

Abstract
Emerging viral diseases pose a unique risk to public health, and thus there is a need to develop therapies. A current focus of funding agencies, and hence research, is the development of broad-spectrum antivirals, and in particular, those targeting common cellular pathways. The scope of this article is to review screening strategies and recent advances in this area, with a particular emphasis on antivirals targeting the step of viral entry for emerging lipid-enveloped viruses such as Ebola virus and SARS-coronavirus.
Keywords: antiviral therapies, broad-spectrum viral entry inhibitors, emerging viruses, high-throughput screening, viral entry
Since the emergence of HIV in the early 1980s, it has been widely assumed that there would be further pandemics of new and dangerous viral pathogens. The SARS-coronavirus (SARS-CoV) and the Henipavirus genus of paramyxoviruses are two examples of such agents that have arisen in the past 20 years. Fortunately, these and other emerging agents such as Ebola virus (EBOV) and Marburg virus (MARV) have failed to demonstrate the transmissibility or animal reservoirs required to become true pandemic threats. However, changes in the virus through mutation or bioweaponization, exposure to new reservoirs through human encroachment or alteration of human susceptibility through coinfections could rapidly change that status. While the direct human and economic cost of these agents has been significant, probably their greatest impact has been in creating public fear. This was most dramatically demonstrated in the SARS outbreak at the beginning of the 21st century. It is also reflected in the governmental responses to real or imagined biowarfare threats. Rapid responses to new or changing pandemic threats would be greatly helped by the presence of an arsenal of antiviral drugs, ideally with overlapping therapeutic indications that at least offered the potential for treatment of newly arising agents. Few, if any, antivirals can claim to be broad-spectrum, with interferon and ribavirin being the closest to that definition [1]; however, they have demonstrated marginal, if any, effectiveness against the majority of emerging pathogens [2–6]. A number of novel broad-spectrum antiviral [7–9] or virucidal [10] compounds and strategies have been suggested recently, but have yet to be proven. Thus, new drugs, both broad-spectrum and pathogen-specific, are required to combat the threat posed by emerging viruses. In this review, the authors discuss a number of potential avenues of research for meeting these challenges, as well as highlighting some exciting new inhibitors of the various stages of entry for several emerging viruses.
Inhibitors of viral entry
The viral life cycle presents numerous potential targets for antiviral intervention, with viral enzymes such as polymerases and proteases representing the canonical targets for therapeutics. While once neglected, viral entry pathways also represent promising emerging targets for antivirals. As with many antivirals, HIV has been the testing ground for a number of these concepts. A handful of new drugs, both on the market and undergoing trials, target different steps of viral entry [11]. Targets include attachment, coreceptor binding and virus–cell fusion [12]. In addition, entry inhibitors could play a particularly exciting role as topical microbicides to prevent virus transmission between individuals, for example, in sexual transmission [13]. Inhibitors of viral entry are likely to provide a rational basis for antivirals, particularly ones that can be used in prophylactic situations as they block at the very first step of viral replication and have the potential to be less toxic because membrane permeability may not be necessary, although in practice such properties are also probably required for effective oral availability of a drug. However, the predominant reason that entry inhibitors have begun to appear for emerging viruses may be due to the fact that many of these pathogens require a high level of biocontainment. Thus, surrogate assays have been developed for performing high-throughput screening (HTS) that include the use of pseudotyping [14,15] and cell–cell fusion that focus on the entry process and require a lower level of containment. Viral entry is often a multiple-step process that involves complicated viral–host interactions that can serve as targets for inhibitors. Targets include the use of common cellular components, such as lipids and lipid rafts, or cellular functionalities, such as endocytosis. We will review the steps of entry and how each step may be a target for inhibitors (Table 1), with particular reference to the potential for broad-spectrum inhibitors capable of blocking members of multiple virus families.
Table 1.
Summary of studies on antivirals targeting viral entry.
| Entry steps | Viruses | Inhibitors | Targeting | Ref. |
|---|---|---|---|---|
| Attachment | EBOV and HIV | Cyanovirin-N | Carbohydrate molecules on the GP | [43] |
| Multiple viruses | Heparin and carrageenan | Heparin sulfate | [38] | |
| Endocytosis | Multiple pH-dependent viruses | Chlorpromazine, sucrose and bafilomycin A1 | Clathrin-mediated endocytosis | [56] |
| Macropinocytosis | EBOV and NiV | Pak1, CtBP/BARS, EIPA, cytochalasin D, PKC inhibitor and siRNAs | Macropinocytosis | [59,60,64] |
| Signaling | EBOV | PI3K inhibitors LY294002 and wortmannin, Akt inhibitor and Rac1 inhibitor | PI3K-Akt/Rac1 signaling pathway | [60,62] |
| Activation and fusion | Multiple class I viruses | Fusion peptides derived from the heptad repeats | Membrane fusion | [91,92] |
| Filo- and corona-viruses | Cathepsin B inhibitors and cathepsin L inhibitors | Endosomal cysteine proteases cathepsin B/L | [65,68,69,81] | |
| EBOV | Benzylpiperazine adamantane diamide derivatives | Targeting endosomal membrane protein NPC1 by interfering with binding of cathepsin-cleaved GP to NPC1 | [26] | |
| Multiple-enveloped viruses | Aryl methyldiene rhodanine derivative, LJ001 | Intercalates into viral lipid membranes and inhibits membrane flexibility | [105] |
EBOV: Ebola virus; GP: Glycoprotein; NiV: Nipah virus; NPC1: Niemann-Pick C1.
High-throughput screening assays for entry inhibitors
The development of HTS assays to specifically search for inhibitors of viral entry has been the biggest factor in the recent development of candidate antivirals targeting entry of emerging viruses. The glycoproteins from emerging viruses have been used to pseudotype onto retroviral, lentiviral or rhabdoviral pseudotyping systems [16–18]. The assays incorporate the envelope glycoproteins of the test virus onto the core of a reporter virus such as HIV, in such a manner that infection is directed in the same way as for the authentic virus, but typically only a single round of infection occurs and infection can be detected using an enzymatic or fluorescent readout. If multiple rounds of infection are desired, in order to fully test the efficacy with which inhibitors block entry, target cells expressing the relevant viral envelope can be used with rhabdovirus vectors to allow multiple rounds of infection [15]. These methods can be relatively easily adapted to HTS in order to screen for inhibitors of only the steps of viral entry mediated by the viral glycoprotein [15,19]. This pseudotyping strategy is beginning to pay dividends for many emerging viruses in terms of identifying inhibitors of viral entry. For example, this method was used to identify EI-1, a small-molecule inhibitor of HCV entry that blocks somewhere between attachment and the requirement for endosomal acidification [20].
The addition of a second (and potentially a third) pseudotype with a different reporter system into the screen adds an extra dimension to this standard HTS [14]. This allows two or more different envelopes to be screened at one time in an assay termed the dual envelope pseudovirion assay. Not only does this save costs in terms of both labor and materials, but it also affords the possibility for each pseudotype to act as an internal control for the other if specific inhibitors are the goal. Alternately, if broad-spectrum inhibitors are being sought then deliberately divergent envelopes and pseudotype systems can be chosen in order to identify inhibitors of multiple systems.
As an alternative to pseudotyping, cell–cell fusion assays driven by surface expression of the glycoprotein [21] or specific fluorescent or enzymatic entry assays based on virus-like particles [22] can be used to potentially reduce nonspecific hits directed against postentry steps of the pseudotype infection cycle.
Viral attachment
Entry begins with relatively nonspecific interactions between the virus and attachment factors on the cell surface, usually followed by engagement of more specific cellular receptors. However, attachment factors may possibly be sufficient to direct some viruses, such as the flavi- and alpha-viruses that have a strong requirement for acidic pH triggering of their envelope proteins [23,24], to endosomes without engagement of more specific receptors. Similarly, for EBOV, the endosome, rather than the cell surface, appears to be the site of specific receptor engagement [25–27]. Thus, it may be that relatively nonspecific attachment factors (that unlike receptors do not trigger changes in the viral glycoproteins) are the main method of targeting for some viruses. For example, attachment factors, such as calcium-dependent lectins, may help direct many viruses to important target organs and cell types such as the liver, lungs and monocyte/macrophages via engagement of carbohydrate moieties on viral glycoproteins [28–35]. The authors have also recently demonstrated that like many other viruses [36,37], EBOV relies on interactions with the glycosaminoglycan chains of proteoglycans for efficient initial contacts with cells [Salvador B & Simmons G, Unpublished data].
Inhibitors of viral attachment
Interference of viral interactions with attachment factors would probably disrupt efficient infection, and thus this step of entry represents a good target for inhibition. However, many of these interactions are carbohydrate- (lectins) or charge-based (glycosaminoglycans [GAGs]); therefore, inhibitors may be relatively non-specific and thus run the risk of harmful off-target interactions. A number of molecules able to inhibit interactions through GAGs have been identified, including using soluble polysaccharides such as heparin or the potential microbicide carrageenan [38]. Small-molecule inhibitors of GAG interactions such as surfen, as well as several peptide-based inhibitors, have also been identified [39–41]. While these antivirals are predominantly being suggested as topical microbicides, they could act as a model for broad-spectrum antivirals against emerging viruses.
Cyanovirin-N (CV-N), a naturally occurring lectin, inhibits attachment of HIV by binding to N-linked high-mannose oligosaccharides on the viral glycoprotein and is currently in preclinical development as a topical microbicide [42]. Recently, CV-N was found to bind to the EBOV viral surface glycoprotein (GP) and inhibits infectivity in both in vitro binding assays and mouse models [43]. A likely mode of action involves simply binding to the same high-mannose moieties as cell surface lectins and blocking viral interactions with cells by steric hindrance [44]. Indeed, CV-N, and other soluble lectins such as griffithsin [45], are active against several emerging viruses [46–48] including in vivo efficacy against influenza [49].
Receptor engagement
For many emerging viruses, the specific receptors are either unknown, or suggested receptors are still controversial. However, there are notable exceptions. For example, angiotensin-converting enzyme 2 (ACE2) is the functional receptor for SARS-CoV [50], while ephrinB2 acts as the cellular receptor for Nipah virus (NiV) [51]. Recently, T-cell immunoglobulin and mucin domain (TIM)-1 has been shown to function as a receptor for EBOV on mucosal epithelia [52]. It is not yet clear whether TIM-1 functions as a true receptor or an attachment factor, nor whether it is likely to play a major role in vivo due to its restricted expression. However, as a possible target for antivirals, it may protect important cellular targets and reduce pathogenesis. A more universal receptor for filoviruses appears to be NPC1, a cholesterol transporter present in late endosomal membranes [25–27]. This unusual circumstance is the first recognized incidence of a specific receptor interaction occurring within the cell, rather than at the plasma membrane.
Inhibitors of receptor engagement
Antagonizing virus/receptor interactions is an obvious antiviral strategy, and has been validated by maraviroc, a CCR5 receptor antagonist that has proved to be a successful HIV inhibitor [11]. Pockets or grooves on viral glycoproteins required for receptor recognition may be ideal for small molecules to specifically bind, although it may be difficult for small molecules to inhibit large proteins coming together. Thus, peptides, antibodies or other soluble protein therapeutics may provide a more likely route of direct receptor binding antagonism. However, several small molecules capable of inhibiting such interactions have been suggested. For example, emodin is a compound identified from Chinese medicinal herbs in screens against SARS-CoV, which demonstrated the ability to inhibit the SARS-CoV S protein from binding to ACE2 protein, as well as S-protein-mediated pseudovirus infection in Vero E6 cells [53]. However, caution is required in terms of attributing the mechanism until thorough studies have been completed. Initially reported to inhibit receptor interactions, the HIV inhibitor BMS-378806 [54] may actually act through an indirect mechanism by binding to the viral glycoprotein and preventing the subsequent conformational changes required to both efficiently bind to the receptor and mediate membrane fusion [55].
Although the CCR5 receptor antagonists are an obvious exception, it would be less ideal to target the receptor itself for antagonism due to possible effects on the natural function of the cellular protein. However, at least one molecule, a benzylpiperazine adamantane diamide-derived compound, has been identified that targets NPC1 and can directly inhibit EBOV interactions with this receptor and hence infection [26]. Assuming targeting NPC1 does not result in excessive cellular toxicity, this may thus represent an exciting prospect for EBOV treatment.
Endocytosis & macropinocytosis
For the majority of viruses, following initial attachment of virus to the cell surface, whether through attachment factors or more specific receptor engagement, specific signaling and trafficking events occur to deliver the virus to late endosomes. Typical routes of viral internalization include clathrin-mediated endocytosis, caveolar (lipid raft)-mediated endocytosis and macropinocytosis [56]. Often multiple mechanisms have been suggested for a single virus. For example, although EBOV has been reported to enter through clathrin-mediated endocytosis [57] or via caveoli [58], entry of filamentous particles into important cell types such as macrophages is now thought to predominantly occur through macropinocytosis [59,60], possibly via an atypical dynamin-2-dependent macropinocytic pathway [61].
Inhibitors of internalization
Targeting internalization has pros and cons as an antiviral strategy. It is likely that targeting such an important cellular function will have toxic side effects. However, it is possible that because multiple pathways exist in the cell, specific mechanisms can be blocked for short treatment windows with minimal effects. This would allow treatment of acute infections such as hemorrhagic fevers where death is the likely alternative. The upside to targeting such a cellular function is the possibility of producing a broad-spectrum antiviral capable of inhibiting a large number of emerging viruses dependent on the same mechanism for entry into cells. Indeed, panels of compounds, such as chlorpromazine, are available for inhibiting various pathways of internalization, and they function efficiently to block infection [62,63]. For example, studies of macropinocytosis using EBOV, as well as morphologically similar virus-like particles showed that the inhibitors of macropinocytosis Pak1 and CtBP/BARS, actin-depolymerizing agent cytochalasin D, Na+/H+ ion exchange inhibitor EIPA and the PKC inhibitor staurosporine all reduced infection or entry [59,60]. Many of the compounds have also been shown to target NiV entry [64], despite the virus being largely pH-independent, suggesting the possibility that such a drug target may be broad-spectrum for at least a subset of emerging viruses. However, such compounds, in their current formulations, are unlikely to be useful antivirals, even though some already have other clinical uses. For example, the concentrations of drugs such as chloroquine or chlorpromazine required to have any clinical antiviral benefit against pH-dependent viruses, or other viruses requiring internalization, is likely significantly higher than the plasma levels at which major side effects would be noted. However, as examples for highlighting the potential for targeting particular pathways for antiviral treatments, they should not be overlooked.
A somewhat more specific target for intervention may be the signaling events required in order to trigger internalization and trafficking to the correct cellular destination. The phosphoinositide-3 kinase (PI3K) cell signaling pathway plays a very important regulatory role for multiple cellular activities including cell growth, migration, survival and vesicular trafficking. Recently, this pathway has been implicated in regulating cellular entry for EBOV virus [60,62]. Inhibitors of PI3K (LY294002 and wortmannin) block EBOV viral entry in a dose-dependent manner. The results again suggest that such pathways could be a target for drug intervention, if toxicity issues can be managed. This is a particularly good avenue of research given that such PI3K inhibitors are already being pursued, given the role this pathway plays in tumor growth.
Glycoprotein activation
Once in late endosomes or lysosomes, EBOV GP is sequentially proteolytically cleaved by the acid-dependent cysteine proteases cathepsin B and, to a lesser extent, L (CatB and CatL, respectively), to remove the heavily glycosylated N-terminus of GP and expose the receptor binding domains [2,65–68]. These cleavage events probably destabilize the prefusion complex of GP, allowing the sequential conformational changes required in GP to occur and hence membrane fusion to proceed [2,65]. EBOV (Zaire ebolavirus) together with the related species Tai Forest ebolavirus and Bundibugyo ebolavirus, are strongly dependent on CatB. Conversely, the Sudan and Reston species, as well as MARV, are not dependent on CatB, but rather have a requirement for as yet unidentified proteases [68]. At least four coronaviruses, including SARS-CoV, share similar entry pathways with EBOV, although for these viruses it is CatL that is predominantly required for entry into cell lines [69–72]. In the case of SARS-CoV, several soluble and membrane-bound proteases can substitute for CatL, particularly in airway cells [73–77]. CatL also plays an important role in processing the envelope glycoproteins of NiV and Hendra viruses. In this instance, proteolysis occurs during recycling of the proteins from the cell surface in the infected producer cells, rather than during viral entry [78–80].
Inhibition of glycoprotein activation
For filo-, henipa- and, to a lesser extent, coronaviruses, general inhibitors of cysteine proteases, or more specific CatB or CatL inhibitors, have proven to be highly effective at blocking infection in vitro due to the viruses’ dependence on these proteases for glycoprotein activation. For example, one of our previous studies identified MDL28170 (also known as calpain inhibitor III, or Z-Val-Phe-CHO) as an efficient inhibitor of CatL and SARS-CoV entry inhibitor in cell lines [69], and another study identified a small-molecule oxocarbazate (CID23631927) as a CatL inhibitor that blocked SARS-CoV and EBOV pseudovirus entry in human cells [81]. The fact that this inhibitor efficiently blocks EBOV, despite being over 700-fold more potent against CatL compared to CatB in vitro, highlights a common theme that many of these inhibitors are less specific in cellular assays. This may actually be advantageous, given the width of protease specificities demonstrated by EBOV [68], as CatB and CatL inhibitors can still inhibit all species of EBOV, but not MARV [82]. As with all inhibitors, escape mutants can be generated, and in the case of CatB-inhibitor resistant EBOV envelopes, the virus is no longer dependent on CatB or CatL, but still requires an unidentified endosomal protease activity, possibly the same activity utilized by MARV [2].
The fact that cathepsin inhibitors appear to lack specificity also opens up the possibility that because these compounds are protease inhibitors, some may act as dual-action antivirals and are able to target both viral entry and viral maturation. Indeed, the authors have identified a class of dipeptidyl epoxyketone molecules (Table 2) able to both target coronavirus entry [14] and inhibit the SARS-CoV-encoded protease, 3Clpro[83].
Table 2.
WRR182, WRR183 and derivatives.
| Compounds | Structure | IC50 (nM)† | IC90 (nM)‡ |
|---|---|---|---|
| WRR182 | 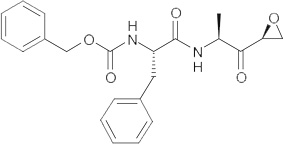 |
0.09 ± 0.01 | 2.97 ± 1.12 |
| WRR183 | 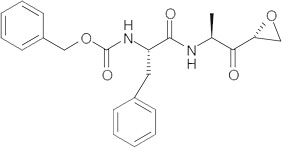 |
32.84 ± 3.52 | 715.07 ± 1.34 |
| WRR495 | 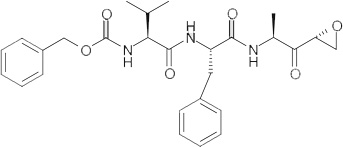 |
3.36 ± 1.06 | 54.31 ± 1.99 |
| CA-074§ | 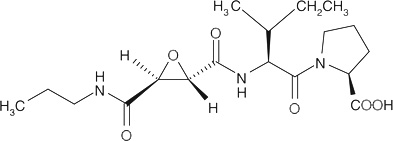 |
>1000 | >1000 |
| Z-Phe-Phe-FMK§ | 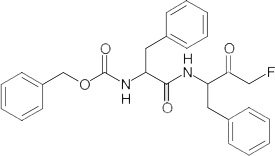 |
4.47 ± 1.05 | 156.22 ± 4.20 |
Assays were performed in triplicate and the values are representative of three or more independent experiments.
IC50 and
IC90 concentration of the compound that produced 50 and 90% decrease in SARS-coronavirus pseudotyped virus infection in 293T-ACE2 cells, respectively.
CA-074 is a commercially available CatB inhibitor and Z-Phe-Phe-FMK is a commercially available CatL inhibitor.
Glycoprotein conformational changes & membrane fusion
Probably the best target for intervention in the multistep process of viral entry for enveloped viruses is the step of activation and triggering of the viral glycoprotein, required to mediate membrane fusion. Regardless of the viral system, the viral glycoprotein has to undergo significant conformational rearrangements in order to drive the process of fusing the viral and cellular membranes. This leaves it open to inhibition at several stages, both by small molecules and peptide and antibody-based therapies. Receptor engagement or the low pH environment of late endosomes/lysosomes are the predominant triggers of conformational changes in viral glycoproteins [84–87].
Viral glycoproteins from the filo-, henipa- and corona-viruses resemble canonical viral glycoproteins from HIV and influenza (termed class I viruses for their fusion machinery) in that they exist as trimers of heterodimers consisting of surface and transmembrane (TM) subunits. The trimers are held in a so-called metastable state, primed for fusion, but requiring activation energy to trigger conformational changes leading to the final, or postfusion, conformational state [86]. Triggers include receptor binding or acidic pH. Initially, conformational changes expose a highly hydrophobic fusion peptide that is able to insert itself into target membrane. Subsequently, the TM subunit folds back on itself, allowing two α-helical regions within the subunit (termed heptad repeats [HR]1 and 2) to interact with each other. HR1 and HR2 from each glycoprotein in the trimer come together to form a six-helix bundle. It is the formation of this stable structure that drives fusion of the two membranes [86,87].
Alternately, the class II viruses such as alpha- and flavi-viruses utilize a different mechanism in order to mediate membrane fusion, although the ultimate structures look somewhat similar in that they form hairpins [88]. The glycoproteins driving membrane fusion are packed in a much more highly ordered, icosahedral structure on the virion surface, with each present as a homodimer in the case of the flaviviruses [85]. Acidic pH then triggers rearrangements leading to the formation of a homotrimer and the resulting exposure of the fusion peptide, which then inserts into the target membrane. The glycoprotein then folds back on itself to form a hairpin and draw the membranes together, although in this case a six-helix bundle is not formed.
Membrane fusion inhibitors
Several steps along the cascade of events leading to membrane fusion are appealing targets for antiviral intervention, particularly the prevention of conformational changes within the glycoprotein. For example, as described for the HIV entry inhibitor BMS-378806 [55], allosteric inhibitors that trap a metastable protein in an inactive state would be excellent lead candidates for drugs [89]. Further downstream of the initial conformational changes are the HR interactions, the inhibition of which would prevent the final conformational changes leading to membrane fusion from occurring. Indeed, the prototypical viral fusion inhibitor is enfuvirtide, a peptide corresponding to the HR2 domain from HIV, which efficiently inhibits infection by preventing completion of six-helix bundle formation [11,90]. Similarly, synthetic peptides derived from the HRs result in inhibition of the membrane fusion step during viral entry of a number of emerging viruses including the Henipaviruses, filoviruses and SARS-CoV [91–93]. As many of these viruses undergo the conformational changes required to expose HRs in endosomes, methods of directing the peptides to internal compartments, including fusing them to targeting domains such as HIV Tat, greatly enhance their potency [91]. Conjugating cholesterol to influenza hemagglutinin-derived peptides successfully targeted the peptide to the endosomal site of viral fusion [94]. Similarly, in the case of NiV, adding cholesterol to HR2 peptides targets them to the lipid raft microdomains utilized by this virus for entry [93].
A benzodiazepine derivative was identified as an entry inhibitor for EBOV and MARV from a HTS of a compound library, utilizing HIV/EBOV-GP pseudotyped viruses [95]. Computational and mutational studies using the EBOV-GP crystal structure [96] suggested a hydrophobic pocket close to the interface between the surface subunits (termed GP1) and TM (GP2) subunits as a potential site for this compound to bind [95]. Similarly, a novel group of small molecules, related to 3,5-disubstituted isoxazoles, were identified using a pseudotyping screening strategy. These compounds also showed selective inhibition of EBOV and MARV-GP-mediated infection of human cells [97]. The mechanism of inhibition for both families of anti-EBOV compounds is not yet fully elucidated, but it is possible that these molecules prevent conformational changes or membrane fusion.
New World arenaviruses, such as Junín, utilize transferrin receptor 1 [98] as a receptor and fuse with the aid of acid pH within endosomes. A number of related compounds have been identified that bind to a pocket on the viral glycoprotein of arenaviruses [99–101]. These compounds have a spectrum of inhibition, with some able to broadly inhibit all arenaviruses, while others are more specific. The fact that the inhibitors share a binding site also suggests a common mode of action, although the exact nature of this is not fully elucidated [102]. However, like other small-molecule entry inhibitors targeting viral glycoproteins [55], it appears that the underlying mechanism of inhibition may be preventing conformational changes critical for glycoprotein transition from the metastable state into an activated one [100].
Somewhat similarly to EBOV utilizing the cholesterol transporter NPC1 in late endosomes, dengue requires a lipid specific to endosomes (bis[monoacylglycero]phosphate) for efficient membrane fusion [103]. This dependence is unlikely to be unique to dengue [104] and may represent an excellent target for broad-spectrum and targeted drug design.
A good example of a promising broad-spectrum antiviral compound targeting entry of enveloped viruses is a small molecule termed LJ001 [105]. An aryl methyldiene rhodanine derivative, LJ001, was identified during a HTS for anti-NiV entry using vesicular stomatitis virus-based pseudoviruses. The compound subsequently proved to effectively inhibit viral entry of numerous enveloped viruses including influenza, HIV, filo-, pox-, arena-, bunya-, paramyxo- and flavi-viruses. LJ001 specifically intercalates into viral lipid membranes and probably inhibits viral entry at a step after virus binding, possibly by affecting the rigidity and curvature of lipid membranes and thus preventing efficient virus–cellular membrane fusion. This type of inhibitor represents some of the unexpected targets revealed by large batch HTS assays using pseudotype viruses, and exemplified how new compounds can be identified with similar mechanisms of action to older drugs. Arbidol (Arb) is a small indole-derivative first marketed in Russia in the 1990s as an influenza treatment [106]. Russian clinical trials are reported to demonstrate a reduction of the length of symptoms in influenza-infected patients [107], although efficacy studies in animals failed to demonstrate any reduction in influenza lung titers or lung consolidation [108]. In addition to influenza A and B, Arb is active in vitro against a number of other enveloped and nonenveloped respiratory viruses [108], as well as HCV [109]. Although various modes of action have been ascribed to Arb, such as immune modulation [110] and binding to influenza hemagglutinin [111], it appears the most likely mechanism of inhibition is through the ability of Arb to integrate into membranes via interactions with the polar head groups of phospholipids [112]. Like LJ001, the broad-spectrum activity of Arb can be explained by the ability to perturb membrane fluidity and flexibility.
Expert commentary & five-year view
Historically, many emerging viruses have been neglected in terms of research, partly due to a lack of funding, but also because of difficulties in obtaining reagents and safety concerns in handling potentially biohazardous samples. However, as many emerging viruses have at least the theoretical potential to be bioweaponized, research on such viruses has profited from governmental investment in biodefense over the last 10 years. While this is controversial [113], and clearly has directed funding away from areas that have a more obvious medical need in the here-and-now, there is no denying that huge strides have been made in understanding what were previously under-researched pathogens. Thus, the outlook over the next 5 years for new antiviral therapies targeting emerging viruses is exciting. In particular, in vivo efficacy studies are being initiated for many of the lead compounds identified over the last few years. In the United States, the latest round of funding for the regional centers of excellence for biodefense and emerging infectious diseases is rapidly approaching and it may be that much of the money invested in this area will begin to dissipate, with a more focused approach replacing the somewhat haphazard, unselective attitude to funding over the last 10 years. However, this should not dissuade investigators with novel ideas for screening drug targets or promising antivirals from applying for funding. More research in particular is warranted for the less popular viruses such as Lassa and chikungunya viruses that have been relatively neglected, but actually infect large numbers of individuals every year, in favor of viruses that grab the public imagination, but are unlikely to ever be a threat – terrorist, or otherwise.
Most antiviral drugs typically target a single virus. This makes it expensive and virtually impossible to stockpile antivirals able to treat all possible emerging viruses and potential weaponized biothreats. Another drawback of the high specificity of traditional antivirals is that the virus can rapidly adapt and develop drug resistance due to accumulating mutations. However, the few current broad-spectrum antivirals are less prone to developing drug resistance, but have side effects, limited potency and are expensive for widespread use. Therefore, one of the important goals in antiviral therapy has become the identification of broad-spectrum antivirals. In all of these regards, targeting viral entry for the discovery of novel small molecules is a valid exercise. Safe and rapid HTS assays exist. The use of multiple envelope screening assays to identify broad-spectrum inhibitors is an intriguing idea, and with new luciferase reporter genes, up to three unrelated envelopes at one time could be included in a single well. Alternately, if specific cellular functions have been identified as being utilized by subgroups of viruses, that particular target can be specifically screened for antivirals. For example, following the identification of cathepsins as important factors in the entry of corona-, filo-, paramyxo- and retro-viruses, we and others screened specific libraries of cathepsin and other protease inhibitors for antivirals [14,81]. Both cellular and viral targets can be pursued, as can both specific and broad-spectrum inhibitors.
Key issues.
Changes in the virus through mutation, exposure to new reservoirs through human encroachment, alteration of human susceptibility through coinfections and the increase in world travel are all risk factors for potential new viral pandemics to arise.
There is a great need to develop antivirals to fight diverse emerging virus diseases, as well as serve potential biodefense needs.
Currently, most antiviral drugs typically target a single virus, with few options for the majority of emerging viruses, and the few broad-spectrum antivirals available have limited potency, often with side effects.
In this article, we have reviewed recent advances in antivirals targeting viral entry for emerging viruses such as Ebola virus and SARS-coronavirus, and discussed potential broad-spectrum antiviral strategies.
Acknowledgements
The authors thank Sean M Amberg (SIGA Technologies) for reading the manuscript and providing helpful comments and insight.
The authors were supported by grant R01AI074986 from the National Institute of Allergy and Infectious Diseases
Footnotes
Financial & competing interests disclosure
The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
No writing assistance was utilized in the production of this manuscript.
References
Papers of special note have been highlighted as:
• of interest
•• of considerable interest
- 1.Snell NJ. Ribavirin – current status of a broad spectrum antiviral agent. Expert Opin. Pharmacother. 2001;2(8):1317–1324. doi: 10.1517/14656566.2.8.1317. [DOI] [PubMed] [Google Scholar]
- 2.Wong AC, Sandesara RG, Mulherkar N, Whelan SP, Chandran K. A forward genetic strategy reveals destabilizing mutations in the Ebolavirus glycoprotein that alter its protease dependence during cell entry. J. Virol. 2010;84(1):163–175. doi: 10.1128/JVI.01832-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhaori G. Antiviral treatment of SARS: can we draw any conclusions? CMAJ. 2003;169(11):1165–1166. [PMC free article] [PubMed] [Google Scholar]
- 4.Chong HT, Kamarulzaman A, Tan CT, et al. Treatment of acute Nipah encephalitis with ribavirin. Ann. Neurol. 2001;49(6):810–813. doi: 10.1002/ana.1062. [DOI] [PubMed] [Google Scholar]
- 5.Jahrling PB, Geisbert TW, Geisbert JB, et al. Evaluation of immune globulin and recombinant interferon-alpha2b for treatment of experimental Ebola virus infections. J. Infect. Dis. 1999;179(Suppl. 1):S224–S234. doi: 10.1086/514310. [DOI] [PubMed] [Google Scholar]
- 6.Ignat’ev GM, Strel’tsova MA, Agafonov AP, Kashentseva EA, Prozorovskii NS. Experimental study of possible treatment of Marburg hemorrhagic fever with desferal, ribavirin, and homologous interferon. Vopr. Virusol. 1996;41(5):206–209. [PubMed] [Google Scholar]
- 7.Hoffmann HH, Kunz A, Simon VA, Palese P, Shaw ML. Broad-spectrum antiviral that interferes with de novo pyrimidine biosynthesis. Proc. Natl Acad. Sci. USA. 2011;108(14):5777–5782. doi: 10.1073/pnas.1101143108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wong JP, Christopher ME, Salazar AM, et al. Broad-spectrum and virus-specific nucleic acid-based antivirals against influenza. Front. Biosci.(Schol. Ed) 2010;2:791–800. doi: 10.2741/s102. [DOI] [PubMed] [Google Scholar]
- 9.Furuta Y, Takahashi K, Shiraki K, et al. T-705 (favipiravir) and related compounds: novel broad-spectrum inhibitors of RNA viral infections. Antiviral Res. 2009;82(3):95–102. doi: 10.1016/j.antiviral.2009.02.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chamoun AM, Chockalingam K, Bobardt M, et al. PD 404,182 is a virocidal small molecule that disrupts hepatitis C virus and human immunodeficiency virus. Antimicrob. Agents Chemother. 2012;56(2):672–681. doi: 10.1128/AAC.05722-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pöhlmann S, Reeves JD. Cellular entry of HIV: evaluation of therapeutic targets. Curr. Pharm. Des. 2006;12(16):1963–1973. doi: 10.2174/138161206777442155. [DOI] [PubMed] [Google Scholar]
- 12.Singh IP, Chauthe SK. Small molecule HIV entry inhibitors: part II. Attachment and fusion inhibitors: 2004–2010. Expert Opin. Ther. Pat. 2011;21(3):399–416. doi: 10.1517/13543776.2011.550876. [DOI] [PubMed] [Google Scholar]
- 13.Emau P, Tian B, O’Keefe BR, et al. Griffithsin, a potent HIV entry inhibitor, is an excellent candidate for anti-HIV microbicide. J. Med. Primatol. 2007;36(4–5):244–253. doi: 10.1111/j.1600-0684.2007.00242.x. [DOI] [PubMed] [Google Scholar]
- 14. Zhou Y, Agudelo J, Lu K, et al. Inhibitors of SARS-CoV entry – identification using an internally-controlled dual envelope pseudovirion assay. Antiviral Res. 2011;92(2):187–194. doi: 10.1016/j.antiviral.2011.07.016. •• Recent development of a novel high-throughput screening assay for identifying entry inhibitors.
- 15.Talekar A, Pessi A, Glickman F, et al. Rapid screening for entry inhibitors of highly pathogenic viruses under low-level biocontainment. PLoS ONE. 2012;7(3):e30538. doi: 10.1371/journal.pone.0030538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wool-Lewis RJ, Bates P. Characterization of Ebola virus entry by using pseudotyped viruses: identification of receptor-deficient cell lines. J. Virol. 1998;72(4):3155–3160. doi: 10.1128/jvi.72.4.3155-3160.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Simmons G, Reeves JD, Rennekamp AJ, Amberg SM, Piefer AJ, Bates P. Characterization of severe acute respiratory syndrome-associated coronavirus (SARS-CoV) spike glycoprotein-mediated viral entry. Proc. Natl Acad. Sci. USA. 2004;101(12):4240–4245. doi: 10.1073/pnas.0306446101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Matsuura Y, Tani H, Suzuki K, et al. Characterization of pseudotype VSV possessing HCV envelope proteins. Virology. 2001;286(2):263–275. doi: 10.1006/viro.2001.0971. [DOI] [PubMed] [Google Scholar]
- 19.Basu A, Mills DM, Bowlin TL. High-throughput screening of viral entry inhibitors using pseudotyped virus. Curr. Protoc. Pharmacol. 2010;Chapter 13(Unit 13B.3) doi: 10.1002/0471141755.ph13b03s51. [DOI] [PubMed] [Google Scholar]
- 20.Baldick CJ, Wichroski MJ, Pendri A, et al. A novel small molecule inhibitor of hepatitis C virus entry. PLoS Pathog. 2010;6(9) doi: 10.1371/journal.ppat.1001086. e1001086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Filone CM, Heise M, Doms RW, Bertolotti-Ciarlet A. Development and characterization of a Rift Valley fever virus cell–cell fusion assay using alphavirus replicon vectors. Virology. 2006;356(1–2):155–164. doi: 10.1016/j.virol.2006.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wolf MC, Wang Y, Freiberg AN, Aguilar HC, Holbrook MR, Lee B. A catalytically and genetically optimized beta-lactamase-matrix based assay for sensitive, specific, and higher throughput analysis of native henipavirus entry characteristics. Virol. J. 2009;6:119. doi: 10.1186/1743-422X-6-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kaufmann B, Chipman PR, Holdaway HA, et al. Capturing a flavivirus pre-fusion intermediate. PLoS Pathog. 2009;5(11) doi: 10.1371/journal.ppat.1000672. e1000672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Li L, Jose J, Xiang Y, Kuhn RJ, Rossmann MG. Structural changes of envelope proteins during alphavirus fusion. Nature. 2010;468(7324):705–708. doi: 10.1038/nature09546. •• Recent description of the structural changes undertaken by a typical class II fusion protein during activation.
- 25. Carette JE, Raaben M, Wong AC, et al. Ebola virus entry requires the cholesterol transporter Niemann-Pick C1. Nature. 2011;477(7364):340–343. doi: 10.1038/nature10348. • Description of NPC1 as the major cellular receptor for Ebola virus.
- 26. Côté M, Misasi J, Ren T, et al. Small molecule inhibitors reveal Niemann-Pick C1 is essential for Ebola virus infection. Nature. 2011;477(7364):344–348. doi: 10.1038/nature10380. •• Identification of a new class of Ebola virus entry inhibitors targeting the cellular receptor, NPC1.
- 27.Miller EH, Obernosterer G, Raaben M, et al. Ebola virus entry requires the host-programmed recognition of an intracellular receptor. EMBO J. 2012;31(8):1947–1960. doi: 10.1038/emboj.2012.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Becker S, Spiess M, Klenk HD. The asialoglycoprotein receptor is a potential liver-specific receptor for Marburg virus. J. Gen. Virol. 1995;76(Pt 2):393–399. doi: 10.1099/0022-1317-76-2-393. [DOI] [PubMed] [Google Scholar]
- 29.Dominguez-Soto A, Aragoneses-Fenoll L, Martin-Gayo E, et al. The DC-SIGN-related lectin LSECtin mediates antigen capture and pathogen binding by human myeloid cells. Blood. 2007;109(12):5337–5345. doi: 10.1182/blood-2006-09-048058. [DOI] [PubMed] [Google Scholar]
- 30.Lin G, Simmons G, Pöhlmann S, et al. Differential N-linked glycosylation of human immunodeficiency virus and Ebola virus envelope glycoproteins modulates interactions with DC-SIGN and DC-SIGNR. J. Virol. 2003;77(2):1337–1346. doi: 10.1128/JVI.77.2.1337-1346.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Simmons G, Reeves JD, Grogan CC, et al. DC-SIGN and DC-SIGNR bind Ebola glycoproteins and enhance infection of macrophages and endothelial cells. Virology. 2003;305(1):115–123. doi: 10.1006/viro.2002.1730. [DOI] [PubMed] [Google Scholar]
- 32.Takada A, Fujioka K, Tsuiji M, et al. Human macrophage C-type lectin specific for galactose and N-acetylgalactosamine promotes filovirus entry. J. Virol. 2004;78(6):2943–2947. doi: 10.1128/JVI.78.6.2943-2947.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Navarro-Sanchez E, Altmeyer R, Amara A, et al. Dendritic-cell-specific ICAM3-grabbing non-integrin is essential for the productive infection of human dendritic cells by mosquito-cell-derived dengue viruses. EMBO Rep. 2003;4(7):723–728. doi: 10.1038/sj.embor.embor866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tassaneetrithep B, Burgess TH, Granelli-Piperno A, et al. DC-SIGN (CD209) mediates dengue virus infection of human dendritic cells. J. Exp. Med. 2003;197(7):823–829. doi: 10.1084/jem.20021840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang ZY, Huang Y, Ganesh L, et al. pH-dependent entry of severe acute respiratory syndrome coronavirus is mediated by the spike glycoprotein and enhanced by dendritic cell transfer through DC-SIGN. J. Virol. 2004;78(11):5642–5650. doi: 10.1128/JVI.78.11.5642-5650.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Spillmann D. Heparan sulfate: anchor for viral intruders? Biochimie. 2001;83(8):811–817. doi: 10.1016/s0300-9084(01)01290-1. • Review of the role that glycosaminoglycans play in viral entry and their potential as drug targets.
- 37.Barth H, Schafer C, Adah MI, et al. Cellular binding of hepatitis C virus envelope glycoprotein E2 requires cell surface heparan sulfate. J. Biol. Chem. 2003;278(42):41003–41012. doi: 10.1074/jbc.M302267200. [DOI] [PubMed] [Google Scholar]
- 38.González ME, Alarcón B, Carrasco L. Polysaccharides as antiviral agents: antiviral activity of carrageenan. Antimicrob. Agents Chemother. 1987;31(9):1388–1393. doi: 10.1128/aac.31.9.1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Krepstakies M, Lucifora J, Nagel CH, et al. A new class of synthetic peptide inhibitors blocks attachment and entry of human pathogenic viruses. J. Infect. Dis. 2012;205(11):1654–1664. doi: 10.1093/infdis/jis273. [DOI] [PubMed] [Google Scholar]
- 40.Donalisio M, Rusnati M, Civra A, et al. Identification of a dendrimeric heparan sulfate-binding peptide that inhibits infectivity of genital types of human papillomaviruses. Antimicrob. Agents Chemother. 2010;54(10):4290–4299. doi: 10.1128/AAC.00471-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schuksz M, Fuster MM, Brown JR, et al. Surfen, a small molecule antagonist of heparan sulfate. Proc. Natl Acad. Sci. USA. 2008;105(35):13075–13080. doi: 10.1073/pnas.0805862105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Botos I, Wlodawer A. Cyanovirin-N: a sugar-binding antiviral protein with a new twist. Cell. Mol. Life Sci. 2003;60(2):277–287. doi: 10.1007/s000180300023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Barrientos LG, O’Keefe BR, Bray M, Sanchez A, Gronenborn AM, Boyd MR. Cyanovirin-N binds to the viral surface glycoprotein, GP1,2 and inhibits infectivity of Ebola virus. Antiviral Res. 2003;58(1):47–56. doi: 10.1016/s0166-3542(02)00183-3. [DOI] [PubMed] [Google Scholar]
- 44.Barrientos LG, Lasala F, Otero JR, Sanchez A, Delgado R. In vitro evaluation of cyanovirin-N antiviral activity, by use of lentiviral vectors pseudotyped with filovirus envelope glycoproteins. J. Infect. Dis. 2004;189(8):1440–1443. doi: 10.1086/382658. [DOI] [PubMed] [Google Scholar]
- 45.O’Keefe BR, Giomarelli B, Barnard DL, et al. Broad-spectrum in vitro activity and in vivo efficacy of the antiviral protein griffithsin against emerging viruses of the family Coronaviridae. J. Virol. 2010;84(5):2511–2521. doi: 10.1128/JVI.02322-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.O’Keefe BR, Smee DF, Turpin JA, et al. Potent anti-influenza activity of cyanovirin-N and interactions with viral hemagglutinin. Antimicrob. Agents Chemother. 2003;47(8):2518–2525. doi: 10.1128/AAC.47.8.2518-2525.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.van der Meer FJ, de Haan CA, Schuurman NM, et al. Antiviral activity of carbohydrate-binding agents against Nidovirales in cell culture. Antiviral Res. 2007;76(1):21–29. doi: 10.1016/j.antiviral.2007.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Helle F, Wychowski C, Vu-Dac N, Gustafson KR, Voisset C, Dubuisson J. Cyanovirin-N inhibits hepatitis C virus entry by binding to envelope protein glycans. J. Biol. Chem. 2006;281(35):25177–25183. doi: 10.1074/jbc.M602431200. [DOI] [PubMed] [Google Scholar]
- 49.Smee DF, Bailey KW, Wong MH, et al. Treatment of influenza A (H1N1) virus infections in mice and ferrets with cyanovirin-N. Antiviral Res. 2008;80(3):266–271. doi: 10.1016/j.antiviral.2008.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li W, Moore MJ, Vasilieva N, et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature. 2003;426(6965):450–454. doi: 10.1038/nature02145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Negrete OA, Levroney EL, Aguilar HC, et al. EphrinB2 is the entry receptor for Nipah virus, an emergent deadly paramyxovirus. Nature. 2005;436(7049):401–405. doi: 10.1038/nature03838. [DOI] [PubMed] [Google Scholar]
- 52.Kondratowicz AS, Lennemann NJ, Sinn PL, et al. T-cell immunoglobulin and mucin domain 1 (TIM-1) is a receptor for Zaire Ebolavirus and Lake Victoria Marburgvirus. Proc. Natl Acad. Sci. USA. 2011;108(20):8426–8431. doi: 10.1073/pnas.1019030108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ho TY, Wu SL, Chen JC, Li CC, Hsiang CY. Emodin blocks the SARS coronavirus spike protein and angiotensin-converting enzyme 2 interaction. Antiviral Res. 2007;74(2):92–101. doi: 10.1016/j.antiviral.2006.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lin PF, Blair W, Wang T, et al. A small molecule HIV-1 inhibitor that targets the HIV-1 envelope and inhibits CD4 receptor binding. Proc. Natl Acad. Sci. USA. 2003;100(19):11013–11018. doi: 10.1073/pnas.1832214100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Si Z, Madani N, Cox JM, et al. Small-molecule inhibitors of HIV-1 entry block receptor-induced conformational changes in the viral envelope glycoproteins. Proc. Natl Acad. Sci. USA. 2004;101(14):5036–5041. doi: 10.1073/pnas.0307953101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mercer J, Schelhaas M, Helenius A. Virus entry by endocytosis. Annu. Rev. Biochem. 2010;79:803–833. doi: 10.1146/annurev-biochem-060208-104626. [DOI] [PubMed] [Google Scholar]
- 57.Sanchez A. Analysis of filovirus entry into vero e6 cells, using inhibitors of endocytosis, endosomal acidification, structural integrity, and cathepsin (B and L) activity. J. Infect. Dis. 2007;196(Suppl. 2):S251–S258. doi: 10.1086/520597. [DOI] [PubMed] [Google Scholar]
- 58.Empig CJ, Goldsmith MA. Association of the caveola vesicular system with cellular entry by filoviruses. J. Virol. 2002;76(10):5266–5270. doi: 10.1128/JVI.76.10.5266-5270.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Saeed MF, Kolokoltsov AA, Albrecht T, Davey RA. Cellular entry of Ebola virus involves uptake by a macropinocytosis-like mechanism and subsequent trafficking through early and late endosomes. PLoS Pathog. 2010;6(9) doi: 10.1371/journal.ppat.1001110. e1001110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nanbo A, Imai M, Watanabe S, et al. Ebolavirus is internalized into host cells via macropinocytosis in a viral glycoprotein-dependent manner. PLoS Pathog. 2010;6(9) doi: 10.1371/journal.ppat.1001121. e1001121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mulherkar N, Raaben M, de la Torre JC, Whelan SP, Chandran K. The Ebola virus glycoprotein mediates entry via a non-classical dynamin-dependent macropinocytic pathway. Virology. 2011;419(2):72–83. doi: 10.1016/j.virol.2011.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Saeed MF, Kolokoltsov AA, Freiberg AN, Holbrook MR, Davey RA. Phosphoinositide-3 kinase–Akt pathway controls cellular entry of Ebola virus. PLoS Pathog. 2008;4(8) doi: 10.1371/journal.ppat.1000141. e1000141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Krishnan MN, Sukumaran B, Pal U, et al. Rab 5 is required for the cellular entry of dengue and West Nile viruses. J. Virol. 2007;81(9):4881–4885. doi: 10.1128/JVI.02210-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pernet O, Pohl C, Ainouze M, Kweder H, Buckland R. Nipah virus entry can occur by macropinocytosis. Virology. 2009;395(2):298–311. doi: 10.1016/j.virol.2009.09.016. [DOI] [PubMed] [Google Scholar]
- 65.Chandran K, Sullivan NJ, Felbor U, Whelan SP, Cunningham JM. Endosomal proteolysis of the Ebola virus glycoprotein is necessary for infection. Science. 2005;308(5728):1643–1645. doi: 10.1126/science.1110656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Schornberg K, Matsuyama S, Kabsch K, Delos S, Bouton A, White J. Role of endosomal cathepsins in entry mediated by the Ebola virus glycoprotein. J. Virol. 2006;80(8):4174–4178. doi: 10.1128/JVI.80.8.4174-4178.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kaletsky RL, Simmons G, Bates P. Proteolysis of the Ebola virus glycoproteins enhances virus binding and infectivity. J. Virol. 2007;81(24):13378–13384. doi: 10.1128/JVI.01170-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Misasi J, Chandran K, Yang JY, et al. Filoviruses require endosomal cysteine proteases for entry but exhibit distinct protease preferences. J. Virol. 2012;86(6):3284–3292. doi: 10.1128/JVI.06346-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Simmons G, Gosalia DN, Rennekamp AJ, Reeves JD, Diamond SL, Bates P. Inhibitors of cathepsin L prevent severe acute respiratory syndrome coronavirus entry. Proc. Natl Acad. Sci. USA. 2005;102(33):11876–11881. doi: 10.1073/pnas.0505577102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Qiu Z, Hingley ST, Simmons G, et al. Endosomal proteolysis by cathepsins is necessary for murine coronavirus mouse hepatitis virus type 2 spike-mediated entry. J. Virol. 2006;80(12):5768–5776. doi: 10.1128/JVI.00442-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hofmann H, Simmons G, Rennekamp AJ, et al. Highly conserved regions within the spike proteins of human coronaviruses 229E and NL63 determine recognition of their respective cellular receptors. J. Virol. 2006;80(17):8639–8652. doi: 10.1128/JVI.00560-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kawase M, Shirato K, Matsuyama S, Taguchi F. Protease-mediated entry via the endosome of human coronavirus 229E. J. Virol. 2009;83(2):712–721. doi: 10.1128/JVI.01933-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Glowacka I, Bertram S, Müller MA, et al. Evidence that TMPRSS2 activates the severe acute respiratory syndrome coronavirus spike protein for membrane fusion and reduces viral control by the humoral immune response. J. Virol. 2011;85(9):4122–4134. doi: 10.1128/JVI.02232-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Matsuyama S, Nagata N, Shirato K, Kawase M, Takeda M, Taguchi F. Efficient activation of the severe acute respiratory syndrome coronavirus spike protein by the transmembrane protease TMPRSS2. J. Virol. 2010;84(24):12658–12664. doi: 10.1128/JVI.01542-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Shulla A, Heald-Sargent T, Subramanya G, Zhao J, Perlman S, Gallagher T. A transmembrane serine protease is linked to the severe acute respiratory syndrome coronavirus receptor and activates virus entry. J. Virol. 2011;85(2):873–882. doi: 10.1128/JVI.02062-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bertram S, Glowacka I, Müller MA, et al. Cleavage and activation of the severe acute respiratory syndrome coronavirus spike protein by human airway trypsin-like protease. J. Virol. 2011;85(24):13363–13372. doi: 10.1128/JVI.05300-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Simmons G, Bertram S, Glowacka I, et al. Different host cell proteases activate the SARS-coronavirus spike-protein for cell–cell and virus–cell fusion. Virology. 2011;413(2):265–274. doi: 10.1016/j.virol.2011.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Diederich S, Thiel L, Maisner A. Role of endocytosis and cathepsin-mediated activation in Nipah virus entry. Virology. 2008;375(2):391–400. doi: 10.1016/j.virol.2008.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Pager CT, Craft WW, Jr, Patch J, Dutch RE. A mature and fusogenic form of the Nipah virus fusion protein requires proteolytic processing by cathepsin L. Virology. 2006;346(2):251–257. doi: 10.1016/j.virol.2006.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Pager CT, Dutch RE. Cathepsin L is involved in proteolytic processing of the Hendra virus fusion protein. J. Virol. 2005;79(20):12714–12720. doi: 10.1128/JVI.79.20.12714-12720.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Shah PP, Wang T, Kaletsky RL, et al. A small-molecule oxocarbazate inhibitor of human cathepsin L blocks severe acute respiratory syndrome and Ebola pseudotype virus infection into human embryonic kidney 293T cells. Mol. Pharmacol. 2010;78(2):319–324. doi: 10.1124/mol.110.064261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Gnirss K, Kühl A, Karsten C, et al. Cathepsins B and L activate Ebola but not Marburg virus glycoproteins for efficient entry into cell lines and macrophages independent of TMPRSS2 expression. Virology. 2012;424(1):3–10. doi: 10.1016/j.virol.2011.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Goetz DH, Choe Y, Hansell E, et al. Substrate specificity profiling and identification of a new class of inhibitor for the major protease of the SARS coronavirus. Biochemistry. 2007;46(30):8744–8752. doi: 10.1021/bi0621415. [DOI] [PubMed] [Google Scholar]
- 84.Connolly SA, Jackson JO, Jardetzky TS, Longnecker R. Fusing structure and function: a structural view of the herpesvirus entry machinery. Nat. Rev. Microbiol. 2011;9(5):369–381. doi: 10.1038/nrmicro2548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Vaney MC, Rey FA. Class II enveloped viruses. Cell. Microbiol. 2011;13(10):1451–1459. doi: 10.1111/j.1462-5822.2011.01653.x. • Recent review on class II enveloped viruses.
- 86.Colman PM, Lawrence MC. The structural biology of type I viral membrane fusion. Nat. Rev. Mol. Cell Biol. 2003;4(4):309–319. doi: 10.1038/nrm1076. [DOI] [PubMed] [Google Scholar]
- 87. Melikyan GB. Membrane fusion mediated by human immunodeficiency virus envelope glycoprotein. Curr. Top. Membr. 2011;68:81–106. doi: 10.1016/B978-0-12-385891-7.00004-0. •• In-depth review of the latest aspects related to the canonical class I fusion protein, HIV gp160.
- 88.Kielian M, Rey FA. Virus membrane-fusion proteins: more than one way to make a hairpin. Nat. Rev. Microbiol. 2006;4(1):67–76. doi: 10.1038/nrmicro1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lee GM, Craik CS. Trapping moving targets with small molecules. Science. 2009;324(5924):213–215. doi: 10.1126/science.1169378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Doms RW. Viral entry denied. N. Engl.J. Med. 2004;351(8):743–744. doi: 10.1056/NEJMp048058. [DOI] [PubMed] [Google Scholar]
- 91.Miller EH, Harrison JS, Radoshitzky SR, et al. Inhibition of Ebola virus entry by a C-peptide targeted to endosomes. J. Biol. Chem. 2011;286(18):15854–15861. doi: 10.1074/jbc.M110.207084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Bosch BJ, Martina BE, Van Der Zee R, et al. Severe acute respiratory syndrome coronavirus (SARS-CoV) infection inhibition using spike protein heptad repeat-derived peptides. Proc. Natl Acad. Sci. USA. 2004;101(22):8455–8460. doi: 10.1073/pnas.0400576101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Porotto M, Yokoyama CC, Palermo LM, et al. Viral entry inhibitors targeted to the membrane site of action. J. Virol. 2010;84(13):6760–6768. doi: 10.1128/JVI.00135-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Lee KK, Pessi A, Gui L, et al. Capturing a fusion intermediate of influenza hemagglutinin with a cholesterol-conjugated peptide, a new antiviral strategy for influenza virus. J. Biol. Chem. 2011;286(49):42141–42149. doi: 10.1074/jbc.M111.254243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Basu A, Li B, Mills DM, et al. Identification of a small-molecule entry inhibitor for filoviruses. J. Virol. 2011;85(7):3106–3119. doi: 10.1128/JVI.01456-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Lee JE, Fusco ML, Hessell AJ, Oswald WB, Burton DR, Saphire EO. Structure of the Ebola virus glycoprotein bound to an antibody from a human survivor. Nature. 2008;454(7201):177–182. doi: 10.1038/nature07082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Yermolina MV, Wang J, Caffrey M, Rong LL, Wardrop DJ. Discovery, synthesis, and biological evaluation of a novel group of selective inhibitors of filoviral entry. J. Med. Chem. 2011;54(3):765–781. doi: 10.1021/jm1008715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Radoshitzky SR, Abraham J, Spiropoulou CF, et al. Transferrin receptor 1 is a cellular receptor for New World haemorrhagic fever arenaviruses. Nature. 2007;446(7131):92–96. doi: 10.1038/nature05539. • Transferrin receptor 1 is the major cellular receptor for New World arenaviruses.
- 99.Larson RA, Dai D, Hosack VT, et al. Identification of a broad-spectrum arenavirus entry inhibitor. J. Virol. 2008;82(21):10768–10775. doi: 10.1128/JVI.00941-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.York J, Dai D, Amberg SM, Nunberg JH. pH-induced activation of arenavirus membrane fusion is antagonized by small-molecule inhibitors. J. Virol. 2008;82(21):10932–10939. doi: 10.1128/JVI.01140-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Thomas CJ, Casquilho-Gray HE, York J, et al. A specific interaction of small molecule entry inhibitors with the envelope glycoprotein complex of the Junín hemorrhagic fever arenavirus. J. Biol. Chem. 2011;286(8):6192–6200. doi: 10.1074/jbc.M110.196428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Nunberg JH, York J. The curious case of arenavirus entry, and its inhibition. Viruses. 2012;4(1):83–101. doi: 10.3390/v4010083. • Review of recent developments in arenavirus entry and inhibition by broad-arenavirus inhibitors.
- 103.Zaitseva E, Yang ST, Melikov K, Pourmal S, Chernomordik LV. Dengue virus ensures its fusion in late endosomes using compartment-specific lipids. PLoS Pathog. 2010;6(10) doi: 10.1371/journal.ppat.1001131. e1001131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Roth SL, Whittaker GR. Promotion of vesicular stomatitis virus fusion by the endosome-specific phospholipid bis(monoacylglycero)phosphate (BMP) FEBS Lett. 2011;585(6):865–869. doi: 10.1016/j.febslet.2011.02.015. [DOI] [PubMed] [Google Scholar]
- 105. Wolf MC, Freiberg AN, Zhang T, et al. A broad-spectrum antiviral targeting entry of enveloped viruses. Proc. Natl Acad. Sci. USA. 2010;107(7):3157–3162. doi: 10.1073/pnas.0909587107. •• Recent description of a compound family able to inhibit membrane fusion for a broad spectrum of enveloped viruses.
- 106.Glushkov RG. Arbidol. Drugs of the Future. 1992;17:1079–1081. [Google Scholar]
- 107.Boriskin YS, Leneva IA, Pécheur EI, Polyak SJ. Arbidol: a broad-spectrum antiviral compound that blocks viral fusion. Curr. Med. Chem. 2008;15(10):997–1005. doi: 10.2174/092986708784049658. [DOI] [PubMed] [Google Scholar]
- 108.Brooks MJ, Burtseva EI, Ellery PJ, et al. Antiviral activity of arbidol, a broad-spectrum drug for use against respiratory viruses, varies according to test conditions. J. Med. Virol. 2012;84(1):170–181. doi: 10.1002/jmv.22234. [DOI] [PubMed] [Google Scholar]
- 109.Pécheur EI, Lavillette D, Alcaras F, et al. Biochemical mechanism of hepatitis C virus inhibition by the broad-spectrum antiviral arbidol. Biochemistry. 2007;46(20):6050–6059. doi: 10.1021/bi700181j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Glushkov RG, Gus’kova TA, Krylova LIu, Nikolaeva IS. Mechanisms of arbidole’s immunomodulating action. Vestn. Akad. Med. Nauk SSSR. 1999;1999(3):36–40. [PubMed] [Google Scholar]
- 111.Leneva IA, Russell RJ, Boriskin YS, Hay AJ. Characteristics of arbidol-resistant mutants of influenza virus: implications for the mechanism of anti-influenza action of arbidol. Antiviral Res. 2009;81(2):132–140. doi: 10.1016/j.antiviral.2008.10.009. [DOI] [PubMed] [Google Scholar]
- 112.Teissier E, Zandomeneghi G, Loquet A, et al. Mechanism of inhibition of enveloped virus membrane fusion by the antiviral drug arbidol. PLoS ONE. 2011;6(1) doi: 10.1371/journal.pone.0015874. e15874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Altman S, Bassler BL, Beckwith J, et al. An open letter to Elias Zerhouni. Science. 2005;307(5714):1409–1410. doi: 10.1126/science.307.5714.1409c. [DOI] [PubMed] [Google Scholar]