

Abstract
Diffusion magnetic resonance imaging (MRI) provides a sensitive indicator of cerebral hypoxia. We investigated if apparent diffusion coefficient (ADC) and transverse relaxation (T2) predict symptoms of acute mountain sickness (AMS), or merely indicate the AMS phenotype irrespective of symptoms. Fourteen normal subjects were studied in two groups; unambiguous AMS and no-AMS at 3,800 m altitude (intermediate AMS scores were excluded). T2 relaxation was estimated from a T2 index of T2-weighted signal normalized by cerebrospinal fluid signal. Measurements were made in normoxia and repeated after 2 days sustained hypoxia (AMS group symptomatic and no-AMS group asymptomatic) and after 7 days hypoxia (both groups asymptomatic). Decreased ADC directly predicted AMS symptoms (P<0.05). Apparent diffusion coefficient increased in asymptomatic subjects, or as symptoms abated with acclimatization. This pattern was similar in basal ganglia, white matter, and gray matter. Corpus callosum behaved differently; restricted diffusion was absent (or rapidly reversed) in the splenium, and was sustained in the genu. In symptomatic subjects, T2,index decreased after 2 days hypoxia and further decreased after 7 days. In asymptomatic subjects, T2,index initially increased after 2 days, but decreased after 7 days. T2,index changes were not predictive of AMS symptoms. These findings indicate that restricted diffusion, an indicator of diminished cerebral energy status, directly predicts symptoms of AMS in humans at altitude.
Keywords: apparent diffusion coefficient, acute mountain sickness, high altitude, hypoxia, magnetic resonance imaging, T2
Introduction
Acute exposure to hypoxia can induce a constellation of symptoms of acute mountain sickness (AMS), characterized by headache in addition to gastrointestinal and neurologic symptoms, sleep disturbance, and fatigue within 6 to 12 hours of rapid exposure to hypoxia at high altitude.1 Despite being a common response to hypoxia, the pathophysiologic mechanisms of AMS remain poorly understood. Acute mountain sickness itself is usually benign and self-limiting; however, it shares many clinical characteristics with high-altitude cerebral edema (HACE) which when untreated is fatal.2 Thus, the current consensus is that AMS and HACE may be part of the same spectrum of illness.
An overlap in pathophysiology of AMS and HACE is also supported by imaging data; in a study of HACE, Hackett and Roach3 reported increased transverse relaxation (T2) of the magnetic resonance imaging (MRI) signal in seven of nine subjects, particularly in the central white matter of the corpus callosum. Matsuzawa et al4 showed similar changes of increased T2 signal intensity in the white matter in the sickest four of seven subjects with AMS after 24 hours of altitude exposure.
Several studies have also looked for ‘subclinical edema' changes in uncomplicated AMS, looking at changes in T2 or the ADC—a physiologic parameter that characterizes the self-diffusion of water in tissue. Fischer et al5 found no changes in cerebral T2 or ADC after 10 hours at 4,500 m simulated altitude in a hypobaric chamber. Schoonman et al6 exposed subjects to 4,500 m simulated altitude under normobaric hypoxia for 6 hours and reported no edema on T2 images, but ∼2.5% increase in ADC in white matter (indicating less restricted water diffusion—a pattern often observed with hydrostatic edema). Kallenberg et al7 exposed subjects to 16 hours of 4,500 m simulated altitude under normobaric hypoxia, and observed a trend toward increasing ADC and T2 in asymptomatic hypoxia, but lower ADC in subjects with AMS.
Despite this mixed picture, a pattern of alterations in cerebral parenchymal microarchitecture is emerging in response to hypoxia. What remains unclear is the natural history of any T2 or ADC changes beyond the acute period. Specifically, (1) whether the changes in cerebral physiology track with symptoms of AMS, or whether they are present in all AMS-susceptible individuals irrespective of symptoms and (2) how ADC and T2 changes vary across cerebral regions with different hypoxia sensitivities.
We hypothesized that alterations in ADC and T2 (if present) would track with symptoms of AMS and would abate as symptoms resolved over the course of acclimatization to altitude. To test this, we examined the time course of ADC and T2 changes during sustained hypoxia at altitude. Cerebral MRI measurements were made at sea level, and after 2 days at 3,800 m altitude (when symptoms of AMS, if present, were at a maximum), and again after 7 days at altitude (when all subjects were fully acclimatized and asymptomatic).
Materials and methods
Subjects
In all, 18 healthy, nonsmoking, sea-level residents were recruited; 8 male (age 28±9.3 years) and 10 female (age 30±9.8 years). Ethical approval for these studies was granted by the Human Research Protection Program of the University of California San Diego. Participants were informed of the experimental procedures and possible risks involved in the study and written informed consent was obtained before participation.
Study Design
Diffusion-weighted MRI measurements were made in the cerebrum at sea level and from these we examined regional ADC of cerebral water, as well as signal changes on T2-weighted images. End-tidal CO2 tension (PETCO2), arterial oxygen saturation of hemoglobin (SaO2), and hematocrit were also measured. The measurements were repeated after 2 days and 7 days of sustained hypoxia at altitude. For hypoxic exposure, subjects resided at high altitude at the White Mountain Research Station (3,800 m altitude, PiO2 90 Torr). The diffusion and T2 changes were compared for AMS and no-AMS groups during normoxia and after the two durations of sustained hypoxia.
Hypoxic Exposure
Exposure to high altitude was rapid. Subjects were driven to the research station, with the majority of the ascent (600 to 3,800 m altitude) completed in ∼2 hours. Subjects spent two or seven nights at altitude and then returned to San Diego for MRI. After allowing ∼2 months for deacclimatization, subjects made a second 2- or 7-day trip. (The ordering of the 2-day or 7-day hypoxic exposures was random between subjects.) For each subject, the SaO2 achieved after 40 hours of hypoxic exposure at altitude (for 2-day trip) or after 160 hours at altitude (for 7-day trip) was maintained throughout the ∼8 hour transportation and the MRI measurements. During transportation, this was accomplished via a venturi mask with variable %N2 in the inspiratory port (to maintain consistent SaO2 despite changing altitude and barometric pressure). Within the MRI scanner subjects breathed a premixed 90 Torr hypoxic mixture (12.5% O2, balance N2) via a close-fitting low-deadspace non-rebreathing mask (Hans Rudolph 7900/2600 Kansas City, MO, USA). SaO2 was intermittently monitored while resident at altitude and continuously monitored during transportation and MRI. Subjects remained hypoxic at the same SaO2 level experienced on their last day at altitude until all MRI measurements were completed.
Acute Mountain Sickness Groups
To maximize our ability to detect potential physiologic differences between subjects at altitude, the Lake Louise Score (LLS), an AMS self-report questionnaire,8 was used to divide subjects into two distinct groups: Those with no symptoms of AMS (no-AMS group), and those with unambiguous AMS (AMS group). The Lake Louise AMS questionnaire is based on responses regarding five different symptoms—headache, gastrointestinal symptoms, fatigue, dizziness, and difficulty sleeping, each graded 0 to 3 in severity. Difficulty sleeping was not included as a criterion for AMS on the first night (to avoid confounds because of the rapid ascent8), but was scored on subsequent days. Subjects with an LLS of ⩽2, or with no headache, were considered as AMS nonsufferers (no-AMS group). Those with an LLS of ⩾5 and a headache plus symptoms of nausea, fatigue, dizziness, or difficulty sleeping were considered as unambiguous AMS sufferers (AMS group). Subjects with an intermediate score (LLS 3 to 4) and a headache were grouped into a third ‘intermediate' group, and not used when comparing AMS versus no-AMS. For correlation analysis between LLS and ADC (or T2,index), all subjects were included. Lake Louise scores were determined in each subject on both day 1 and day 2 (each immediately after a night at altitude, before any daily exercise). Beyond day 3 LLSs rapidly normalized with acclimatization and no longer indicated AMS susceptibility. The mean of the day 1 and day 2 scores was used to characterize subjects into AMS and no-AMS groups.
Physiologic Measurements
Arterial O2 saturation was measured and logged using a Nonin 3100 wrist pulse oximeter (at altitude and during transportation) and a Nonin 8600FO MRI-compatible pulse oximeter (Nonin Medical, Plymouth, MN, USA) (during MRI measurement) that was calibrated in each subject against an arterial blood sample. Inspired and expired partial pressures of O2 and CO2 were continuously monitored during MRI measurements using a Perkin-Elmer 1100 medical gas spectrometer (Perkin-Elmer Inc., Waltham, MA, USA). Hematocrit was determined from direct measurements of packed cell height in a capillary tube after centrifuging.
Magnetic Resonance Imaging Measurements
All MRI data were collected at 3 T on a General Electric scanner (GE Medical Systems, Milwaukee, WI, USA).
Diffusion-Weighted Images
Apparent diffusion coefficient maps and T2-weighted images were generated from diffusion-weighted images of the cerebrum (double spin-echo echo-planar imaging acquisition, echo time=85 ms, repetition time=7 seconds, b=1,000 s/mm2, field of view=24 × 24 cm, 128 × 128 acquisition matrix (zero padded to 256 × 256 during reconstruction), 4.4 mm slices, 1 minute).
3D T1-Weighted Anatomical Magnetic Resonance Imaging
A high-resolution FSPGR T1-weighted 3D-gradient echo image was also acquired to allow coregistration of the subjects' regions of interest (ROIs) data across the multiple imaging sessions (echo time=4.2 ms, repetition time=10.1 ms, TI=450 ms (effective repetition time 460.1 ms), bandwidth 20.83 kHz, field of view 25 × 25 × 16 cm, matrix 256 × 256 × 128, ∼1 × 1 × 1.3 mm resolution, 5.5 minutes).
Data Analysis
Regions of interest analysis
Apparent diffusion coefficient and T2 signal were evaluated in five anatomic regions: (1) gray matter (right and left insular, occipital, and frontal cortex); (2) white matter (right and left anterior and posterior areas of centrum semiovale); (3) basal ganglia, as this region is known to be especially hypoxia sensitive (right and left putamen, caudate, and globus pallidus); (4) the genu of the corpus callosum, and (5) splenium of the corpus callosum, as AMS and HACE have a predilection for these central white-matter tracts. Global signal changes were evaluated for all regions combined. The ROI location and mean pixel counts for each area are detailed in Figure 1. T2 signal was also measured in the lateral ventricles for the T2,index calculation. To calculate mean signal intensity in each region, the histogram of the raw MRI signals in each ROI was fitted to a Gaussian, and the mean of this distribution was used as the mean ROI signal.
Figure 1.

Location of regions of interest used for analysis. Green/horizontal lines=gray matter, blue/vertical lines=white matter, red/cross hatch lines=basal ganglia, and yellow/diagonal lines=corpus callosum (genu anterior and splenium posterior). Mean and standard deviation voxel counts for 18 subjects in the 5 primary regions of interest (Tables 2 and 3, Figures 5 and 6) were gray matter 12,822±3,354, white matter 6,574±70, basal ganglia 3,298±733, splenium of corpus callosum 1,424±602, and genu of corpus callosum 1,450±558. Voxel counts for all regions combined (Figures 3 and 4) 31,275±2,943. Voxel volume is 3.87 mm3. The color reproduction of this figure is available at the Journal of Cerebral Blood Flow and Metabolism journal online.
To ensure that the same ROIs were used across the different imaging sessions, the high-resolution 3D anatomic MRI and the diffusion-weighted MRI were coregistered within each imaging session. The key ROIs were initially defined on the baseline (normoxia) high-resolution 3D T1-weighted anatomic MRI (Amira, Visage Imaging, San Diego, CA, USA). These anatomic MRI from each of the three measurement sessions (baseline normoxia, 2 days hypoxia, and 7 days hypoxia) were then coregistered, which allowed us to determine the rotation matrix for coregistering MRI scans between each imaging session. The ROIs were then rotated using this rotation matrix, and aligned to the diffusion-weighted MRI data for each session. This approach of rotating only the ROI, and not the images ensured that measurements were only made from raw MRI data (which had not been rotated or smoothed) thus removing potential bias across imaging sessions.
T2 index
T2-weighted images were acquired from the diffusion data with the diffusion gradients off (b=0 s/mm2). To correct for changes in absolute MRI signal across imaging sessions, the signal for each ROI on T2-weighted images was normalized by the signal in pure cerebrospinal fluid (CSF) in lateral ventricles, thus generating an index of normalized T2 signal for each ROI.
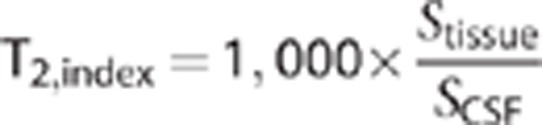 |
The relationship between T2,index and calculated T2 relaxation is shown for one subject in Figure 2. For white-matter and gray-matter regions, the index provides a good surrogate of the actual T2 relaxation time (R2=0.51, P<10−5), allowing comparison with prior studies.
Figure 2.

Scatter plot of T2,index versus T2 relaxation for one subject. Imaging sequence used spin-echo echo-planar imaging as described in text with b=0 s/mm2. For T2,index echo time (TE)=82.9 ms, for T2 relaxation calculation TE1=37.1 ms, TE2=82.9 ms. Voxels identified in image as gray matter, white matter, and cerebrospinal fluid (CSF) using FAST automated segmentation (FSL, Oxford, UK). Image resolution down sampled to 7 × 7 × 4.4 mm to improve signal-to-noise ratio (SNR). Solid line is best fit to gray-matter and white-matter voxels. Y=7.2X+113.9. R2=0.51 P<10−5.
Apparent Diffusion Coefficient
Apparent diffusion coefficient was calculated per pixel from the log slope of the signal attenuation between b=0 s/mm2 and b=1,000 s/mm2 images. Mean ADC was then calculated from the histogram fit for each ROI.
Correlation Analysis
We used the LLS, change in SaO2, change in hematocrit, and change in ETCO2 as parametric variables to address potential corollaries with the changes in ADC and T2 (between 2 days hypoxia and baseline normoxia).
Voxel-Based Analysis
The images were also analyzed using a voxel-based approach, to evaluate areas with signal differences related to AMS symptoms. For each subject in the AMS or no-AMS group, the ADC and T2,index images were transformed to Talairach space (AFNI software, NIMH, Bethesda, MD, USA), and voxels were down sampled to 4 mm3 isotropic and spatially smoothed (Gaussian filter, full width half max=5 mm). For each subject, the images from day 7 (no AMS symptoms) were subtracted from the images for day 2 (maximal AMS Symptoms) to create a map of ADC (or T2,index changes). These difference images were then averaged for each group. Voxels with very large, unphysiologic, values (>100) were treated as misregistration errors and ignored. The averaged map for the no-AMS group was subtracted from averaged map for the AMS group. In this final map, the voxels were colored by pixel intensity and superimposed on a grayscale background image of the average ADC (or T2,index) grayscale image from all the subjects.
Statistical Analysis
Data were analyzed with repeated measures ANOVA of our primary outcome variables (ADC, T2,index), with two grouping variables (AMS and no-AMS) and three measurement levels (normoxia, 2 days hypoxia, and 7 days hypoxia) (StatView 5.0.1, SAS Institute, Cary, NC, USA). Data were expressed as mean±s.d. Changes were significant at P<0.05 two tailed.
Results
Acute Mountain Sickness
To highlight any physiologic changes in ADC and T2,index that accompany the development and recovery of AMS symptoms, we only included subjects who were virtually asymptomatic (no-AMS group) or who developed unambiguous AMS (AMS group). Of the 18 subjects recruited into the study, 6 developed criteria for AMS (LLS ⩾5 and headache; 1M, 5F). A further eight subjects met the criteria for no AMS (no-AMS) (LLS ⩽2 or no headache; 5M, 3F). The remaining four subjects were characterized as intermediate, and were not included in further analysis.
All of the 14 subjects selected for analysis had measurements for the 3 repeated time points, with the exception of 2 missing data points; 2-day ADC and T2-weighted images corrupted in one subject (male, no-AMS group) and 7-day ADC and T2-weighted images corrupted in one subject (female, no-AMS group).
Hypoxia
After 2 days at 3,800 m altitude (PiO2=90 Torr), all subjects showed decreased arterial saturation (98.2±0.8% to 83.3±3.7%, P<0.0005) and increased ventilatory drive with reduced PETCO2 (38.5±3.1 Torr to 31.9±3.2 Torr, P<0.0005), consistent with prolonged hypoxia. The AMS group showed a slightly lower mean arterial saturation (82.2±5.2%) than the no-AMS group (84.1±2.1%), but the difference between groups was not significant. Hematocrit was increased in all subjects relative to normoxia (38±7% to 40±6%), but this did not reach significance.
After 7 days at altitude, all subjects showed moderate increases in arterial saturation relative to their 2-day values (83.3±3.7% to 87.1±2.5%, P<0.0005) in keeping with altitude acclimatization. There was no significant difference in arterial saturation between AMS groups. PETCO2 at 7 days further decreased relative to 2 days hypoxia (31.9±3.2 Torr to 30.6±2.3 Torr, P=NS), consistent with prolonged hypoxia, but this did not reach significance. Hematocrit showed further small increases in AMS and no-AMS groups relative to 2 day hypoxia (40±6% to 42±6%), but this too did not reach significance. Results are summarized in Table 1.
Table 1. Physiologic changes during hypoxia.
| Normoxia | Two days hypoxia | Seven days hypoxia | ||||
|---|---|---|---|---|---|---|
| no-AMS |
AMS |
no-AMS |
AMS |
no-AMS |
AMS |
|
| |
(n=8) |
(n=6) |
(n=8) |
(n=6) |
(n=8) |
(n=6) |
| SaO2 | 98.3±1.0 | 98.0±0.5 | 84.1±2.1 | 82.2±5.2 | 87.2±1.6 | 87.0±3.6 |
| 97.6–99.0 | 97.6–98.4 | 82.6–85.5 | 78.1–86.4 | 86.0–88.3 | 84.1–89.8 | |
| PETCO2 | 38.1±3.9 | 38.9±1.8 | 31.1±3.0 | 33.0±3.2 | 30.4±2.2 | 31.0±2.7 |
| 35.4–40.8 | 37.5–40.4 | 29.0–33.2 | 30.4–35.6 | 28.9–31.9 | 28.8–33.2 | |
| (n=5) | (n=5) | |||||
| Hct | 40±8 | 36±6 | 40±6 | 40±7 | 41±6 | 44±7 |
| 33–46 | 31–41 | 36–44 | 34–46 | 37–45 | 38–40 | |
Physiologic data during normoxia and after 2 days and 7 days sustained hypoxia at 3,800 m altitude (PiO2=90 Torr). Subjects were grouped by acute mountain sickness (AMS) from Lake Louise Scores (LLS) based on being virtually symptom free (no-AMS group) (LLS ⩽2, or no headache) or having unambiguous AMS symptoms (AMS group) (LLS⩾5, with headache). Data are mean±1 s.d., and 95% confidence limits. Units of arterial saturation (SaO2), in %saturation, end-tidal CO2 tension (PETCO2) in Torr, Hematocrit (Hct) in %.
Apparent Diffusion Coefficient and T2,index
Comparing changes between 2-day hypoxia versus normoxia in all ROI grouped together (putamen, globus pallidus, caudate, gray matter, white matter, splenium of corpus callosum, and genu of corpus callosum), there was a decrease in ADC relative to normoxia in the AMS group (ΔADC=−13.3±26.1 × 10−6 mm2/s), but an increase in ADC (ΔADC=+19.9±29.2 × 10−6 mm2/s) in the no-AMS group (P<0.05 between AMS groups after 2 days). After 7 days of hypoxia, the ADC change (relative to normoxia) remained elevated in the no-AMS group (ΔADC=+21.0±27.5 × 10−6 mm2/s). Apparent diffusion coefficient increased above normoxia levels in the AMS group too (ΔADC=+2.3±28.8 × 10−6 mm2/s) (P=NS between AMS groups after 7 days). During the 2-day to 7-day interval, although ADC increased in both AMS and no-AMS groups, there was a considerably larger increase in ADC in the AMS group. See Figure 3.
Figure 3.

Changes in apparent diffusion coefficient (ADC) for acute mountain sickness (AMS) and no-AMS groups across all cerebral regions. (A) Change in ADC between 2 days hypoxia and normoxia (2d-N) (at this time point, AMS subjects are symptomatic and no-AMS subjects are asymptomatic), (B) between 7 days hypoxia and normoxia (7d-N) (at this time point, all subjects are asymptomatic), (C) between 2 days and 7 days hypoxia (7d-2d) (shows changes occurring during hypoxia acclimatization period). The no-AMS group is characterized by increased ADC on both days (A, B), and little change in ADC during hypoxia acclimatization period. AMS group are characterized by reduced ADC at 2 days when symptomatic, and increased ADC at 7 days when asymptomatic. Large increase in ADC during hypoxia acclimatization period as symptoms abate. Data are mean changes. Error bar=1s.d. Significant differences in ADC between 2 days hypoxia and normoxia (P<0.05, main effect of AMS). At 7 days hypoxia, no significant differences in relative ADC (P=NS, main effect of AMS). During acclimatization period (2 days to 7 days hypoxia) changes in ADC were significant between groups (P<0.05, main effect of AMS).
For T2,index; after 2 days hypoxia, there was a decrease in T2,index relative to normoxia in the AMS group (ΔT2,index=−26.5±26.5), but an increase in the no-AMS group (ΔT2,index=+12.3±34.3) (P<0.05 between AMS groups after 2 days). After 7 days hypoxia, the decrease in T2,index in the AMS group persisted (ΔT2,index=−47.9±35.9), but values for the no-AMS group had almost completely normalized to normoxia levels for the no-AMS group (ΔT2,index=−6.9±31.6) (P<0.05 between AMS groups after 7 days). During the 2-day to 7-day interval, T2,index decreased equally in both AMS and no-AMS groups. See Figure 4.
Figure 4.

Changes in T2,index for acute mountain sickness (AMS) and no-AMS groups across all cerebral regions (A, B, and C are same time periods as Figure 1). The no-AMS group have increased T2,index at 2 days and reduced T2,index by day-7 (A, B). The AMS group have reduced T2,index at day-2 and further reduction in T2,index at day-7 (A, B). Both groups show similar reduction in T2,index during acclimatization to hypoxia from day-2 to day-7 despite differences in symptoms (C). Data are mean changes. Error bar=1s.d. Significant differences in ADC at both 2 days and 7 days hypoxia relative to normoxia (P<0.05, main effect of AMS).
The changes in ADC and T2,index were further analyzed by brain region. The ADC changes for basal ganglia (putamen, caudate, and globus pallidus), gray matter, and white matter followed the same trend described above. The ADC changes in corpus callosum showed a different pattern in the AMS group. In the splenium, there was an increase in ADC after 2 days hypoxia (rather than the decreases seen in other regions). After 7 days, ADC had increased further. In the genu, there was a larger decrease in ADC after 2 days hypoxia. Unlike other cerebral regions, the decreased ADC in the genu persisted after 7 days hypoxia (P<0.01 for region × AMS interaction). (Table 2; Figure 5). T2,index changes were also different in the genu of the corpus callosum; after 2 days hypoxia there was an initial decrease in T2,index (similar to other regions), but this decreased considerably more after 7 days hypoxia (P<0.0001 for region × AMS interaction) (Table 3; Figure 6).
Table 2. ADC changes during hypoxia.
| Normoxia | Two days hypoxia | Seven days hypoxia | ||||
|---|---|---|---|---|---|---|
| no-AMS |
AMS |
no-AMS |
AMS |
no-AMS |
AMS |
|
| |
(n=8) |
(n=6) |
(n=8) |
(n=6) |
(n=8) |
(n=6) |
| Basal ganglia | 681±21 | 685±33 | 693±20 | 677±41 | 695±18 | 691±25 |
| 667–696 | 659–711 | 679–707 | 644–709 | 683–708 | 671–712 | |
| Gray matter | 672±56 | 715±29 | 705±40 | 687±51 | 688±54 | 712±39 |
| 633–710 | 692–738 | 677–733 | 646–728 | 651–725 | 681–743 | |
| White matter | 669±48 | 709±51 | 684±37 | 704±35 | 684±33 | 725±55 |
| 636–703 | 668–750 | 659–709 | 676–732 | 661–706 | 680–769 | |
| CC splenium | 640±81 | 626±60 | 650±55 | 644±63 | 665±20 | 664±37 |
| 584–696 | 578–675 | 612–688 | 594–694 | 651–679 | 635–694 | |
| CC genu* | 675±50 | 783±118 | 721±88 | 731±48 | 725±45 | 729±62 |
| 640–710 | 688–877 | 660–782 | 693–769 | 694–756 | 679–788 | |
MRI, magnetic resonance imaging. MRI measures of apparent diffusion coefficient (ADC) × 10−6 mm2/s during normoxia and after 2 days and 7 days sustained hypoxia for five cerebral regions (see Figure 1 for details of regions, CC splenium and CC genu=splenium and genu of the corpus callosum). Data are mean±1 s.d., and 95% confidence limits. Data analyzed using ANOVA for repeated measures. *Significant ADC changes in genu of corpus callosum (P<0.05, for main effect of duration of hypoxia). Similar trend in basal ganglia (P=0.06). Other differences in ADC were not significant.
Figure 5.

Changes in apparent diffusion coefficient (ADC) for acute mountain sickness (AMS) and no-AMS groups across different cerebral regions (basal ganglia, gray matter, white matter, splenium of corpus callosum, genu of corpus callosum—see text for details. Panels (A), (B), and (C) are same time periods as Figure 1). The no-AMS group shows a broadly similar pattern in all regions with increased ADC at 2 days hypoxia relative to normoxia (A), which is still elevated at 7 days hypoxia (B). The AMS group show decreased ADC when symptomatic at 2 days hypoxia in basal ganglia, gray matter, and white matter (A), with normalization or increased ADC at 7 days when asymptomatic (B). Different pattern of ADC change is seen in the AMS group in corpus callosum: in the splenium (CC splen.) ADC is initially increased despite symptoms of AMS (A), which increases further at 7 days (B). In the genu (CC Genu), the ADC is initially reduced at 2 days hypoxia relative to normoxia, and shows no change by 7 days despite recovery of symptoms (B, C). Data are mean changes. Error bar=1s.d. Significant differences in ADC at both 2 days and 7 days hypoxia relative to normoxia (P<0.05, main effect of AMS), and between regions at 7 days (P<0.01, AMS × region interaction).
Table 3. T2,index changes during hypoxia.
| Normoxia | Two days hypoxia | Seven days hypoxia | ||||
|---|---|---|---|---|---|---|
| no-AMS |
AMS |
no-AMS |
AMS |
no-AMS |
AMS |
|
| |
(n=8) |
(n=6) |
(n=8) |
(n=6) |
(n=8) |
(n=6) |
| Basal ganglia | 272±44 | 254±34 | 266±57 | 242±13 | 250±38 | 243±22 |
| 241–303 | 227–281 | 227–305 | 232–253 | 224–276 | 225–261 | |
| Gray matter | 480±55 | 526±121 | 518±132 | 464±82 | 477±75 | 443±85 |
| 442–518 | 430–623 | 426–609 | 398–530 | 425–529 | 375–511 | |
| White matter* | 283±56 | 294±54 | 318±86 | 269±33 | 290±60 | 268±31 |
| 245–322 | 251–338 | 258–378 | 243–295 | 248–332 | 243–292 | |
| CC splenium | 327±38 | 317±50 | 340±93 | 299±28 | 333±47 | 288±24 |
| 301–354 | 277–357 | 276–405 | 277–322 | 300–366 | 269–307 | |
| CC genu* | 413±80 | 453±59 | 432±111 | 409±90 | 421±61 | 290±63 |
| 358–468 | 406–500 | 356–509 | 337–481 | 378–463 | 239–340 | |
MRI, magnetic resonance imaging; ROI, region of interest.
MRI measures of T2,index during normoxia and after 2 days and 7 days sustained hypoxia for five cerebral regions (index is quotient of T2 signal in each ROI and T2 signal in cerebrospinal fluid (CFS) in ventricles—see text for details. CC splenium and CC genu=splenium and genu of the corpus callosum). Data are mean±1 s.d., and 95% confidence limits. Data analyzed using ANOVA for repeated measures. *Significant T2 changes in genu of corpus callosum (P<0.05 for main effect of duration of hypoxia, and duration × AMS interaction), and in white matter (P<0.05 for duration × AMS interaction). Other differences in T2,index were not significant.
Figure 6.

Changes in T2,index for acute mountain sickness (AMS) and no-AMS groups for different cerebral regions (regions are same as Figure 3. Panels (A), (B), and (C) are same time periods as Figure 1). Broadly similar pattern in all regions except basal ganglia. The no-AMS group have increased T2,index at 2 days, which has returned to approximately normoxia apparent diffusion coefficient (ADC) levels by 7 days (A, B). The AMS group has reduced T2,index at 2 days and further reduction in T2,index at 7 days (A, B). Genu of the corpus callosum (CC Genu) shows especially large reductions in T2,index during 2-day to 7-day acclimatization period (C). In basal ganglia, the no-AMS group shows an initial decrease in T2,index at 2 days relative to normoxia (A). The AMS group shows an initial decrease in T2,index with no further reduction by 7 days. Error bar=1s.d. Significant differences in T2,index at both 2 days and 7 days hypoxia relative to normoxia (P<0.05, main effect of AMS), and between regions at 7 days (P<0.0005, main effect of region, and P<0.0001 AMS × region interaction).
Because of a technical failure with the Talairach transform with the T2,index images, only the ADC voxel-based maps are presented here. The spatial extent of the pattern of ADC change seen in Figure 3 is shown in maps in Figure 7. This showed a similar pattern of increased ADC with symptoms of AMS in regions of frontal, temporal, and parietal gray matter, and in basal ganglia. This pattern is also seen more diffusely in areas of white matter.
Figure 7.

Voxel-wise changes in apparent diffusion coefficient (ADC), showing voxels with a signal change corresponding to symptoms of acute mountain sickness (AMS). Color scale indicates ADC change between symptomatic (AMS group) and asymptomatic (no-AMS group) evaluated as the difference between ADC at 2 days hypoxia (when AMS subjects are symptomatic, no-AMS subject are asymptomatic) and 7 days hypoxia (when all subject are asymptomatic). Voxels with ADC change <10 × 10−6 s/mm2 (i.e., insignificant difference between AMS and no-AMS group) or ADC change >100 × 10−6 s/mm2 (i.e., artifactually large changes, likely because of misregistration subtraction errors) are not colored. Map indicates that basal ganglia, frontal gray matter, and regions of parietal and temporal cortex, as well as scattered areas of white matter in centrum semiovale show ADC changes that increase with AMS symptoms. The color reproduction of this figure is available at the Journal of Cerebral Blood Flow and Metabolism journal online.
When the ADC and T2 changes were analyzed for correlations with changes in other physiologic parameters during sustained hypoxia (Lake Louise AMS score, SaO2, hematocrit, and ETCO2), we observe a significant positive correlation between the change in Hct after 2 days hypoxia, and the T2,index changes in the basal ganglia (r=0.66, P=0.03). There were no other significant correlations between the physiologic changes and the T2,index change for any other brain region. Some regions did show a trend toward a negative correlation between T2,index and LLS: for gray matter r=−0.41, P=0.09; for white matter r=−0.42, P=0.08. In addition, there was a trend toward a positive correlation between the change in ETCO2 with 2 days sustained hypoxia, and the ADC change in the basal ganglia (r=0.42, P=0.08). There were no other significant correlations with the changes in ADC for any other brain region.
Discussion
The primary findings in the current study are different diffusion MRI characteristics in those subjects with symptoms of AMS and those without, with a direct correlation between ADC changes in the brain and symptoms of AMS. A decrease in ADC in gray matter, white matter, and basal ganglia is associated with symptoms of AMS at altitude. Conversely, an increase in ADC (either in subjects who are not susceptible to symptoms of AMS, or in susceptible subjects after subsequent acclimatization to altitude) is associated with an absence, or recovery from AMS symptoms. In addition, we observed an initial increase in T2,index in subjects without symptoms of AMS, but an initial decrease in T2,index in symptomatic subjects. After 7 days hypoxia, when all subjects were asymptomatic, T2,index was reduced below normoxia levels in everyone.
Edema or other increases in extracellular water are detectable on T2-weighted MRI as an increase in the MRI signal, because of the prolonged T2 relaxation time. Previous studies have used simple visual inspection to assess changes in T2-weighted signal,5, 6 but this is both subjective and lacks precision. For this study, we quantified changes in extracellular water from changes in the MRI signal intensity on T2-weighted images. To remove biases from shifts in the global MRI signal by gain differences across different scan sessions, we normalized the tissue signal by the CSF signal in lateral ventricles (CSF signal is not expected to change in hypoxic conditions, and varies only with differences in amplifier gain during MRI acquisition). This quotient is robust across repeated scan sessions and allows a more precise quantitation of changes in T2-weighted signal than visual observation, or uncorrected signals, and the normalized index equates well with true T2 relaxation. One shortcoming of our use of T2-weighted images and T2 index rather than a multiecho T2 relaxation arose when we did voxel-based analysis. The T2-weighted images did not transform well into Talairach space. We suspect that this is related to B1 sensitivity in the images. This was not an issue with the ADC images, as any signal biases present on both b=0 and b=1,000 images tend to cancel. We would expect a similar B1 insensitivity in true maps of T2 relaxation where biases present in images from multiple echoes will also tend to cancel out.
Prior studies in severe AMS3, 9 have shown that the sickest patients may have increased T2, particularly in the splenium of the corpus callosum. Thus, our finding of a reduction in T2 signal specifically in subjects with symptoms of AMS was unexpected. The reduction in T2 signal would suggest a decrease in measurable extracellular water compared with normoxia conditions. In ischemia, the earliest T2 effect is a decrease in the relaxation time. Grohn et al,10, 11 showed that this is because of a negative blood oxygenation level-dependent effect directly related to decreased perfusion (without significant increase in CMRO2), and suggested that in this context, reduced T2 might indicate a period of ischemia in which tissue injury is fully reversible. In the current studies, perfusion was increased during sustained hypoxia,12 and the arterial and venous compartments contain increased deoxyhemoglobin, so blood oxygenation level-dependent changes will add to the observed reduction in T2,index. From Zhao et al,13 for a hematocrit of 0.44 the transverse relaxation rate, R2, can be approximated in terms of oxygen saturation, Y, as:
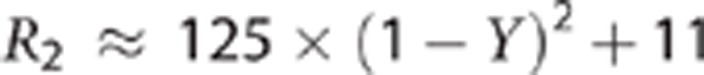 |
Since R2=1/T2, from this we can model the expected T2 change in gray matter because of the decrease in intravascular oxygen saturation of hemoglobin. Assuming a voxel is composed of 4% blood (split 1% arterial and 3% venous), and that the T2 of the remaining 96% cellular material is not affected by the change in oxygen saturation of hemoglobin, we can approximate the total signal as:
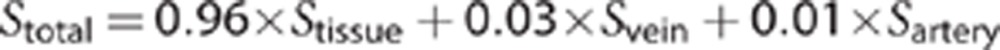 |
For a change in SaO2 from 98% (normoxia) to 83% (hypoxia), and a corresponding SvO2 change from 83% (normoxia) to 49% (hypoxia),12 we would expect a decrease in T2 in a voxel of gray matter of ∼0.7% if the T2 was affected only by intravascular desaturation. Since the observed T2 changes in gray matter were 9% increase in non-AMS subjects and 12% decrease in AMS sufferers, the impact of the altered oxygen saturation of hemoglobin alone on the T2 is small, and other factors must be considered—in particular, changes in extracellular water. One confound that needs to be considered is the hydration status of the subjects, and whether a mild global dehydration is contributing to the apparent reduction in cerebral extracellular water. Unlike prior studies of ADC (and T2) in a hypoxic chamber,5, 6, 7 the current study was conducted at high altitude. The prevailing conditions were those of the ambient environment, with relative humidity typically <5% and high daytime temperatures. All subjects with AMS symptoms experienced headache, nausea, and mild-to-moderate anorexia; thus, it is possible that subjects with AMS may have been less able to maintain adequate hydration status than subjects without symptoms. Hematocrit levels were elevated in all subjects consistent with a normal erythropoietic response to hypoxia. The elevated hematocrit may also represent reduced plasma volume from dehydration. We noted that hematocrit was more elevated in subjects with AMS after 7 days at altitude than those without symptoms (although this difference was not significant). Previous studies conducted in hypoxia chambers under less extreme environmental conditions have failed to find any changes in T2 with hypoxia,5, 6 thus the high altitude environment needs to be considered when interpreting these extracellular fluid shifts.
Of the regions studied here, basal ganglia and gray matter are especially hypoxia sensitive, with white matter being more hypoxia resistant.14 This regional hypoxia sensitivity did not predict the magnitude of the changes in ADC or T2 index. The ADC changes in the corpus callosum are however intriguing. The splenium has previously been observed to have a unique response to sustained hypoxia, and HACE has a predilection for this region.3 In the present study, we noted that the ADC changes in the splenium and genu of the corpus callosum differed from the responses in other cerebral areas. In the genu, there is a larger decrease in ADC during acute hypoxia, than in other cerebral regions in symptomatic subjects. This exaggerated decrease in ADC was also described during acute studies.7 After acclimatization the ADC remains low and there is no subsequent rise in ADC as symptoms abate. Conversely, the splenium does not show the usual pattern of decreased ADC and restricted diffusion in symptomatic subjects, but instead shows a pattern of increased ADC indicating less restriction to diffusion when symptoms are present. After 7 days hypoxia, when symptoms have resolved, ADC in the splenium remains elevated. This different pattern may represent a different mode of response in the splenium (increased rather than decreased ADC), or possibly a similar response to other regions, but at a faster timescale. Of note, the magnitude of the increase in ADC seen in the splenium between day-2 and day-7 in symptomatic subjects is similar to that seen in other regions, which would favor a typical response, but a very early decrease (before we made our 2-day measurements) and subsequent increase in ADC during sustained hypoxia. Our speculation of a typical, but faster response in the splenium is further supported by acute ADC measurements,7 where earlier ADC measurements in symptomatic subjects (at 16 hours of hypoxia) did find small decreases in ADC. Previous reports of MRI in severe AMS and HACE describe a reversible vasogenic edema in the splenium.3, 15 This correlates with our findings in the current study that during AMS symptoms, the only cerebral area with a vasogenic pattern of increased ADC is the splenium of the corpus callosum.
In the basal ganglia (but not other cerebral regions), the change in T2,index with 2 days sustained hypoxia was positively correlated with a change in hematocrit. The mechanisms underpinning this observation remain unclear. It does not relate to the usual intravascular relationship of shorter T2 times as Hct increases.13 We speculate that there could be a local fluid shift between intracellular and extracellular compartments in the basal ganglia during hypoxia, with increased extracellular fluid tending to increase local T2. This observation warrants further investigation. The trend toward a negative correlation between AMS score and T2,index is also intriguing, with shorter T2,index times seen in those subjects with the most AMS symptoms. As discussed above, since nausea and poor appetite are significant symptoms of AMS, the AMS group as a whole would be expected to eat and drink less. Combined with low ambient humidity, a greater propensity to mild dehydration in the AMS group is very likely under these conditions. These observations become significant when the intermediate group is omitted (gray matter: R=−0.55, P=0.04; white matter R=−0.57, P=0.03). Thus, analyzing subjects as two polarized groups (AMS/no-AMS) improves our ability to detect changes that may be associated with physiologic differences during sustained hypoxia.
Despite being a common condition, the pathophysiologic mechanisms of AMS remain poorly understood. A leading hypothesis is that increased intracranial pressure because of brain swelling is the cause of the symptoms of AMS.16 In this hypothesis, the variability between individuals is because of differences in the capacity of the CSF system to accommodate brain swelling, with a smaller capacity for changes in CSF volume producing a ‘stiffer' less-compliant brain and a greater risk of increased intracranial pressure for the same level of brain swelling. The first step down the path to brain swelling is hypothesized to be increased cerebral blood flow and associated increased cerebral blood volume, which directly produces brain swelling—leading to increased capillary pressure and cerebral edema. We previously found that increased cerebral blood flow is a common outcome of ascent to high altitude,17 as is cerebral swelling and reduced CSF volume,18 but neither was associated with a greater propensity to develop AMS. An alternate hypothesis for brain swelling and symptoms of AMS may relate to compromised cerebral energy status. In early experiments with mitochondrial preparations, Wilson et al19 found that the oxygen metabolic rate could be maintained even at very low PO2 values. However, even at PO2 levels well above these metabolic limits, the phosphorylation potential gradually deteriorated as PO2 was decreased. In a study in dogs, Nioka et al20 measured the phosphorylation potential for different levels of hypoxia. Applying a model by Buxton21 for estimating tissue PtO2 in cerebral tissues to Nioka's data, we observe that PtO2 had reduced to 27% of normal when the phosphorylation potential reduced to 75% of normal (RB Buxton, personal communication). At 90 Torr PiO2 (as in the current study) under acute conditions, we calculated reductions in human cerebral tissue PtO2 to ∼40% of normal;22 suggesting that for the degree of hypoxia encountered in the current study, subjects are approaching the level at which phosphorylation potential can be compromised.
Restricted diffusion in cerebral tissues with reduced ADC is an early change during cerebral ischemia, and precedes T2 increases by 6 to 48 hours.23 The hypothesis that hyperacute decrease in ADC is associated with cytotoxic edema24 has been supported by a number of studies, but the degree to which the ADC change involves intracellular or extracellular water remains controversial.25, 26 Several studies indicate a link between altered ADC and cellular energy status; results from 31P phosphorous spectroscopy studies link reduced ADC to a lowered phosphorylation potential.27 In experimental models, there is a decrease in the energy-dependent Na+-K+ ATPase activity that maintains ionic gradients within minutes after the onset of ischemia.28 Decreased diffusion may also be reproduced by intraparenchymal infusions of ouabain, an inhibitor of Na+-K+ ATPase, or by infusions of glutamate or N-methyl-aspartate into the brain,29 which activate N-methyl-aspartate receptors, mediators of ischemic neurotoxicity. Reversal of ischemia sufficiently early causes reversal of the ADC changes that is not greater than a threshold value and prevents infarction.30 Apparent diffusion coefficient does not decrease until oxygen delivery decreases below levels critical for maintenance of Na+-K+ ATPase activity.31 Reversible decreases in ADC are seen in the tissues immediately surrounding an infarct (the penumbra)23 and these represent areas of brain in which there is temporary interruption of Na+-K+ ATPase, but not sufficient to cause Na+-K+ ATPase failure and disrupted ionic homeostasis.32 We speculate that a similar mechanism seen in the penumbra underlies the reversible ADC changes seen here during symptomatic hypoxia.
The exact biophysical mechanism linking cellular energy status with ADC remains uncertain. One possible mechanism to consider is intracellular streaming. Wheatley33 argued that an intracellular circulatory system is necessary for metabolic function in most cells and that simple diffusion cannot provide adequate intracellular transport at the molecular level. This motion perfuses the interior of the cell by streaming of the fluid compartment of the cytoplasm and is energy dependent. During disruptions to cellular energy status this energy-dependent facilitated transport is lost, and ADC will be reduced as water motion changes from the high ADC of facilitated transport, to a lower value representing simple Brownian diffusion.
No direct assessment of causality was undertaken as part of this study; thus, the precise temporal relationship between AMS symptoms and ADC changes, and the mechanisms underpinning this relationship, still need to be established. However, we speculate that the changes in cellular microarchitecture that result in restricted diffusion are responsible for the development of AMS symptoms. Our findings of ADC changes that track symptoms of AMS support compromised cellular energy as the critical factor leading to AMS rather than a hyperdynamic circulation. We have previously observed increased CMRO2 during sustained hypoxia in the same subject population as the current study.12 Thus, the difference between AMS-susceptible and AMS-resistant subjects may lie in the degree to which their cerebral phosphorylation potential is impacted by the hypoxia.
Acknowledgments
The authors are grateful to Richard B Buxton for helpful discussions.
The authors declare no conflict of interest.
Footnotes
This study was supported by National Institutes of Health Grants R01-NS053934 and R21-NS075812 (DJD).
References
- Basnyat B, Murdoch DR. High-altitude illness. Lancet. 2003;361:1967–1974. doi: 10.1016/S0140-6736(03)13591-X. [DOI] [PubMed] [Google Scholar]
- Hackett PH, Roach RC. High-altitude illness. N Engl J Med. 2001;345:107–114. doi: 10.1056/NEJM200107123450206. [DOI] [PubMed] [Google Scholar]
- Hackett PH, Yarnell PR, Hill R, Reynard K, Heit J, McCormick J. High-altitude cerebral edema evaluated with magnetic resonance imaging: clinical correlation and pathophysiology. JAMA. 1998;280:1920–1925. doi: 10.1001/jama.280.22.1920. [DOI] [PubMed] [Google Scholar]
- Matsuzawa Y, Kobvayashi T, Fujimoto K, Schinozaki S.Cerebral edema in acute mountain sicknessIn: Reeves JT, Sekiguchi M, (eds). High-Altitude Medicine Shinshu University Matsumoto, Japan; 1992300–304. [Google Scholar]
- Fischer R, Vollmar C, Thiere M, Born C, Leitl M, Pfluger T, et al. No evidence of cerebral oedema in severe acute mountain sickness. Cephalalgia. 2004;24:66–71. doi: 10.1111/j.1468-2982.2004.00619.x. [DOI] [PubMed] [Google Scholar]
- Schoonman GG, Sandor PS, Nirkko AC, Lange T, Jaermann T, Dydak U, et al. Hypoxia-induced acute mountain sickness is associated with intracellular cerebral edema: a 3 T magnetic resonance imaging study. J Cereb Blood Flow Metab. 2007;28:198–206. doi: 10.1038/sj.jcbfm.9600513. [DOI] [PubMed] [Google Scholar]
- Kallenberg K, Bailey DM, Christ S, Mohr A, Roukens R, Menold E, et al. Magnetic resonance imaging evidence of cytotoxic cerebral edema in acute mountain sickness. J Cereb Blood Flow Metab. 2007;27:1064–1071. doi: 10.1038/sj.jcbfm.9600404. [DOI] [PubMed] [Google Scholar]
- Roach RC, Bärtsch P, Oelz O, Hackett PH.The Lake Louise acute mountain sickness scoring systemIn: Sutton JR, Houston CS, Coates G, (eds). Hypoxia and Mountain Medicine Queen City Printers Inc Burlington, VT; 1993272–274. [Google Scholar]
- Levine BD, Yoshimura K, Kobayashi T, Fukushima M, Shibamoto T, Ueda G. Dexamethasone in the treatment of acute mountain sickness. N Engl J Med. 1989;321:1707–1713. doi: 10.1056/NEJM198912213212504. [DOI] [PubMed] [Google Scholar]
- Grohn OH, Kettunen MI, Penttonen M, Oja JM, van Zijl PC, Kauppinen RA. Graded reduction of cerebral blood flow in rat as detected by the nuclear magnetic resonance relaxation time T2: a theoretical and experimental approach. J Cereb Blood Flow Metab. 2000;20:316–326. doi: 10.1097/00004647-200002000-00013. [DOI] [PubMed] [Google Scholar]
- Grohn OH, Lukkarinen JA, Oja JM, van Zijl PC, Ulatowski JA, Traystman RJ, et al. Noninvasive detection of cerebral hypoperfusion and reversible ischemia from reductions in the magnetic resonance imaging relaxation time, T2. J Cereb Blood Flow Metab. 1998;18:911–920. doi: 10.1097/00004647-199808000-00012. [DOI] [PubMed] [Google Scholar]
- Smith ZM, Krizay E, Guo J, Shin DD, Scadeng M, Dubowitz DJ.Sustained high-altitude hypoxia increases cerebral oxygen metabolism J Appl Physiol 2012. doi: 10.1152/japplphysiol.00703.2012(e-pub ahead of print). [DOI] [PMC free article] [PubMed]
- Zhao JM, Clingman CS, Narvainen MJ, Kauppinen RA, van Zijl PC. Oxygenation and hematocrit dependence of transverse relaxation rates of blood at 3T. Magn Reson Med. 2007;58:592–597. doi: 10.1002/mrm.21342. [DOI] [PubMed] [Google Scholar]
- Dubowitz DJ, Bluml S, Arcinue E, Dietrich RB. MR of hypoxic encephalopathy in children after near drowning: correlation with quantitative proton MR spectroscopy and clinical outcome. AJNR Am J Neuroradiol. 1998;19:1617–1627. [PMC free article] [PubMed] [Google Scholar]
- Wong SH, Turner N, Birchall D, Walls TJ, English P, Schmid ML. Reversible abnormalities of DWI in high-altitude cerebral edema. Neurology. 2004;62:335–336. doi: 10.1212/01.wnl.0000103440.64964.86. [DOI] [PubMed] [Google Scholar]
- Krasney JA. A neurogenic basis for acute altitude illness. Med Sci Sport Exer. 1994;26:195–208. doi: 10.1249/00005768-199402000-00010. [DOI] [PubMed] [Google Scholar]
- Dyer EA, Hopkins SR, Perthen JE, Buxton RB, Dubowitz DJ. Regional cerebral blood flow during acute hypoxia in individuals susceptible to acute mountain sickness. Respir Physiol Neurobiol. 2008;160:267–276. doi: 10.1016/j.resp.2007.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubowitz DJ, Dyer EA, Theilmann RJ, Buxton RB, Hopkins SR. Early brain swelling in acute hypoxia. J Appl Physiol. 2009;107:244–252. doi: 10.1152/japplphysiol.90349.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson DF, Erecinska M, Drown C, Silver IA. The oxygen dependence of cellular energy metabolism. Arch Biochem Biophys. 1979;195:485–493. doi: 10.1016/0003-9861(79)90375-8. [DOI] [PubMed] [Google Scholar]
- Nioka S, Smith DS, Chance B, Subramanian HV, Butler S, Katzenberg M. Oxidative phosphorylation system during steady-state hypoxia in the dog brain. J Appl Physiol. 1990;68:2527–2535. doi: 10.1152/jappl.1990.68.6.2527. [DOI] [PubMed] [Google Scholar]
- Buxton RB. Interpreting oxygenation-based neuroimaging signals: the importance and the challenge of understanding brain oxygen metabolism. Front Neuroenerg. 2010;2:1–16. doi: 10.3389/fnene.2010.00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith ZM, Hunt JS, Li E, Guo J, Shinn DD, Buxton RB, et al. Elevated CO2 mitigates the rise in CMRO2 during acute hypoxia and improves cerebral tissue oxygenation. Proc Int Soc Magn Res Med. 2011;1:767. [Google Scholar]
- Warach S, Gaa J, Siewert B, Wielopolski P, Edelman RR. Acute human stroke studied by whole brain echo planar diffusion-weighted magnetic resonance imaging. Ann Neurol. 1995;37:231–241. doi: 10.1002/ana.410370214. [DOI] [PubMed] [Google Scholar]
- Moseley ME, Kucharczyk J, Mintorovitch J, Cohen Y, Kurhanewicz J, Derugin N, et al. Diffusion-weighted MR imaging of acute stroke: correlation with T2-weighted and magnetic susceptibility-enhanced MR imaging in cats. AJNR Am J Neuroradiol. 1990;11:423–429. [PMC free article] [PubMed] [Google Scholar]
- Ackerman JJ, Neil JJ. The use of MR-detectable reporter molecules and ions to evaluate diffusion in normal and ischemic brain. NMR Biomed. 2010;23:725–733. doi: 10.1002/nbm.1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duong TQ, Ackerman JJH, Ying HS, Neil JJ. Evaluation of extra- and intracellular apparent diffusion in normal and globally ischemic rat brain via 19F NMR. Magn Reson Med. 1998;40:1–13. doi: 10.1002/mrm.1910400102. [DOI] [PubMed] [Google Scholar]
- Thornton JS, Ordidge RJ, Penrice J, Cady EB, Amess PN, Punwani S, et al. Temporal and anatomical variations of brain water apparent diffusion coefficient in perinatal cerebral hypoxic-ischemic injury: relationships to cerebral energy metabolism. Magn Reson Med. 1998;39:920–927. doi: 10.1002/mrm.1910390609. [DOI] [PubMed] [Google Scholar]
- Mintorovitch J, Baker LL, Yang GY, Shimizu H, Weinstein PR, Moseley ME, et al. Diffusion-weighted hyperintensity of early cerebral ischemia: correlation with brain water content and ATPase activity. Proc Int Soc Magn Res Med. 1991;10:329. [Google Scholar]
- Benveniste H, Hedlund LW, Johnson GA. Mechanism of detection of acute cerebral ischemia in rats by diffusion-weighted magnetic resonance microscopy. Stroke. 1992;23:746–754. doi: 10.1161/01.str.23.5.746. [DOI] [PubMed] [Google Scholar]
- Mintorovitch J, Moseley ME, Chileuitt L, Shimizu H, Cohen Y, Weinstein PR. Comparison of diffusion- and T2-weighted MRI for the early detection of cerebral ischemia and reperfusion in rats. Magn Reson Med. 1991;18:39–50. doi: 10.1002/mrm.1910180106. [DOI] [PubMed] [Google Scholar]
- Busza AL, Allen KL, King MD, van Bruggen N, Williams SR, Gadian DG. Diffusion-weighted imaging studies of cerebral ischemia in gerbils. Potential relevance to energy failure. Stroke. 1992;23:1602–1612. doi: 10.1161/01.str.23.11.1602. [DOI] [PubMed] [Google Scholar]
- Astrup J, Siesjo BK, Symon L. Thresholds in cerebral ischemia—the ischemic penumbra. Stroke. 1981;12:723–725. doi: 10.1161/01.str.12.6.723. [DOI] [PubMed] [Google Scholar]
- Wheatley DN. Mini-review. On the possible importance of an intracellular circulation. Life Sci. 1985;36:299–307. doi: 10.1016/0024-3205(85)90114-6. [DOI] [PubMed] [Google Scholar]