kg i.p. every hour for 10 hours). A–C: representative pictures at 10x field (H and E stain). A) normal pancreas from saline injected mice. B) Presence of significant degree of acinar injury, edema and neutrophil infiltration in wild type mice. C) Less acinar injury in T−/− mice. D) Quantification of necrosis by morphometry. N=18 per group from 3 independent experiments. p<0.00001 for WT AP Vs T−/−AP.
Trypsinogen activation is not required for progression of local and systemic inflammation during AP
The extent of local inflammation was analyzed by measuring MPO activity in the pancreas which provides an estimate of the degree of neutrophil infiltration. Surprisingly, T−/− mice showed similar extent of local inflammatory response as compared to WT mice (figure 5a).
Figure 5. Comparable local and systemic inflammation during acute pancreatitis in WT and T−/− mice.



Neutrophil infiltration in (A) pancreas (n=18 per group from 3 independent experiments) and (B) lungs (n=12 per group from 3 independent experiments). C–E: Representative pictures of lungs at 10x field (H and E stain). C) control, D) WT and E) T−/− mice with pancreatitis, demonstrating similar parenchymal damage, edema and inflammatory infiltrate in the lungs.
Similarly, inflammatory response in the lungs during acute pancreatitis has been classically used as a marker of systemic inflammatory response. Absence of trypsinogen activation in T−/− mice had no effect on the extent of systemic inflammation demonstrated by similar degree of neutrophil infiltration (figure 5b) and histological changes in lungs (figure 5d–e) of WT and T−/−mice.
NFkB is activated early during AP independent of trypsinogen activation
Activation of NFkB is an early event in pancreatitis paralleling trypsinogen activation (19, 48). However the relationship between these two early events has been a matter of hot debate (19, 49–51). Figure 6a shows activation of NFkB in the presence (WT) and absence (T−/−) of trypsinogen activation during in-vivo pancreatitis. To establish that NFkB activation was indeed taking place in the acinar cells, we conducted experiments using acinar cell preparation in-vitro. As demonstrated in figure 6b, caerulein supramaximal stimulation led to NFkB activation in acinar cells from WT as well as T−/− mice. The use of cathepsin B knockout mice (CB−/−) which have been previously shown to lack significant trypsinogen activation provided parallel evidence (figure 6a and b, last 2 lanes). IkBα degradation immediately follows NFkB activation and reduction in IkBα levels to similar extent in WT and T−/− mice (figure 6c) further confirms that intra-acinar NFkB activation during acute pancreatitis is independent of trypsinogen activation.
Figure 6. NFkB is activated early in parallel but independent of trypsinogen activation during acute pancreatits.

A and B: NFkB activation was assessed by EMSA. A) NFkB is activated in the pancreas of mice receiving caerulein (50μg/kg i.p.) as assessed after 30 minutes. Pictures are representative of results from 3 independent experiments. B) NFkB activation in acinar cells in-vitro. Acini from WT, T−/− and CB−/− mice were incubated with or without 100nM caerulein for 30 minutes. Pictures are representative of results from 3 independent experiments. C) IkBα degradation demonstrated in the cytosolic fraction of pancreas from mice (WT and T−/−) after 30 minutes of caerulein injection (50μg/kg i.p).
Discussion
In this study, we report generation of a novel knockout mice lacking trypsinogen isoform-7 (mouse paralog of human cationic trypsinogen (PRSS1)) and have characterized its phenotype. Using this novel and simple yet rigorous tool, we show that cationic trypsinogen is the isoform that is involved in pathologic intra-acinar activation during acute pancreatitis, and explore the role of this pathologic process. We establish that intracellular trypsinogen activation leads to acinar cell death during early pancreatitis and this process is responsible for half of eventual pancreatic injury in acute pancreatitis. The progression of local and systemic inflammation during acute pancreatitis does not require trypsinogen activation to occur. We further show that NFkB is activated in acinar cells early during pancreatitis independent of trypsinogen activation and this may be responsible for local inflammation contributing to the remainder of pancreatic injury and for the development of the widespread systemic inflammatory response. Based on these data, we propose a novel paradigm for the pathogenesis of acute pancreatitis (figure 7).
Figure 7. A simplified schematic describing the pathogenesis of acute pancreatitis.

Intra-acinar trypsinogen activation and NFkB activation are activated independently and in parallel early during pancreatitis. Trypsinogen activation contributes to acinar injury during early pancreatitis, which accounts for about half of total pancreatic damage. NFkB activation, known to be associated with several inflammatory diseases, leads to production of inflammatory mediators which elicits severe local inflammation causing further pancreatic damage (the other half of the pancreatic damage). The production of inflammatory mediators from acini as well as from incumbent inflammatory infiltrate leads to the widespread systemic inflammation during acute pancreatitis.
The findings of this study challenge the century old trypsin-central dogma of pancreatitis. Ever since the observation of Chiari (3) more than a century back, pancreatitis has been considered as an autodigestive process resulting from premature activation of digestive enzymes in the acini triggered by activation of trypsinogen. We show that while trypsinogen activation is important, it is only a partial player. It leads to acinar cell death as conceived in the trypsin-central paradigm. However, it is responsible for only half of eventual pancreatic damage thus contesting the belief that pancreatitis is merely an autodigestive phenomena. The fact that markers of local and systemic injury during pancreatitis were not affected by the absence of trypsinogen activation underscores the importance of so far under-appreciated trypsin-independent events in acute pancreatitis.
Recently, Gaiser et al (52) demonstrated that expression of active trypsin in pancreas was sufficient for induction of AP. In this study, a rat anionic trypsinogen PRSS2 construct was modified to result in continuous intra-acinar expression of moderate-low levels of active trypsin, diverging from the known pattern of transient but high level trypsin activation which was an important limitation of this model. Nevertheless, the conclusions from this model represent important advance in this field although it raises a very fundamental dilemma- whether trypsin activation is a prerequisite for AP? Our findings clearly establish that trypsinogen activation is required for early pancreatic damage during AP. However, the mechanism of trypsin induced cell death may be more complex than as yet widely believed autodigestive phenomenon and remains to be explored further.
Previous studies have shown reduction in pancreatic injury with inhibition of trypsinogen activation using cathepsin B inhibitors (18, 20), in cathepsin B knockout mice (17) or using protease inhibitors (27). Notably, significant pancreatic injury was persistent in these studies consistent with our findings (17, 18, 20). Further, levels of MPO in the pancreas and lungs remained unaffected in some of these studies (17, 18, 20). Though suggestive, these experiments lacked concrete interpretability due to lack of a direct trypsin-specific genetic model or by off-target effects of chemical inhibitors (17, 18, 20, 27). Another study observed lack of correlation between the extent of trypsinogen activation and pancreatic injury in cathepsin L knockout mice which have much higher trypsinogen activation but significantly reduced pancreatic injury (30). Based on this, an alternate hypothesis was put forward suggesting trypsinogen activation may even have a protective role during pancreatitis by degrading trypsin(ogen) and possibly other proteases thus preventing or retarding activation of intra-acinar digestive enzymes (28–31). These much debated questions in the pathogenesis of pancreatitis have been definitively answered in the present study.
It has been generally accepted that trypsinogen activation is the first step ultimately triggering the cascade of other digestive enzyme activation both in physiological conditions in the duodenum and in the pathological process within the acini (47). However, some investigators have suggested the possibility that some digestive enzymes may be activated independent of trypsinogen activation. The finding that chymotrypsinogen activation was completely abrogated in the absence of trypsinogen activation in T−/− mice (figure 1c) confirms that trypsinogen activation is indeed the key step during intra-acinar zymogen activation.
The relationship between key early events- trypsinogen activation and NFkB activation has been well-debated for over a decade (19, 48–51, 53–58). One study reported inhibition of NFkB activation in-vitro using non-specific protease inhibitors, hence concluding that trypsinogen activation is essential for NFkB activation (51). However, adenoviral mediated expression of active trypsin in acinar cells in-vitro failed to induce NF-kB activation (22). In this study, presence of early NFkB activation of comparable magnitude in the presence (WT mice) and absence (T−/− mice) of trypsinogen activation (figure 6) both in-vivo and in-vitro clearly establishes that intra-acinar NFkB activation is independent of trypsinogen activation during pancreatitis.
Intra-acinar NFkB activation has been previously shown to result in local pancreatic damage as well as systemic inflammation (53, 54). Thus NFkB activation may be the key early event responsible for progression systemic injury and the trypsin-independent component of local injury observed in T−/− mice. This is further supported by another important study which demonstrated partial reduction in acinar necrosis during pancreatitis in mice lacking p50 unit of NFkB (p50−/−) (55). Though this study lacked quantification, the observed reduction of acinar injury in p50−/− mice is directly attributable to NFkB activation. But the persistent acinar injury may be accounted for by trypsinogen activation, consistent with our findings. In contrast, NFkB-related component of injury is persistent while trypsin-dependent component of acinar injury is absent in T−/− mice. Although activation of other inflammatory cascades have been described in AP and may theoretically lead to trypsin-independent and NF-kB independent injury which need to explored in further studies, these pathways are generally known to be minor players compared to NFkB pathway.
Another important highlight of this study is the establishment of cationic trypsinogen as the trypsinogen isoform that is involved in pathologic activation during AP. The association of gain of function mutations exclusively in the cationic trypsinogen isoform (PRSS1) with hereditary pancreatitis was suggestive but no direct proof existed until now of the role of various trypsinogen isoforms in pancreatitis (24, 25, 35, 59). Sahin-toth et al (41) compared activation and degradation kinetics of cationic trypsinogen with anionic trypsinogen in calcium and pH simulating physiology (as in duodenum) as well as in pathology. In physiological conditions, both demonstrated similar behavior (41) implying that anionic trypsinogen is as functional as the cationic isoform for physiological function. This is well demonstrated by observation of intact physiology in T−/− mice further indicating anionic trypsinogen is in fact sufficient for physiological functions. This is not totally surprising given the known huge functional reserve of the exocrine pancreas.
In contrast, the behavior of major trypsinogens is very different in pathologic conditions. In conditions simulating colocalization compartments, activation of anionic trypsinogen was considerably inhibited relative to cationic trypsinogen while the autocatalytic degradation of anionic trypsinogen and trypsin derived from it was 10–20 folds higher (41) indicating that cationic trypsinogen and its trypsin are important players in pathological conditions. In fact, upregulation of anionic trypsinogen reported to occur in pancreatic diseases (45, 46, 60) could be a protective response to limit enzyme activation (41) although upregulation of anionic trypsinogen has also been cited to argue otherwise (52). Using knockout approach, we report that the absence of cationic trypsinogen abrogates pathologic trypsinogen activation, thus clearly establishing that cationic trypsinogen is the isoform that is involved in pathologic intra-acinar enzyme activation during pancreatitis. The slower activation kinetics of anionic trypsinogen and much faster degradation of its trypsin in pathologic conditions also explain lack of pathologic trypsinogen activation in T−/− mice despite presence of non-cationic trypsinogens. Other alternate possibilities could be prompt inactivation by trypsin inhibitors (in fact, we found that SPINK-3 levels were not altered in T−/− mice, data not shown) and potential differences in affinity for cathepsin B mediated activation. These biochemical aspects need to be explored in future studies.
In conclusion, we have established that cationic trypsinogen is the isoform involved in pathologic activation and this leads to acinar cell death during early pancreatitis. Progression of inflammation in acute pancreatitis is independent of trypsinogen activation. NFkB activation is a key early event independent of trypsinogen activation and may be responsible for progression of local and systemic inflammation. These findings redefine our current understanding of the pathogenesis of pancreatitis.
Supplementary Material
Acknowledgments
Grant support: R01 DK 058694, R01 DK 092145 and R01 DK 093047 (to A.K.S)
Abbreviations
- AP
Acute pancreatitis
- T7
Trypsinogen isoform-7
- T−/−
trypsinogen isoform-7 knock out mice
- PRSS1
Protease Serine 1
- WT
wild type
- NFkB
Nuclear Factor kappa B
Footnotes
Conflict of interest: None to disclose.
References
- 1.Banks PA, Freeman ML. Practice guidelines in acute pancreatitis. Am J Gastroenterol. 2006;101(10):2379–400. doi: 10.1111/j.1572-0241.2006.00856.x. [DOI] [PubMed] [Google Scholar]
- 2.Fagenholz PJ, Fernandez-del Castillo C, Harris NS, et al. Direct medical costs of acute pancreatitis hospitalizations in the United States. Pancreas. 2007;35(4):302–7. doi: 10.1097/MPA.0b013e3180cac24b. [DOI] [PubMed] [Google Scholar]
- 3.Chiari H. ÜberdieSelbstverdauung des menschlichenPankreas. ZeitschriftfürHeilkunde. 1896;17:69–96. [Google Scholar]
- 4.Hofbauer B, Saluja AK, Lerch MM, et al. Intra-acinar cell activation of trypsinogen during caerulein-induced pancreatitis in rats. Am J Physiol. 1998;275(2 Pt 1):G352–62. doi: 10.1152/ajpgi.1998.275.2.G352. [DOI] [PubMed] [Google Scholar]
- 5.Saluja AK, Bhagat L, Lee HS, et al. Secretagogue-induced digestive enzyme activation and cell injury in rat pancreatic acini. Am J Physiol. 1999;276(4 Pt 1):G835–42. doi: 10.1152/ajpgi.1999.276.4.G835. [DOI] [PubMed] [Google Scholar]
- 6.Saluja A, Saluja M, Villa A, et al. Pancreatic duct obstruction in rabbits causes digestive zymogen and lysosomal enzyme colocalization. J Clin Invest. 1989;84(4):1260–6. doi: 10.1172/JCI114293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Saluja AK, Donovan EA, Yamanaka K, et al. Cerulein-induced in vitro activation of trypsinogen in rat pancreatic acini is mediated by cathepsin B. Gastroenterology. 1997;113(1):304–10. doi: 10.1016/s0016-5085(97)70108-2. [DOI] [PubMed] [Google Scholar]
- 8.Saluja AK, Lerch MM, Phillips PA, et al. Why does pancreatic overstimulation cause pancreatitis? Annu Rev Physiol. 2007;69:249–69. doi: 10.1146/annurev.physiol.69.031905.161253. [DOI] [PubMed] [Google Scholar]
- 9.Lerch MM, Gorelick FS. Early trypsinogen activation in acute pancreatitis. Med Clin North Am. 2000;84(3):549–63. viii. doi: 10.1016/s0025-7125(05)70239-x. [DOI] [PubMed] [Google Scholar]
- 10.Grady T, Mah’Moud M, Otani T, et al. Zymogen proteolysis within the pancreatic acinar cell is associated with cellular injury. Am J Physiol. 1998;275(5 Pt 1):G1010–7. doi: 10.1152/ajpgi.1998.275.5.G1010. [DOI] [PubMed] [Google Scholar]
- 11.Kloppel G, Dreyer T, Willemer S, et al. Human acute pancreatitis: its pathogenesis in the light of immunocytochemical and ultrastructural findings in acinar cells. Virchows Arch A Pathol Anat Histopathol. 1986;409(6):791–803. doi: 10.1007/BF00710764. [DOI] [PubMed] [Google Scholar]
- 12.Willemer S, Kloppel G, Kern HF, et al. Immunocytochemical and morphometric analysis of acinar zymogen granules in human acute pancreatitis. Virchows Arch A Pathol Anat Histopathol. 1989;415(2):115–23. doi: 10.1007/BF00784348. [DOI] [PubMed] [Google Scholar]
- 13.Gorelick FS, Thrower E. The acinar cell and early pancreatitis responses. Clin Gastroenterol Hepatol. 2009;7(11 Suppl):S10–4. doi: 10.1016/j.cgh.2009.07.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Singh VP, Saluja AK, Bhagat L, et al. Phosphatidylinositol 3-kinase-dependent activation of trypsinogen modulates the severity of acute pancreatitis. J Clin Invest. 2001;108(9):1387–95. doi: 10.1172/JCI12874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Watanabe O, Baccino FM, Steer ML, et al. Supramaximal caerulein stimulation and ultrastructure of rat pancreatic acinar cell: early morphological changes during development of experimental pancreatitis. Am J Physiol. 1984;246(4 Pt 1):G457–67. doi: 10.1152/ajpgi.1984.246.4.G457. [DOI] [PubMed] [Google Scholar]
- 16.Lerch MM, Saluja AK, Dawra R, et al. The effect of chloroquine administration on two experimental models of acute pancreatitis. Gastroenterology. 1993;104(6):1768–79. doi: 10.1016/0016-5085(93)90658-y. [DOI] [PubMed] [Google Scholar]
- 17.Halangk W, Lerch MM, Brandt-Nedelev B, et al. Role of cathepsin B in intracellular trypsinogen activation and the onset of acute pancreatitis. J Clin Invest. 2000;106(6):773–81. doi: 10.1172/JCI9411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Van Acker GJ, Saluja AK, Bhagat L, et al. Cathepsin B inhibition prevents trypsinogen activation and reduces pancreatitis severity. Am J Physiol Gastrointest Liver Physiol. 2002;283(3):G794–800. doi: 10.1152/ajpgi.00363.2001. [DOI] [PubMed] [Google Scholar]
- 19.Hietaranta AJ, Saluja AK, Bhagat L, et al. Relationship between NF-kappaB and trypsinogen activation in rat pancreas after supramaximal caerulein stimulation. Biochem Biophys Res Commun. 2001;280(1):388–95. doi: 10.1006/bbrc.2000.4120. [DOI] [PubMed] [Google Scholar]
- 20.Van Acker GJ, Weiss E, Steer ML, et al. Cause-effect relationships between zymogen activation and other early events in secretagogue-induced acute pancreatitis. Am J Physiol Gastrointest Liver Physiol. 2007;292(6):G1738–46. doi: 10.1152/ajpgi.00543.2006. [DOI] [PubMed] [Google Scholar]
- 21.Kereszturi E, Sahin-Toth M. Intracellular autoactivation of human cationic trypsinogen mutants causes reduced trypsinogen secretion and acinar cell death. J Biol Chem. 2009;284(48):33392–9. doi: 10.1074/jbc.M109.056812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ji B, Gaiser S, Chen X, et al. Intracellular trypsin induces pancreatic acinar cell death but not NF-kappaB activation. J Biol Chem. 2009;284(26):17488–98. doi: 10.1074/jbc.M109.005520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gaiser S, Ahler A, Gundling F, et al. Expression of mutated cationic trypsinogen reduces cellular viability in AR4–2J cells. Biochem Biophys Res Commun. 2005;334(2):721–8. doi: 10.1016/j.bbrc.2005.06.148. [DOI] [PubMed] [Google Scholar]
- 24.Whitcomb DC. Genetic aspects of pancreatitis. Annu Rev Med. 2010;61:413–24. doi: 10.1146/annurev.med.041608.121416. [DOI] [PubMed] [Google Scholar]
- 25.Teich N, Rosendahl J, Toth M, et al. Mutations of human cationic trypsinogen (PRSS1) and chronic pancreatitis. Hum Mutat. 2006;27(8):721–30. doi: 10.1002/humu.20343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Singh VP, Chari ST. Protease inhibitors in acute pancreatitis: lessons from the bench and failed clinical trials. Gastroenterology. 2005;128(7):2172–4. doi: 10.1053/j.gastro.2005.03.087. [DOI] [PubMed] [Google Scholar]
- 27.Singh VP, Bren GD, Algeciras-Schimnich A, et al. Nelfinavir/ritonavir reduces acinar injury but not inflammation during mouse caerulein pancreatitis. Am J Physiol Gastrointest Liver Physiol. 2009;296(5):G1040–6. doi: 10.1152/ajpgi.90642.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Simon P, Weiss FU, Sahin-Toth M, et al. Hereditary pancreatitis caused by a novel PRSS1 mutation (Arg-122 --> Cys) that alters autoactivation and autodegradation of cationic trypsinogen. J Biol Chem. 2002;277(7):5404–10. doi: 10.1074/jbc.M108073200. [DOI] [PubMed] [Google Scholar]
- 29.Kukor Z, Mayerle J, Kruger B, et al. Presence of cathepsin B in the human pancreatic secretory pathway and its role in trypsinogen activation during hereditary pancreatitis. J Biol Chem. 2002;277(24):21389–96. doi: 10.1074/jbc.M200878200. [DOI] [PubMed] [Google Scholar]
- 30.Wartmann T, Mayerle J, Kahne T, et al. Cathepsin L inactivates human trypsinogen, whereas cathepsin L-deletion reduces the severity of pancreatitis in mice. Gastroenterology. 138(2):726–37. doi: 10.1053/j.gastro.2009.10.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Halangk W, Kruger B, Ruthenburger M, et al. Trypsin activity is not involved in premature, intrapancreatic trypsinogen activation. Am J Physiol Gastrointest Liver Physiol. 2002;282(2):G367–74. doi: 10.1152/ajpgi.00315.2001. [DOI] [PubMed] [Google Scholar]
- 32.Li W, Nakagawa T, Koyama N, et al. Down-regulation of trypsinogen expression is associated with growth retardation in alpha1,6-fucosyltransferase-deficient mice: attenuation of proteinase-activated receptor 2 activity. Glycobiology. 2006;16(10):1007–19. doi: 10.1093/glycob/cwl023. [DOI] [PubMed] [Google Scholar]
- 33.Roach JC, Wang K, Gan L, et al. The molecular evolution of the vertebrate trypsinogens. J Mol Evol. 1997;45(6):640–52. doi: 10.1007/pl00006268. [DOI] [PubMed] [Google Scholar]
- 34.Ozsvari B, Hegyi P, Sahin-Toth M. The guinea pig pancreas secretes a single trypsinogen isoform, which is defective in autoactivation. Pancreas. 2008;37(2):182–8. doi: 10.1097/MPA.0b013e3181663066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen JM, Ferec C. Chronic pancreatitis: genetics and pathogenesis. Annu Rev Genomics Hum Genet. 2009;10:63–87. doi: 10.1146/annurev-genom-082908-150009. [DOI] [PubMed] [Google Scholar]
- 36.Watanabe T, Ogasawara N. Purification and properties of multiple forms of mouse trypsinogen. Biochim Biophys Acta. 1982;717(3):439–44. doi: 10.1016/0304-4165(82)90285-9. [DOI] [PubMed] [Google Scholar]
- 37.Watanabe T. Purification and properties of genetic variants of mouse trypsinogen. Biochem Genet. 1983;21(7–8):761–72. doi: 10.1007/BF00498922. [DOI] [PubMed] [Google Scholar]
- 38.Isobe M, Ogita Z. Electrophoretic analysis of pancreatic proteases and zymogen-activating factors in the mouse. J Exp Zool. 1984;230(3):347–54. doi: 10.1002/jez.1402300303. [DOI] [PubMed] [Google Scholar]
- 39.Canbay A, Guicciardi ME, Higuchi H, et al. Cathepsin B inactivation attenuates hepatic injury and fibrosis during cholestasis. J Clin Invest. 2003;112(2):152–9. doi: 10.1172/JCI17740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kukor Z, Toth M, Pal G, et al. Human cationic trypsinogen. Arg(117) is the reactive site of an inhibitory surface loop that controls spontaneous zymogen activation. J Biol Chem. 2002;277(8):6111–7. doi: 10.1074/jbc.M110959200. [DOI] [PubMed] [Google Scholar]
- 41.Kukor Z, Toth M, Sahin-Toth M. Human anionic trypsinogen: properties of autocatalytic activation and degradation and implications in pancreatic diseases. Eur J Biochem. 2003;270(9):2047–58. doi: 10.1046/j.1432-1033.2003.03581.x. [DOI] [PubMed] [Google Scholar]
- 42.Koshikawa N, Yasumitsu H, Nagashima Y, et al. Identification of one- and two-chain forms of trypsinogen 1 produced by a human gastric adenocarcinoma cell line. Biochem J. 1994;303 ( Pt 1):187–90. doi: 10.1042/bj3030187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen JM, Ferec C. Genes, cloned cDNAs, and proteins of human trypsinogens and pancreatitis-associated cationic trypsinogen mutations. Pancreas. 2000;21(1):57–62. doi: 10.1097/00006676-200007000-00052. [DOI] [PubMed] [Google Scholar]
- 44.Guy O, Lombardo D, Bartelt DC, et al. Two human trypsinogens. Purification, molecular properties, and N-terminal sequences. Biochemistry. 1978;17(9):1669–75. doi: 10.1021/bi00602a014. [DOI] [PubMed] [Google Scholar]
- 45.Rinderknecht H, Renner IG, Carmack C. Trypsinogen variants in pancreatic juice of healthy volunteers, chronic alcoholics, and patients with pancreatitis and cancer of the pancreas. Gut. 1979;20(10):886–91. doi: 10.1136/gut.20.10.886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rinderknecht H, Stace NH, Renner IG. Effects of chronic alcohol abuse on exocrine pancreatic secretion in man. Dig Dis Sci. 1985;30(1):65–71. doi: 10.1007/BF01318373. [DOI] [PubMed] [Google Scholar]
- 47.Barrett KE, Barman SM, Boitano S, et al. Barrett KE, Barman SM, Boitano S, Brooks H, editors. Protein Digestion. Ganong’s Review of Medical Physiology. (23) 2010 http://www.accessmedicine.com/content.aspx?aID=5242356.
- 48.Gukovsky I, Gukovskaya AS, Blinman TA, et al. Early NF-kappaB activation is associated with hormone-induced pancreatitis. Am J Physiol. 1998;275(6 Pt 1):G1402–14. doi: 10.1152/ajpgi.1998.275.6.G1402. [DOI] [PubMed] [Google Scholar]
- 49.Rakonczay Z, Jr, Hegyi P, Takacs T, et al. The role of NF-kappaB activation in the pathogenesis of acute pancreatitis. Gut. 2008;57(2):259–67. doi: 10.1136/gut.2007.124115. [DOI] [PubMed] [Google Scholar]
- 50.Han B, Ji B, Logsdon CD. CCK independently activates intracellular trypsinogen and NF-kappaB in rat pancreatic acinar cells. Am J Physiol Cell Physiol. 2001;280(3):C465–72. doi: 10.1152/ajpcell.2001.280.3.C465. [DOI] [PubMed] [Google Scholar]
- 51.Tando Y, Algul H, Schneider G, et al. Induction of IkappaB-kinase by cholecystokinin is mediated by trypsinogen activation in rat pancreatic lobules. Digestion. 2002;66(4):237–45. doi: 10.1159/000068364. [DOI] [PubMed] [Google Scholar]
- 52.Gaiser S, Daniluk J, Liu Y, et al. Intracellular activation of trypsinogen in transgenic mice induces acute but not chronic pancreatitis. Gut. 2011 doi: 10.1136/gut.2010. gut.2010.226175 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen X, Ji B, Han B, et al. NF-kappaB activation in pancreas induces pancreatic and systemic inflammatory response. Gastroenterology. 2002;122(2):448–57. doi: 10.1053/gast.2002.31060. [DOI] [PubMed] [Google Scholar]
- 54.Baumann B, Wagner M, Aleksic T, et al. Constitutive IKK2 activation in acinar cells is sufficient to induce pancreatitis in vivo. J Clin Invest. 2007;117(6):1502–13. doi: 10.1172/JCI30876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Altavilla D, Famulari C, Passaniti M, et al. Attenuated cerulein-induced pancreatitis in nuclear factor-kappaB-deficient mice. Lab Invest. 2003;83(12):1723–32. doi: 10.1097/01.lab.0000101734.82054.be. [DOI] [PubMed] [Google Scholar]
- 56.Tando Y, Algul H, Wagner M, et al. Caerulein-induced NF-kappaB/Rel activation requires both Ca2+ and protein kinase C as messengers. Am J Physiol. 1999;277(3 Pt 1):G678–86. doi: 10.1152/ajpgi.1999.277.3.G678. [DOI] [PubMed] [Google Scholar]
- 57.Algul H, Treiber M, Lesina M, et al. Pancreas-specific RelA/p65 truncation increases susceptibility of acini to inflammation-associated cell death following cerulein pancreatitis. J Clin Invest. 2007;117(6):1490–501. doi: 10.1172/JCI29882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Aleksic T, Baumann B, Wagner M, et al. Cellular immune reaction in the pancreas is induced by constitutively active IkappaB kinase-2. Gut. 2007;56(2):227–36. doi: 10.1136/gut.2005.084665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Witt H, Sahin-Toth M, Landt O, et al. A degradation-sensitive anionic trypsinogen (PRSS2) variant protects against chronic pancreatitis. Nat Genet. 2006;38(6):668–73. doi: 10.1038/ng1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Itkonen O. Human trypsinogens in the pancreas and in cancer. Scand J Clin Lab Invest. 70(2):136–43. doi: 10.3109/00365511003615317. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.