

Abstract
Macroautophagy is an essential cellular pathway mediating the lysosomal degradation of defective organelles, long-lived proteins and a variety of protein aggregates. Similar to other intracellular trafficking pathways, macroautophagy involves a complex sequence of membrane remodeling and trafficking events. These include the biogenesis of autophagosomes (APs), which engulf portions of cytoplasm at specific subcellular locations, and their subsequent maturation into autophagolysosomes through fusion with the endo-lysosomal compartment. Although the formation and maturation of APs are controlled by molecular reactions occurring at the membrane-cytosol interface, little is known about the role of lipids and their metabolizing enzymes in this process. Historically dominated by studies on class III phosphatidylinositol 3-kinase (PI3K) (also known as Vps34), its product PI3P, as well as on the lipidation of Atg8/LC3-like proteins, this area of research has recently expanded, implicating a variety of other lipids, such as phosphatidic acid and diacylglycerol, and their metabolizing enzymes in macroautophagy. This review summarizes this progress and highlights the role of specific lipids in the various steps of macroautophagy, including the signaling processes underlying macroautophagy initiation, AP biogenesis and maturation.
Introduction
Autophagy pathways play a critical role in physiology, where their main function is to protect cells or organisms against starvation, enabling them to recycle nutrients from digested organelles and macromolecules at times of nutrient scarcity as well as to ensure cell homeostasis by removing damaged organelles and aberrantly folded proteins. Accordingly, perturbation of autophagy has been linked to aging and various disorders, including cancer, neurodegeneration, myopathies, liver and heart diseases [1, 2]. While there are three different types of autophagy (macroautophagy, microautophagy and chaperone-mediated autophagy) (Figure 1), macroautophagy (hereafter referred to as autophagy) has received the most attention. Our mechanistic understanding of the autophagy process was greatly enhanced by the discovery of approximately 30 yeast autophagy genes (i.e., “Atg genes”), most of which are conserved in mammals [3, 4]. The functional characterization of these autophagy genes in various systems has established that the formation of APs requires at least four major protein complexes: (i) the Atg1/Unc-51-like kinase (ULK) complex, which represents a key signaling intermediate primarily integrating inputs from the target of rapamycin (TOR); (ii) the autophagy-specific class III PI3K (Vps34) complexes, which produce a pool of phosphatidylinositol-3-phosphate (PI3P) that is important for AP formation and maturation; (iii) the transmembrane protein Atg9/mAtg9 and its trafficking machinery allowing for membrane addition and retrieval to and from sites of AP biogenesis; and (iv) the ubiquitin-like proteins Atg12 and Atg8/LC3 and their conjugation machinery ultimately leading to the lipidation of Atg8/LC3 with phosphatidylethanolamine (PE), a process required for AP formation and closure [3-7]. These protein complexes largely operate at the interface between the cytosol and various membranous compartments in the cell, which not only include the pre-autophagosomal structures and the AP, but also the endoplasmic reticulum (ER), the mitochondria, the plasma membrane, the Golgi apparatus and the endo-lysosomal system [6, 7] (Figure 1). Thus, although no “Atg” genes reported so far appear to encode lipid enzymes (perhaps with the exception of Atg15, a putative lipase [8]), lipids are increasingly implicated in the control of the biochemical processes and membrane remodeling underlying the biogenesis of APs and, more generally, the autophagy process (Figure 1).
Figure 1. Lipids and their metabolizing enzymes in the regulation of mammalian autophagy.
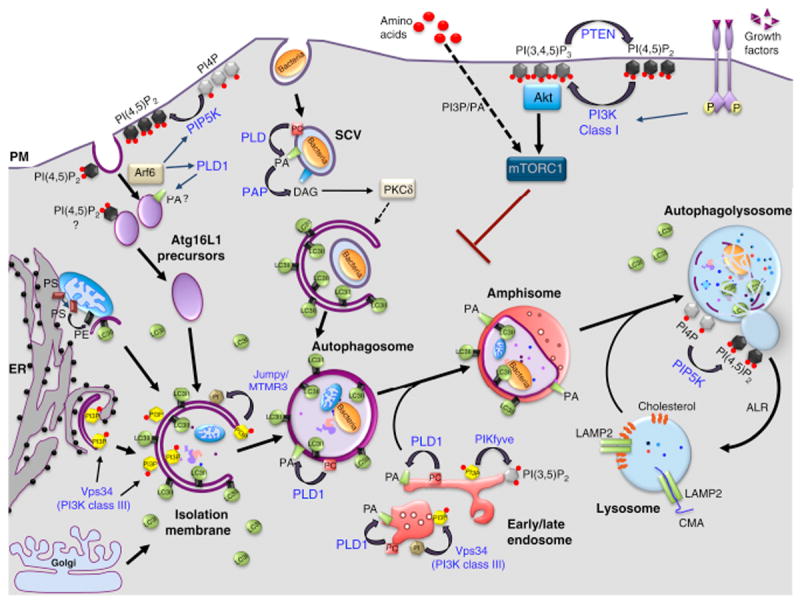
Macroautophagy begins by the formation of an isolation membrane (IM), which sequesters cytoplasmic substrates and seals to form autophagosomes (APs). APs undergo a maturation process through fusion with early and late endosomes, forming amphisomes. These hybrid organelles eventually fuse with lysosomes (autophagolysosomes, AL), where the degradation of sequestered material occurs. At the end of this process, ALs give rise to lysosomes through a recycling process called autophagic lysosome reformation (ARL). Several sources of membrane have been proposed for the IM: endoplasmic reticulum (ER), plasma membrane (PM), mitochondria (M) and Golgi apparatus (GA). A popular model posits that APs are nucleated at specialized sites of the ER referred to as “omegasomes” (Om), which are generated through a Vps34/PI3P-dependent mechanism and disengage to form the IM. Plasma membrane-derived Atg16L-positive vesicles can also contribute to the formation of IMs after their homotypic fusion. The small GTPase Arf6 modulates this process by activating two lipid enzymes, PLD1 and PIP5K, which produce PA and PI(4,5)P2, respectively. LC3 family members are lapidated through a covalent bond to phosphatidylethanolamine (PE) on the IM and APs. The pool of LC3-like molecules located on the outer membrane is released before APs fuse with the lysosomes, while the one located in the inner membrane is degraded alongside the AL cargoes. Other lipids (black) and their corresponding metabolizing enzymes (blue) are shown in the figure, based on their implication in starvation-induce autophagy or non-canonical forms of autophagy, such as that induced by Salmonella. Lysosomes are also essential for chaperone-mediated autophagy (CMA) and microautophagy (not shown on the figure). In CMA, proteins harboring the peptide motif KFERQ are selectively recognized by a cytosolic chaperone, Hsc70, targeting these proteins to the lysosomal membrane, where they can be internalized for degradation through the formation of a complex with LAMP2. In contrast, microautophagy involves the direct engulfment of cytoplasmic material, including proteins, into lysosomes through mechanisms involving the formation of intraluminal vesicles. SCV, Salmonella containing vacuole.
Lipids and lipid metabolizing enzymes mediate the autophagy process by controlling at least four fundamental aspects (Figure 1 and 2). First, they regulate signaling cascades converging onto the mammalian TOR (mTOR) pathway, which, in turn, negatively regulates the initiation of autophagy (Figure 2A). Central to this signaling cascade are class I PI3Ks and their product, phosphatidylinositol-3,4,5-trisphosphate [PI(3,4,5)P3] (Figure 1 and 2B) [9]. Second, lipids act as membrane-bound localized signals controlling membrane dynamics by specifically recruiting cytosolic protein effectors that mediate membrane deformation, expansion and vesicle transport (Figure 2B). The prototypical example of this regulation is PI3P, which controls the biogenesis and maturation of APs through this mechanism (Figure 1 and 2B) [5, 10]. Third, covalent binding of amine-containing phospholipids, such as PE, to Atg8/LC3 family members confers a unique mode of regulation by stably anchoring these critical factors to the membrane of phagophores that mediate their elongation and ultimately, their closure (Figure 2C) [3]. Finally, lipids can control membrane dynamics by directly affecting the physicochemical properties of lipid bilayers independently of protein effectors. Examples of this regulation include cone-shape lipids, such as phosphatidic acid (PA), which prefers or induces negative curvature or cholesterol, which promotes or stabilizes a liquid ordered phase within the bilayers (Figure 2D) [11]. In this review, we summarize our understanding of the roles of lipids in the control of autophagy with an emphasis on phosphoinositides and their metabolizing enzymes as well as the role of other, understudied lipids or lipid families, such as PA, diacyglycerol (DAG) and sphingolipids.
Figure 2. Modes of action of lipids.
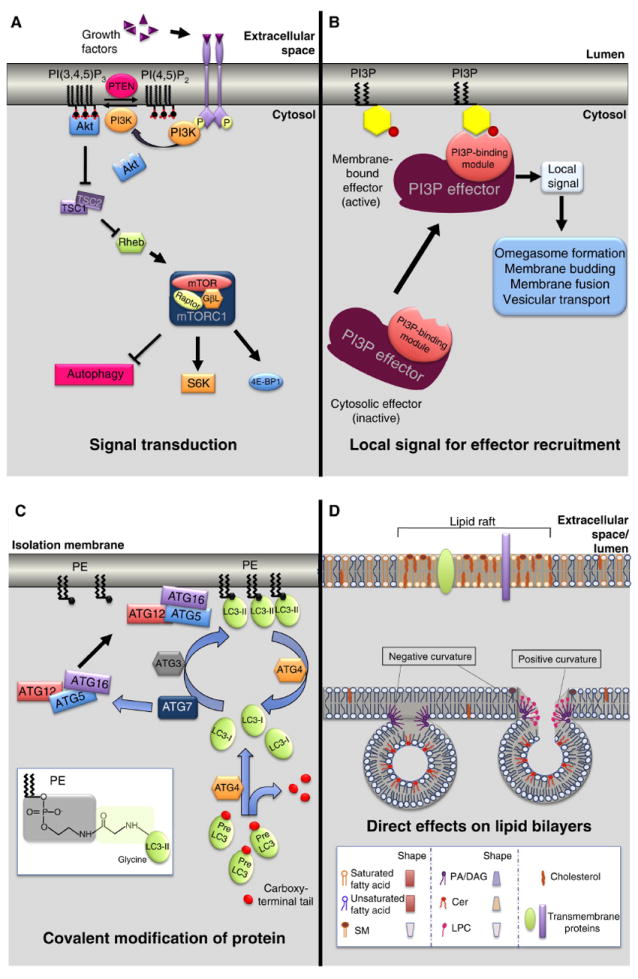
(A) Lipids mediating signal transduction processes. Class I PI3Ks and their product PI(3,4,5)P3 play a central role in mediating the signaling cascade of mTORC1 pathway. Growth factor-activated receptors stimulate PI3K to phosphorylate PI(4,5)P2 in order to produce PI(3,4,5)P3, which activates the Akt signaling pathway. Akt blocks the inhibitory effects of the TSC1/TSC2 complex on the small GTPase Rheb. Activated Rheb, in turn, promotes TORC1 complex signaling.
(B) Lipids mediating the local recruitment of effectors to membranes. PI3P acts as membrane-bound localized signal controlling the assembly of protein scaffolds at the membrane-cytosol interface mediating AP biogenesis through membrane deformation and expansion as well as perhaps AP transport.
(C) Lipids mediating covalent modification of proteins. The Atg8/LC3 family is stably anchored to the membrane of phagophores through a covalent binding to PE. This reaction is mediated by a cohort of proteins (ATG3, ATG4, ATG7, ATG10, ATG12, ATG5) that act as a ubiquitin-like conjugation system. Atg8/LC3 lipidation is critical to mediate isolation membrane elongation and AP closure.
(D) Lipids directly affecting the physicochemical properties of lipid bilayers. Examples of this regulation include the lipids rafts (top): area of the membrane enriched in cholesterol, sphingomyelin (SM) and phospholipids harboring mostly saturated fatty acyl chains which stabilize a liquid ordered phase within the bilayers. For instance, cholesterol-enriched microdomains have been shown to play a role in chaperone-mediated autophagy. Other examples are the cone-shape lipids, such as phosphatidic acid (PA), diacylglycerol (DAG) and ceramide (Cer), which prefer or induce membrane curvature facilitating the budding or fusion of vesicles. Inverted cone shape lipids, such as lysophosphatidylcholine (LPC), favor instead positive curvature.
Phosphoinositides are master regulators of autophagy
Phosphoinositides are a family of seven phosphorylated derivatives of PI that mediate fundamental aspects of cell physiology by controlling the cytosol-membrane interface of most subcellular compartments [12]. A variety of lipid kinases and phosphatases mediate the generation and interconversion of phosphoinositides through phosphorylation/dephosphorylation of the inositol group at three different positions: 3’, 4’ and 5’. Fundamental features of these regulatory lipids are their low abundance, their high turnover (due essentially to the phosphates decorating the inositol head group) and their differential subcellular localization in cells [12]. For instance PI3P is enriched in endosomes and in autophagic structures, while PI(4,5)P2 is concentrated at the plasma membrane [10, 12, 13].
Phosphoinositides exert their cellular actions primarily by recruiting to membranes protein effectors harboring a variety of well-defined protein modules or unstructured peptide motifs that recognize the head groups of these lipids with high selectivity. Electrostatic interaction of the phosphate groups with basic residues within these modules or motifs plays a critical role, although other types of interaction may also be involved [12]. Both PI3P and PI(3,4,5)P3 have been extensively studied in the context of autophagy, although other phosphoinositides have also been implicated and are discussed below.
The Class I PI3K pathway, PI(3,4,5)P3 and mTOR regulation
TOR is a protein kinase that plays a central role in the control of the balance between growth and starvation [9]. Generally, TOR is active in the presence of nutrients, driving growth and anabolic processes, while suppressing autophagy. Conversely, nutrient scarcity inactivates TOR, slowing growth and anabolism as well as stimulating “recycling” programs through autophagy-mediated degradation of proteins and organelles. The internal production of these metabolites ensures cell and organismal survival during prolonged starvation [9]. Importantly, in higher eukaryotes such as mammals, feeding leads to a variety of physiological responses, including the release of insulin, which, in turn, stimulates a class I PI3K-dependent signaling cascade in peripheral tissues that activates mTOR complex 1 (mTORC1) and suppresses autophagy [9, 14]. Conversion of PI(4,5)P2 to PI(3,4,5)P3 by Class I PI3Ks in response to insulin is thus the initial trigger for this signaling cascade (Figure 1, 3 and 4). Nutrients (e.g., amino acids) per se are potent activators of the mTORC1 pathway and thus suppressors of autophagy, but the underlying signaling process at the cellular level does not directly involve the class I PI3K pathway [9]. Conversely, the immunosuppressant macrolide, rapamycin, is a potent activator of autophagy because it inhibits the ability of mTORC1 (i.e., Raptor-bound), but not mTORC2 (i.e., Rictor-bound), to phosphorylate substrates, such as S6 kinase 1 (S6K1) and eIF4E-binding protein 1 (4E-BP1), which control the rate of protein synthesis [6, 9, 14].
Figure 3. Lipid and lipid enzymes implicated in autophagy.
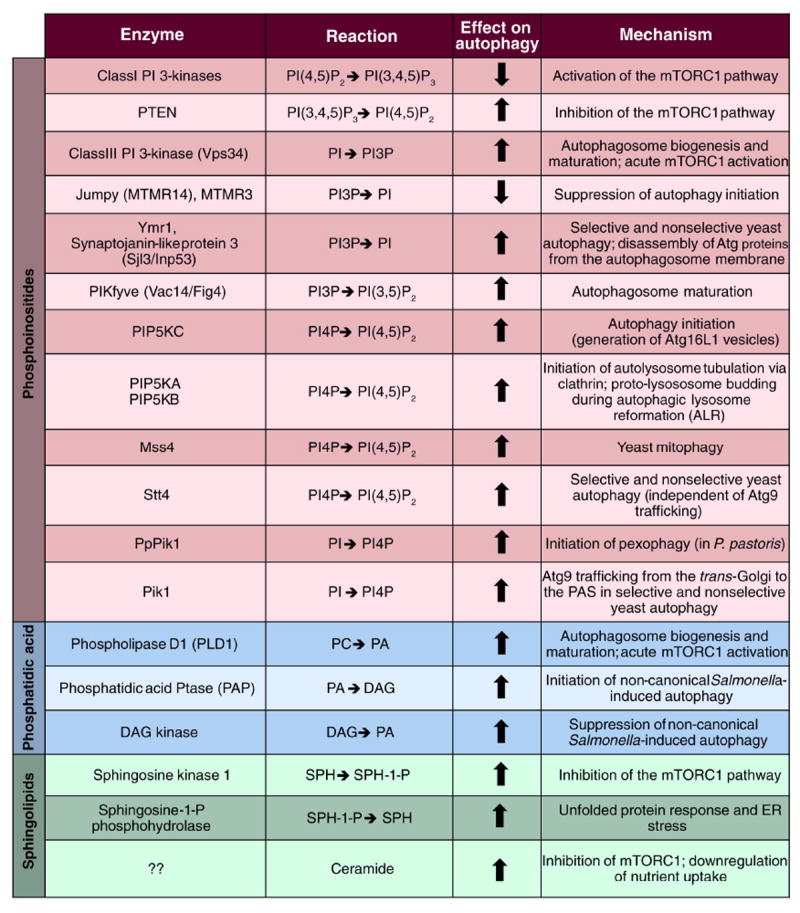
The lipids enzymes involved in the modulation of the autophagy pathway are subdivided into three classes, depending on whether they act on phosphoinositides, phosphatidic acid (PA) or sphingolipids. Their catalytic reactions as well as specific functions in autophagy regulation are indicated. Arrows denote a positive or a negative effect on autophagy. Note that the synaptojanin-like protein 3 is also an inositol 5-phosphatase that can dephosphorylate PI(4,5)P2 but only its 3-phosphatase activity mediated by its Sac1 domain has been implicated in autophagy so far.
Figure 4. Lipid signaling in the mTORC1 pathway.
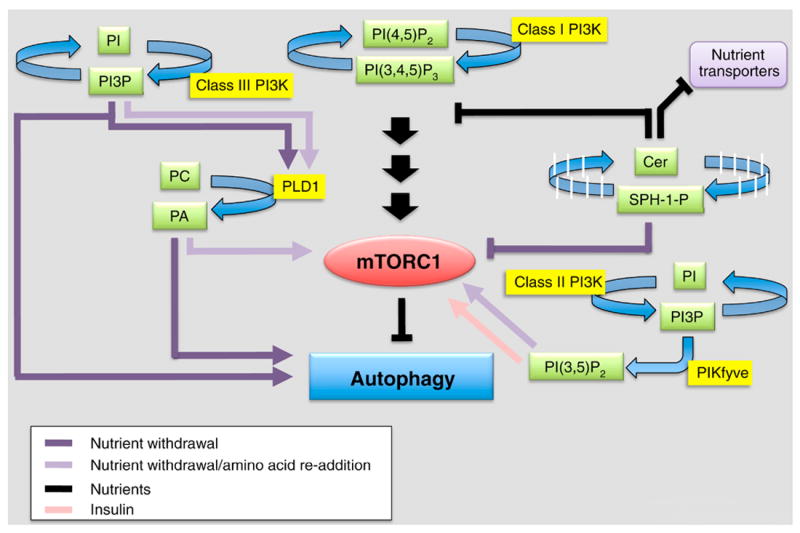
mTORC1 modulates cell response (cell growth and proliferation) to nutrients and is a key suppressor of autophagy. An adequate level of amino acids, glucose and growth factors is required for its activation (black arrows). Lipids (green) and lipid enzymes (yellow) play a major role in mediating the regulation of mTORC1. In the presence of nutrients and mitogens, class I PI3K product PI(3,4,5)P3 indirectly activates mTORC1, however, in absence of nutrients (purple arrows) mTORC1 signaling is shut down and several lipids (e.g., PI3P, PA, DAG, SPH-1-P) act as positive modulators of autophagy. Specifically, SPH-1-P produced by SK1 contributes to mTORC1 inhibition, whereas PI3P, PA and DAG produced by Class III PI3K, PLD1 and PAP, respectively, act downstream (or independently) of mTORC1 and mediate signaling and membrane remodelling to support autophagy activation. Both PI3P and PA have an antagonistic role on mTORC1 regulation: amino acid re-addition after nutrient withdrawal (light purple lane) stimulates PI3P production and activation of PLD1. Locally synthesized PA, binds directly to mTORC1 and contributes to its recruitment back to the lysosomes and recovery of the signaling. Under normal growth conditions (black lane), ceramide upregulation activates autophagy through the downregulation of nutrient (amino acids, glucose) transporters and inhibition of upstream mTORC1 signaling (Akt/PKB). The product of lipid kinase PIKfyve, PI(3,5)P2, binds to mTORC1 and participates to its reactivation in presence of insulin (pink lane) and amino-acids re-addition (light purple lane).
The signaling cascade initiated by PI(3,4,5)P3 elevation in response to insulin (and other growth factors) has been extensively characterized and reviewed elsewhere (Figure 2A) [9, 15, 16]. It involves phosphoinositide-dependent kinase 1 (PDK1)-mediated phosphorylation of Akt, which is recruited to the membrane through the binding of its pleckstrin homology domain to PI(3,4,5)P3 and PI(3,4)P2. Activated Akt, in turn, phosphorylates tuberous sclerosis complex 2 (TSC2), thus inhibiting the GTPase activating protein activity of the TSC1/2 complex towards the small GTPase Rheb (Ras homologue enriched in brain). GTP-bound Rheb ultimately activates mTORC1 and downregulates autophagy (Figure 2A) [9, 16]. Accordingly, treatment of cells with synthetic PI(3,4,5)P3 and inhibiting the PI(3,4,5)P3 3-phosphatase PTEN (Figure 3), which enhances PI(3,4,5)P3 levels, both repress autophagy [17]. mTORC1-mediated inhibition of autophagy occurs in part through phosphorylation of components of the ULK1 complex, which contains ULK1 itself (i.e., the mammalian homolog of yeast Atg1) as well as ATG13, ATG17/ FIP200, and ATG101 [18]. When the ULK1 complex is activated by autophagy-promoting signals, ULK1 dissociates from mTOR kinase and is targeted to preautophagosomal membranes, where it can initiate the autophagy process [18]. Interestingly, while the class IA PI3K is believed to activate the mTORC1 pathway and repress autophagy, the p110-β catalytic subunit of this complex has been recently identified as a positive regulator of autophagy independently of its catalysis, but via stimulation of class III PI3K Vps34 (Figure 3) [19]. Overall, the precise molecular mechanisms controlling autophagy initiation/repression downstream of class I PI3K/mTOR as well as the crosstalk between these lipid kinases and other autophagy-relevant signaling pathways converging onto mTOR regulation are beginning to emerge.
The Class III PI3K pathway, PI3P and its effectors
The main pathway for the synthesis of PI3P involves the phosphorylation of PI on the 3’ position of the inositol ring by class III PI3K or Vps34 [15], which was originally identified in budding yeast as the product of a gene required for the correct sorting of vacuolar hydrolases from the late-Golgi to the vacuole [20, 21]. After an original report showing that standard PI3K inhibitors, such as wortmannin and LY294002, block autophagy [22], three converging studies specifically demonstrated that Vps34 and its PI3P product are implicated in this process (Figure 3). Initially, in the yeast Hansenula polymorpha, the putative Vps34 ortholog was shown to be involved in the selective autophagy of peroxisomes (i.e., pexophagy) [23]. Further studies in budding yeast identified an interaction between Vps34 and Vps30/Atg6 (i.e., the ortholog of Beclin 1), a protein operating both in autophagy and vacuolar protein sorting through distinct and specialized Vps34 complexes [24]. Lastly, it was shown in mammalian cells that addition of the PI3P lipid species alone was sufficient to stimulate autophagy [25].
The Vps34 complexes and their specialized roles in autophagy
Yeast Vps34 was originally shown to form two different complexes, both of which localize to the vacuole as well as one of two subcellular locations, conferring a compartment-specific function to each of the two complexes [24]. Vps34 complex I localizes to the pre-autophagosomal structure (PAS) and functions in the initiation of autophagy. This complex consists of Vps34, Vps30/Atg6, Vps15 (i.e., a myristoylated serine/threonine kinase that is required for the lipid kinase activity of Vps34), and Atg14 (i.e., a protein that is essential for the targeting of Vps34 complex I to the PAS). Vps34 complex II is similar to complex I, except that Atg14 is replaced by Vps38, which targets Vps30/Atg6 to endosomes. Accordingly, Vps34 complex II localizes to endosomes and functions in vacuolar protein sorting, but is not involved in autophagy [24, 26, 27]. In higher eukaryotes, the autophagy-specific Vps34 complex (I) is directed via Atg14L, an Atg14-like protein, to a subdomain of the ER, called the “omegasome” [10, 28]. In fact, it has been suggested that during starvation, Vps34-containing vesicles are targeted to the ER, where they can either fuse with this compartment or enable the synthesis of PI3P in trans onto the omegasome [28]. Hierarchical analysis has demonstrated that Atg14L localizes to the nucleation site upstream and independently of Vps34 and is required for autophagy [5, 29-32].
While yeast Vps34 complex II is not required for autophagy, the mammalian complex II and its variants play a clear role in this process, in part through the greater diversity of Beclin 1 interactors acquired through evolution and a more pronounced role of the endosomal system for the maturation of mammalian APs [5]. One such interactor, UVRAG (i.e., a functional homolog of Vps38), competes with Atg14L for binding to Beclin 1 and has been shown to positively regulate autophagy (but see also [33]), perhaps by recruiting to APs Bif-1/endophilin B1, a BAR domain-containing protein that may sense and induce membrane curvature at sites of membrane expansion [29-31, 34]. UVRAG may also facilitate AP maturation in part via its interaction with the core class C-Vps complex, which mediates the fusion of APs with lysosomes [35]. Additionally, Vps34-associated Beclin 1 interacts with Ambra 1 and Rubicon, which are positive and negative regulators of autophagy, respectively [29-31, 36]. Finally, the interaction of Vps34 with Beclin 1 can be disrupted by the anti-apoptotic protein Bcl-2, which binds to Beclin 1 and prevents its binding to Vps34 [37, 38]. Phosphorylation of Bcl-2 by Jun N-terminal protein kinase 1 (JNK1) dissociates the complex and enables Beclin-1 to stimulate autophagy [39]. Overall, the precise molecular basis governing the participation of these different Vps34 subcomplexes to the biogenesis and maturation of APs, as well as the control of endo-lysosomal function is starting to emerge. There are however additional layers in the regulation of Vps34 function, including postranslational modifications such as phosphorylation and ubiquitination, which may affect the targeting, enzymatic activity and stability of the various Vps34 complexes and thus deserve further investigation [40-42].
Role of PI3P and its effectors in autophagy
PI3P and its effectors play a fundamental role in various aspects of autophagy, including the control of AP biogenesis, maturation and intracellular transport (Figure 5) [6]. One hypothesis states that the local production of PI3P on the isolation membrane promotes its negative curvature and controls AP size. Notably, it has been observed in yeast that PI3P is highly enriched on the inner (concave) surfaces of phagophores as well as on uncharacterized organelles located in the vicinity of phagophores that may supply a pool of PI3P necessary for their growth [43]. PI3P production on the elongating isolation membrane tips has also been visualized in mammalian cells and has been proposed to facilitate expansion and sealing of the spherical structure’s edges [30]. Most models, however, explain the actions of PI3P through effector proteins that harbor PI3P-binding modules, such as FYVE, PX or WD40 domains (Figure 5) [12]. Localized production of PI3P has also been proposed to create a membrane platform to concentrate and spatially coordinate specific effectors necessary for downstream signal transduction and progression of autophagy [5, 6, 27]. Several PI3P effectors have been implicated in autophagy initiation (Figure 5). In particular, DFCP1, an ER resident protein, forms PI3P-positive puncta along the ER upon starvation. Re-localization and concentration of DFCP1 during autophagy enabled the visualization of the omegasome [28, 44]. Although DFCP1 is recruited to these structures, knockdown studies do not produce any overt phenotypes and its function here remains unknown [28]. A better-characterized family of PI3P effector proteins includes the PROPPIN family or WD-repeat proteins interacting with phosphoinositide (WIPI-1-4) proteins in mammals (Figure 5) [6, 45-47]. The PROPPIN family is defined by its WD40 repeats that adopt a seven-bladed ß-propeller fold containing two pseudo-equivalent PI3P binding sites and a FRRG motif that are required for its full autophagic function [48]. Three PROPPINs, WIPI-1, -2, and 4, are implicated in the initiation of mammalian autophagy. WIPI-1 and WIPI-2 are recruited to omegasomes upon autophagy induction, where they play a role in the formation and maturation of the isolation membrane, respectively [49, 50]. These two proteins are phylogenetically similar to yeast Atg18, which has also been implicated in the initiation step, perhaps more indirectly through shuttling Atg9 together with Atg2 in both the cytoplasm-to-vacuole (Cvt) and autophagy pathways [51-53]. While WIPI-4 has not been characterized in mammals, a C. elegans homolog of WIPI-4, egf-6, was also shown to localize to and control the maturation of the omegasome. However, unlike WIPI-2, egf-6/WIPI-4 appears to restrict omegasome expansion and AP size [54]. Additional PI3P effectors operating in the initiation step have been described in yeast (i.e., Atg21 and Ygr223c) and are discussed in Figure 5 [6, 27, 55].
Figure 5. Glossary of PI3P autophagy effectors.
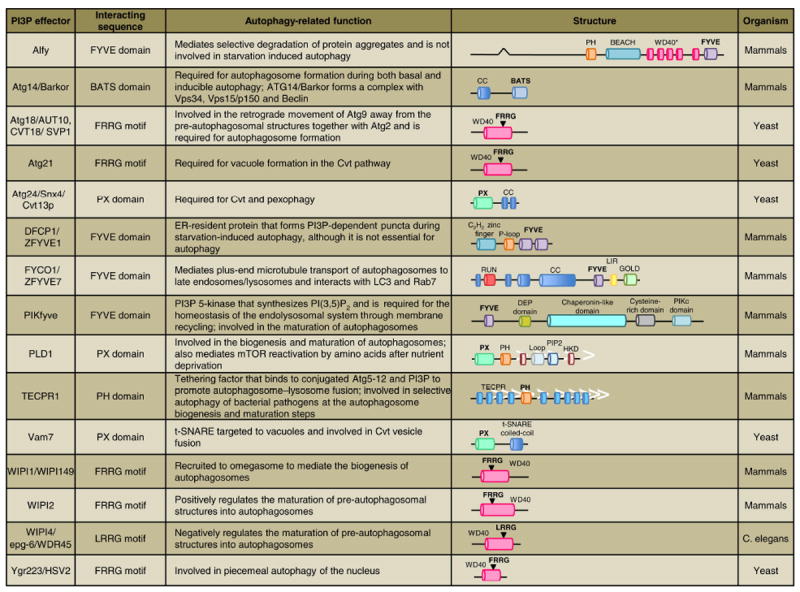
PI3P-binding modules/motifs (shown in bold) include: BATS, biotin and thiamine synthesis associated domain; FYVE, protein present in Fab1, YOTB, Vac1, and EEA1; PH, pleckstrin homology domain; PX, Phox homology domain; WD40, defined as a core unit of approximately 40 amino acids ending with the residues tryptophan and aspartic acid which adopts a 7-bladed beta propeller fold containing a PI3P binding FRRG or LRRG motif. In the case of Alfy, however, the individual WD40 regions are shown (WD40*) because it is unclear whether they adopt a beta propeller fold.
PI3P may also play a role in selective cargo capture by the nascent AP. In particular, Alfy (Autophagy-linked FYVE protein), a nuclear scaffolding protein involved in the selective degradation of ubiquitinated protein aggregates, interacts with Atg5 and p62 via its WD40 domain as well as PI3P through its FYVE domain (Figure 5) [56-58]. Localization of PI3P along the inner AP membrane may provide a docking site for Alfy to promote selective engulfment of protein aggregate cargo by directing the membrane building machinery to the site of p62 inclusion bodies [58]. Remarkably, Alfy is not required for starvation-induced autophagy but is necessary for aggregate clearance, suggesting it plays a role in a selective form of autophagy called aggrephagy [6, 58].
PI3P is also a major regulator of the maturation steps of autophagy. In higher eukaryotes, the maturation step involves the fusion of APs with endosomes, generating amphisomes, which in turn fuse with lysosomes. As such, perturbations of the endo-lysosomal compartment typically result in defects of AP maturation and cargo clearance [59-61]. Recent work in mammals identified the PI3P effector, TECPR1, as a likely tethering factor mediating AP-lysosome fusion (Figure 5). In the case of this protein, PI3P binding not only requires its pleckstrin homology (PH) domain, but also a contribution from conjugated Atg5-12, with which TECPR1 interacts [62, 63]. TECPR1 is also critical for efficient targeting of intracellular bacteria to APs during selective autophagy [62]. While in yeast evidence for an amphisome intermediate is still missing, there are PI3P effectors, such as Atg24 and Vam7, which are necessary for efficient AP-vacuole fusion (Figure 5) [6, 27].
Finally, PI3P is important for the retrograde movement of APs from the cell periphery toward the (+) ends of microtubules via the dynein/dynactin complex. FYCO1 (FYVE and coiled-coil [CC] domain containing 1), a PI3P binding, FYVE-domain containing protein, was recently identified as a novel LC3 and Rab7 interactor (Figure 5). During starvation, FYCO1 relocalizes from a juxtanuclear position to more peripheral APs and amphisomes [64]. The recruitment of FYCO1 associated APs to Rab7 positive endosomes is likely important for proper targeting of the APs as well as promoting Rab7-dependent maturation of these organelles [65, 66].
Although a growing number of PI3P effectors enabling the efficient biogenesis and maturation of APs are being identified, it will be important to establish a hierarchy for these factors in the autophagy process and to better understand their regulation and specific functions. Finally, the molecular links between PI3P effectors and the Atg8/LC3 conjugation system are still unclear.
Revisiting the role of Vps34 in mammalian autophagy
Previous genetic, molecular and pharmacological studies suggested that local production of PI3P by Vps34 is required for AP biogenesis. For instance, in yeast, Vps34 is the sole PI3K and its ablation prevents the formation of APs [24]. Importantly, re-expression of a kinase-dead version of Vps34 in the null background fails to rescue the phenotype, demonstrating the importance of PI3P synthesis [43]. Similarly, in flies not expressing Vps34 or expressing a catalytically-dead version of the lipid kinase, autophagy induction is severely inhibited in fat body and mosaic eye imaginal discs [67]. However, in higher eukaryotes such as mammals, the extent to which autophagy depends on Vps34 is less clear for several reasons. First, evidence obtained from experiments utilizing low specificity PI3K inhibitors, such as wortmannin or 3-MA, is typically regarded as proof for an involvement of Vps34, which can be misleading as these drugs are known to have other targets. Second, functional studies targeting major Vps34 regulators, such as Beclin 1, are often used as indirect proofs for an involvement of Vps34, which is not necessarily the case. Finally, recent mouse genetic studies have provided contradictory evidence regarding the role of Vps34 in autophagy. Although full-body deletion of Vps34 proved embryonically lethal [68], conditional genetics models allowed for the study of autophagy as well as the other cellular functions of murine Vps34 in a cell type- or tissue-specific fashion. In sensory neurons, lack of Vps34 primarily causes defects in the endo-lysosomal system, sparing AP formation and LC3 lipidation, although the functionality of autophagy was not investigated [69]. Another study confirmed this result by electron microscopy in Vps34-deficient T-lymphocytes from this same mouse model [70]. Contrary to this finding, a second, independent Vps34 conditional knockout mouse model reported Vps34 to be essential for AP formation in cultured mouse embryonic fibroblasts as well as in hepatocytes and cardiomyocytes in vivo [71]. This finding agrees with a third, more recently published model, which reported lower levels of autophagy in T-cells lacking Vps34 as evidenced by decreased LC3-II levels and an accumulation of p62 [72]. This disparity in the phenotypes reported may be accounted for by differences in mouse genetic background, gene targeting strategy and cell types used for those studies. Overall, it remains unclear whether a Vps34-independent source of PI3P can promote AP formation and/or whether other lipid signals are implicated in this process. In fact, precedents exist for alternate sources of PI3P, such as class II PI3Ks (PI3K-C2) and inositol 4-phosphatase Inpp4, in contexts other than autophagy [15, 73, 74]. Finally, it will be essential to assess genetically the role of Vps34-derived PI3P in non-canonical, alternative autophagy pathways [75]. These non-canonical pathways, for which evidence is accumulating, refer to mechanisms leading to the formation of functional APs that bypass key autophagy proteins and occur in response to specific stimuli/drugs, such as etoposide [76] or resveratrol [77].
Regulation of autophagy by inositol 3-phosphatases
At the step of AP formation, the balance between the production of PI3P by Vps34 and the turnover of PI3P by phosphatases is critical in regulating both size and rate of production of APs [13]. In fact, the asymmetric distribution of PI3P along the isolation membrane may be achieved not only through spatially-restricted synthesis of PI3P, but also through local changes in the metabolism of PI3P via tightly controlled switches in the balance between kinases and phosphatases. For instance, phosphoinositide 3-phosphatases, such as Jumpy (MTMR14) and MTMR3, have been implicated in the negative regulation of a pool of PI3P relevant for autophagy [13, 78-80]. For instance, knockdown of Jumpy resulted in an increase in WIPI-1 puncta and autophagic flux under both normal and starvation conditions [78]. Similarly, silencing of MTMR3 was shown to increase AP formation, whereas overexpression resulted in fewer and smaller APs [80]. Interestingly, the protein tyrosine phosphatase σ (PTPσ) has also been implicated in the negative regulation of PI3P levels during autophagy, although the underlying mechanism is unknown [81]. Thus, phosphatases are clearly important in controlling PI3P levels at the initiation step of AP formation, but whether this type of regulation occurs at later steps of autophagy is unclear. However, a recent study in yeast, has reported that conversion of PI3P to PI by PI3P phosphatases is required for the disassembly of Atg proteins from the limiting membrane of APs as well as AP-vacuole fusion during selective and nonselective types of autophagy [82]. The PI3P phosphatase mediating PI3P turnover in this instance is Ymr1, the sole myotubularin ortholog in yeast, although another phosphoinositide phosphatase, synaptojanin-like protein 3, contributes to this turnover via its Sac1 phosphatase domain [82] (Figure 3). Overall, this study suggests that PI3P phosphatases also have a positive role in autophagy, in contrast to previous models [13, 82].
PI(3,5)P2 controls late stages in the autophagy process
PI3P can also be phosphorylated to PI(3,5)P2, which has been implicated in autophagy and, specifically, in the maturation and turnover of APs [6, 83, 84]. PI(3,5)P2 is synthesized by a protein complex consisting of three major components: the PI3P 5-kinase Fab1/PIKfyve, the 5-phosphatase Fig 4/Sac3 and the scaffold protein Vac14 [84, 85]. Fab1/PIKfyve binds endosomal PI3P via its FYVE domain and controls trafficking along and recycling from the endo-lysosomal system, as evidenced by the striking increase in vacuoles upon inactivation of PI(3,5)P2 synthesis [84, 86]. Although yeast autophagy appears to be independent of PI(3,5)P2, a role for PI(3,5)P2 synthesis in AP maturation was observed in the fly mutant of Fab1 [87] and confirmed in mammals through both pharmacologic and genetic approaches [83, 88-90] (Figure 3). It has been hypothesized that PI(3,5)P2 may indirectly affect autophagosomal turnover through its more global homeostatic role in the endo-lysosomal system by promoting efficient membrane trafficking and fusion events as well as compartment acidification [86]. Alternatively, PI(3,5)P2 production on the AP membrane may provide a lipid signal necessary for the maturation of these organelles. Lastly, PI(3,5)P2 may be required for the recycling of autophagolysosome membrane after fusion of the AP/amphisome with the lysosome [86] (Figure 1). Whether phosphatases, such as myotubularin family members, are used to negatively regulate P(3,5)P2 levels during AP maturation remains undetermined. Finally, because the PROPPIN family (e.g., Atg18) tends to have dual specificity for PI3P and PI(3,5)P2 [48], PI(3,5)P2 may play an underappreciated role in the control of PROPPINS’ function during autophagy.
Emerging roles for PI4P and PI(4,5)P2 in autophagy
Studies in the yeast Pichia pastoris have originally revealed an important role for PI4P in selective degradation of peroxisomes. Generation of PI4P at the micropexophagy-specific membrane apparatus (MIPA) by the PI 4-kinase (PI4K) PpPik1 concentrates the PI4P effector PpAtg26 through its GRAM and trGRAM-PH domains (Figure 3). Recruitment of PpAtg26 to the nucleation site of pexophagosomes enables the sterol conversion to sterol glucoside necessary for elongation and maturation of the growing membrane structure [91]. However, a recent study reported that Pik1, the homolog of PpPik1 in in S. cerevisiae, has a broader role in autophagy as it functions both in nonselective and selective (e.g., mitophagy) types of autophagy [92]. In this species, Pik1 and its product PI4P typically mediate the exit of secretory vesicles from the trans-Golgi (TGN) under normal conditions (reviewed in ref. [12]). Upon autophagy induction, Pik1 promotes the anterograde traffic of Atg9 from the Golgi complex and/or post-Golgi compartments towards the PAS [92] (Figure 3), a process required for the recruitment of various other essential Atg proteins [3]. The same study showed that a distinct and plasma membrane-localized PI4K isoform, Stt4, also functions in nonselective and selective types of autophagy (Figure 3), although this requirement appears to be independent of the Atg9 traffic regulation [92]. Finally, this study showed that conversion of a plasma membrane-bound pool of PI4P to PI(4,5)P2 by the PI4P 5-kinase (PIP5K), Mss4, is required for mitophagy, but not for nonselective autophagy [92] (Figure 3). While the identification in yeast of autophagic roles for PI4K and PIP5K is an important step forward, elucidating the effectors acting downstream of their products PI4P and PI(4,5)P2, respectively, should provide mechanistic insights into the molecular basis of this involvement. One potential PI4P effector relevant for yeast autophagy is Kes1, a member of the oxysterol-binding protein family, which binds both PI4P and sterol and localizes to TGN/endosomal membranes [93]. Indeed, Kes1 appears to negatively regulate autophagy through its metabolic control of PI3P levels [94].
Remarkably, the PI4P-to-PI(4,5)P2 conversion was also shown to play a key role in a late step of mammalian autophagy called autophagic lysosome reformation (ALR) (Figure 1). This process, which occurs during prolonged starvation, refers to the de novo biogenesis of lysosomes from existing auto(phago)lysosomes through tubulation of their limiting membranes, followed by fission of these tubular protrusions [95]. Clues obtained through proteomic analysis of these purified tubules pointed to clathrin and its associated machinery, including clathrin daptors and PI(4,5)P2-synthesizing enzymes, as candidate mediators of ARL [96]. Further characterization and validation of these proteomic hits through RNA interference indicated that conversion of PI4P to PI(4,5)P2 by PIP5K1B mediates the recruitment of clathrin to the autolysosomes (via its adaptors), while the same reaction catalyzed by a distinct isoform, PIP5K1A, appears to control the fission of these tubules reforming from autolysosomes [96] (Figure 1 and 3). Interestingly, PI4P and PI(4,5)P2 were found to be differentially localized on these organelles, as PI4P was uniformly distributed on autolysosomal membranes, whereas PI(4,5)P2, similar to PIPK51A, was enriched on buds and tubules emerging from autolysosomes [96]. Overall, this study suggests that transient and local synthesis of PI4P and PI(4,5)P2, mediates important steps of ARL through the “reuse” of factors, such as clathrin and its adaptors, who function in other trafficking steps under basal conditions [96]. Additional roles for PIP5Ks in autophagy are discussed below.
Phosphatidic acid and the phospholipase D pathway
While phosphoinositide metabolizing enzymes are the best-known lipid enzymes regulating autophagy, recent work has demonstrated the importance of other pathways, such as phospholipase D (PLD). This enzyme hydrolyzes phosphatidylcholine (PC) to produce choline as well as the bioactive phospholipid PA, which has been implicated in a variety of trafficking and signaling processes [11, 97]. PA has been proposed to exert its biological actions through its direct effects on membranes via its “cone” shape and negative curvature-inducing properties (Figure 2D) or through effector proteins, such as mTOR (Figure 4). Finally, PA can be converted to metabolites, such as diacyglycerol (DAG), which have distinct bioactive properties [11, 97, 98].
PLD1 as a positive modulator of autophagy
Work from our laboratory identified PLD1 as a positive modulator of autophagy in various cell lines as well as in hepatocytes in vivo using a mouse genetic model [99]. Under normal conditions the majority of PLD1 is localized to the endo-lysosomal system; however, during nutrient deprivation, PLD1 partially relocalizes to the outer membrane of AP-related structures, but appears absent from isolation membranes [99] (Figure 1). The subcellular localization of PLD1 during starvation, which depends on its PI3P-binding NH2-terminal PX domain and Vps34 (Figure 5) [99, 100], is consistent with a role of PLD1 in the maturation of APs. However, inhibition of the PLD pathway also decreases the production of LC3 positive structures as well as LC3-II levels, suggesting that the PLD pathway also regulates AP formation. Importantly, genetic ablation of PLD1 leads to a reduction in the size and number of APs in the liver of starved mice, a phenotype consistent with that observed in PLD1-deficient mouse embryonic fibroblasts (MEFs) [99]. Additionally, blockade of PLD with a pharmacological inhibitor results in enhanced levels of p62 and tau aggregates in organotypic brain slices, suggesting that the function of PLD is not restricted to starvation-induced autophagy [99]. Although the catalytic activity of PLD is essential for its autophagic functions, the mechanism of action of PLD and the specific modulatory role(s) of PA in the biogenesis and maturation of APs are unclear.
PLD as a source of DAG for PKC-regulated autophagy
A potential mechanism for the involvement of PLD in the initiation of autophagy was described in a study of Salmonella-induced autophagy, which showed that PLD enzymes are a critical source of DAG to initiate the host cell’s antibacterial autophagy of S. typhimurium [101] (Figure 1). In this non-canonical autophagy pathway (which may be related to LC3-associated phagocytosis [102]), DAG is produced by the dephosphorylation of PLD-produced PA by a PA phosphatase (PAP) and mediates the downstream stimulation of protein kinase Cδ (PKCδ), which can in turn activate autophagy by dissociating the Bcl-2-Beclin 1 complex via JNK and by stimulating NADPH oxidase [101]. Interestingly, the PA/DAG/PKCδ signaling cascade in Salmonella-induced autophagy represents a parallel pathway to the one involving the ubiquitin-binding p62/SQTSM1 adaptor (ubi-p62/SQTSM1) [103]. In fact, blockade of both PA/DAG/PKC and ubi-p62/SQTSM1 pathways additively inhibits antibacterial autophagy [101]. Importantly, this PKC pathway is also required for rapamycin-induced autophagy. In addition to PKCδ, two other mammalian PKC isoforms, PKCα and PKCθ, mediate palmitic acid- and ER stress-induced autophagy, respectively [104, 105]. The autophagic function of PKC is conserved in evolution, as the single yeast ortholog of PKC, Pkc1, is necessary for starvation-induced autophagy in yeast [101]. However, the PLD/PA/DAG/PKC pathway does not appear to be conserved, as the sole PLD ortholog in yeast, Spo14, is not essential for starvation-induced autophagy. In Pichia pastoris, Spo14 is important for the unconventional secretion of Acb1, a phenomenon requiring various elements of the autophagy machinery for the formation of APs containing Acb1 prior to their Spo14-dependent fusion with the plasma membrane [106].
Regulation of ATG16L traffic and autophagy initiation by the Arf6-PLD pathway
The implication of PLD1 in starvation-induced autophagy [99] has brought attention to the upstream regulation of this enzyme. One such regulator, the small GTPase Arf6, has been recently shown to control the initiation of autophagy in a PLD-dependent fashion [107] (Figure 1). Previous studies had shown that the plasma membrane and endocytic processes contribute directly to the formation of Atg16L1-positive AP precursors [108]. Arf6, which mediates a form of clathrin-independent endocytosis as well as recycling of various cargoes from the recycling endosome to the cell surface [109], was implicated in autophagy initiation at this very step [107]. Arf6 is mobilized onto isolation membranes under nutrient starvation [107]. Importantly, it is known to stimulate PI(4,5)P2 and PA synthesis via an activation of PIP5K and PLD, respectively [97]. In fact, a positive feedback loop has been described in various contexts, whereby PA stimulates PIP5Ks and PI(4,5)P2, in turn, stimulates PLD activity [97] (Figure 1). Similarly, the autophagic function of Arf6 requires PIP5KC, which is distinct from the two PIP5K isoforms involved in ARL, as well as PLD (Figure 3). The latter was shown using an Arf6 mutant (N48R) that is deficient in PLD binding, but preserves other critical interactions [107, 110]. Overall, this study on Arf6 provides incentive for additional studies examining the effects on autophagy of various PLD activators, including PKC isoforms, the small GTPase Ral and 5’ adenosine monophosphate-activated protein kinase (AMPK), all of which have already been implicated in autophagy [101, 104, 105, 111-114].
The PLD1/Vps34 paradox: autophagy vs. nutrient sensing via mTORC1
While Vps34 and PLD1 have been implicated in the positive regulation of starvation-induced autophagy, both lipid enzymes also enable amino acid-induced stimulation of the mTORC1 pathway, which suppresses autophagy [9] (Figure 4). PLD-derived PA was originally shown to promote the mitogenic activation of mTOR by binding to its FKBP12-rapamycin binding (FRB) domain in a site that competes with rapamycin [115]. Importantly, mitogens were shown to increase PLD1 activity and this increase required Rheb, a positive regulator of mTORC1, as well as the presence of amino acids [115]. Not only mitogens, but also amino acids and glucose were shown to positively regulate the activity of PLD1 following nutrient withdrawal [100, 116]. In addition to Rheb, Vps34 and its product PI3P appear to be important for PLD1- and amino acid-mediated mTORC1 signaling [40, 98, 100, 116] (Figure 4). Remarkably, in the absence of amino acids, exogenously-added PI3P is sufficient to promote PLD1 activation but fails to activate mTORC1; moreover, in the absence of Vps34, exogenously-added PA or PI3P can reactivate mTORC1 signaling but only in the presence of amino acids, suggesting that the Vps34/PLD1 pathway is necessary but not sufficient to reactivate mTORC1 after nutrient deprivation [98, 100] (Figure 4). Additionally, the lack of obvious phenotypes at the organismal level in mice lacking PLD1 argues against a fundamental role in mTOR signaling [99]. Furthermore, a recent study employed Vps34 knockout MEFs and mice to show that the lipid kinase does not control the basal level of mTORC1 activation, but is critical for acute mTORC1 reactivation by nutrients following amino acid withdrawal (in MEFs) or food restriction (in liver tissue) [71]. Overall, because both PLD1 and Vps34 play a role in the biology of the endo-lysosomal system, their involvement in mTORC1 regulation at these stations is not surprising. However, the molecular basis controlling the sensing of amino acids, and more generally nutrients, by Vps34 and PLD1 as well as that controlling the pro- and anti-autophagic roles of these enzymes need to be clarified. Finally, other lipid enzymes have been shown to control mTORC1 signaling. Recent work in adipocytes has shown that Raptor, a subunit of the mTORC1 complex, binds directly to PI(3,5)P2 through its WD40 domain and that PI3K-C2α and PIKfyve-dependent sequential generation of PI3P and PI(3,5)P2 provides a local signal for the activation of mTORC1 at the plasma membrane by both amino acids and insulin [117] (Figure 4).
Control of autophagy by sphingolipids
Sphingolipids are a class of bioactive molecules implicated in a wide range of physiological processes, such as autophagy, cell growth, apoptosis, stress responses, angiogenesis and inflammatory response [118]. The two main species of sphingolipids involved in autophagy are ceramide and shingosine-1-phosphate (SPH-1-P) (Figure 3). Both these lipids activate autophagy, promoting cell survival and protecting against apoptotic cell death. However, prolonged ceramide signaling can also lead to non-apoptotic cell death [6, 118]. In the past decade, several studies have established that ceramide levels are upregulated when autophagy is induced by stress or a variety of drug treatments (e.g., tamoxifen, resveratrol), and that ceramide itself activates autophagy [118]. For instance, the exogenous application of short-chain ceramide (e.g., C2-ceramide) stimulates autophagy, likely by promoting the de novo synthesis of long chain-ceramide [118, 119]. Long chain ceramides, in turn, can activate autophagy by inhibiting the phosphorylation of Akt/PKB in the class I PI3K pathway, reducing the activation of mTOR and upregulating Beclin 1 function, in part through a JNK1-mediated dissociation of the Beclin 1-Bcl2 complex [118, 120]. Alternate models involve the ceramide-induced downregulation of amino acid transporters, causing a decrease in nutrient uptake and a metabolic stress that stimulates autophagy through reduced mTOR signaling (Figure 4) [121].
Ceramide is also a key intermediate for the generation of SPH-1-P, which plays equally important roles in autophagy [118]. Indeed, ceramidase-mediated hydrolysis of ceramide gives rise to sphingosine, which, in turn, can be phosphorylated to SPH-1-P by sphingosine kinases (SK) 1 and 2. Starvation increases the activity of SK1, promoting the intracellular formation of SPH-1-P (Figure 3) [122]. Moreover, SK1 overexpression stimulates autophagy by inhibiting the mTOR pathway independently of Akt/PKB phosphorylation and protects against starvation-induced cell death. Conversely, depletion or inactivation of SK1 causes autophagic cell death [118, 122]. In agreement with these findings, upregulation of SPH-1-P levels by silencing one of its catabolizing enzymes, sphingosine phosphate phosphatase 1, induces the unfolded protein response, ER stress and a form of autophagy that is insensitive to 3-MA and independent of mTOR regulation [123] (Figure 3 and 4). In summary, while there is a clear implication of sphingolipids in the control of the balance between autophagy and cell death, their precise mechanisms of action need to be further elucidated.
Conclusions and future directions
As highlighted in this review, lipids play pleiotropic roles in the control of autophagy, a process that involves a dramatic remodeling of intracellular membranes. Central to this regulation is the membrane-cytosol interface, where a variety of lipids, primarily phospholipids, either directly sculpt lipid bilayers, or assemble protein scaffolds responsible for the various steps that constitute the autophagy process. These range from signal transduction processes to the biogenesis and maturation of APs and ultimately, to the clearance of the AP cargoes. One of the key questions in the field concerns the precise lipid-based mechanisms controlling the biogenesis of APs [7, 61]. One model posits that lipids may be directly transferred in a non-vesicular fashion to the nascent AP in close proximity to the ER [124, 125] or mitochondria [126], while another model implies vesicular transfer of lipids through bona fide membrane carriers. It can be hypothesized that the local transfer of lipids from the ER or mitochondria to the isolation membrane may help with the initial phase in the growth of the AP, whereas the massive expansion of this organelle into an AP likely relies on vesicular inputs. High-resolution electron microscopy, combined with super-resolution fluorescence microscopy may resolve this issue.
While this review focuses on the regulatory roles of lipids in autophagy, lipids themselves have also been identified as autophagy substrates in a recently discovered pathway that involves the direct consumption of cellular fat in the form of lipid droplets by autophagolysosomes [2]. This phenomenon, which is referred to as macrolipophagy, mediates the breakdown of triglycerides into fatty acids for the purpose of energy homeostasis in such tissues as the liver, although a growing number of cell types and tissues are reported to utilize this pathway, pointing to its fundamental role in cellular and organismal physiology. Further reinforcing the crosstalk between lipids and autophagy, free fatty acids, such as palmitic acid, can also trigger this process [104]. Finally, cholesterol has been implicated in the organization of microdomains within lysosomal membranes that control the efficacy of CMA as well as AP-lysosome fusion [127, 128]. Overall, the five main lipid classes (i.e., fatty acids, phospholipids, glycerolipids, sphingolipids and sterols) have been directly implicated in autophagy, although for most of these classes, the molecular basis underlying their involvement in autophagy is poorly understood. In this respect, systems biology approaches such as lipidomics, a mass spectrometry-based technology, should help understand in an unbiased manner the specific roles of lipids in autophagy [129]. This methodology has been recently used to determine the impact of high fat or high cholesterol diet as well as aging on the lipid composition of lysosomal membranes derived from mouse liver, giving rise to novel lipid-based hypotheses on how these treatments may diminish the efficacy of CMA [128]. It is of note that the profiling of autophagic vacuole membrane by various “omics”-based approaches, including lipidomics, faces the inherent problem of cargoes contributing perhaps significantly to the molecular profiles, requiring a prior separation of these cargoes from the autophagic vacuole membranes for accurate measurements.
Finally, there is intense debate in the field about the potential of autophagy modulation as a therapeutic tool. Exploiting lipid signaling pathways should be seriously considered, based on the highly successful precedents of statins and cyclooxygenase inhibitors in a variety of therapeutic applications. Particularly, targeting autophagy modulators rather than essential components of this pathway may prove beneficial.
Acknowledgments
We thank Richard Kessin, Rebecca Williamson and Ai Yamamoto for critically reading the manuscript as well as John Brumell for helpful suggestions This work was supported by NIH grants R01 NS056049 (to G.D.P.). P50 AG08702 (to Michael Shelanski, G.D.P. and C.D.A. projects) and P30 DK063608 (to Domenico Accili, G.D.P project). K.A.D. is supported by NIH training grant T32 GM07367 (to Michael Shelanski).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069–1075. doi: 10.1038/nature06639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Singh R, Cuervo AM. Autophagy in the cellular energetic balance. Cell Metab. 2011;13:495–504. doi: 10.1016/j.cmet.2011.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mizushima N, Yoshimori T, Ohsumi Y. The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol. 2011;27:107–132. doi: 10.1146/annurev-cellbio-092910-154005. [DOI] [PubMed] [Google Scholar]
- 4.Yang Z, Klionsky DJ. Eaten alive: a history of macroautophagy. Nature cell biology. 2010;12:814–822. doi: 10.1038/ncb0910-814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Simonsen A, Tooze SA. Coordination of membrane events during autophagy by multiple class III PI3-kinase complexes. J Cell Biol. 2009;186:773–782. doi: 10.1083/jcb.200907014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Knaevelsrud H, Simonsen A. Lipids in autophagy: Constituents, signaling molecules and cargo with relevance to disease. Biochim Biophys Acta. 2012;1821:1133–1145. doi: 10.1016/j.bbalip.2012.01.001. [DOI] [PubMed] [Google Scholar]
- 7.Rubinsztein DC, Shpilka T, Elazar Z. Mechanisms of autophagosome biogenesis. Curr Biol. 2012;22:R29–34. doi: 10.1016/j.cub.2011.11.034. [DOI] [PubMed] [Google Scholar]
- 8.Epple UD, Suriapranata I, Eskelinen EL, Thumm M. Aut5/Cvt17p, a putative lipase essential for disintegration of autophagic bodies inside the vacuole. J Bacteriol. 2001;183:5942–5955. doi: 10.1128/JB.183.20.5942-5955.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol. 2011;12:21–35. doi: 10.1038/nrm3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Burman C, Ktistakis NT. Regulation of autophagy by phosphatidylinositol 3-phosphate. FEBS Lett. 2010;584:1302–1312. doi: 10.1016/j.febslet.2010.01.011. [DOI] [PubMed] [Google Scholar]
- 11.Haucke V, Di Paolo G. Lipids and lipid modifications in the regulation of membrane traffic. Current opinion in cell biology. 2007;19:426–435. doi: 10.1016/j.ceb.2007.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Di Paolo G, De Camilli P. Phosphoinositides in cell regulation and membrane dynamics. Nature. 2006;443:651–657. doi: 10.1038/nature05185. [DOI] [PubMed] [Google Scholar]
- 13.Vergne I, Deretic V. The role of PI3P phosphatases in the regulation of autophagy. FEBS Lett. 2010;584:1313–1318. doi: 10.1016/j.febslet.2010.02.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mehrpour M, Esclatine A, Beau I, Codogno P. Overview of macroautophagy regulation in mammalian cells. Cell Res. 2010;20:748–762. doi: 10.1038/cr.2010.82. [DOI] [PubMed] [Google Scholar]
- 15.Vanhaesebroeck B, Guillermet-Guibert J, Graupera M, Bilanges B. The emerging mechanisms of isoform-specific PI3K signalling. Nat Rev Mol Cell Biol. 2010;11:329–341. doi: 10.1038/nrm2882. [DOI] [PubMed] [Google Scholar]
- 16.Toker A. Phosphoinositide 3-kinases-a historical perspective. Subcell Biochem. 2012;58:95–110. doi: 10.1007/978-94-007-3012-0_4. [DOI] [PubMed] [Google Scholar]
- 17.Arico S, Petiot A, Bauvy C, Dubbelhuis PF, Meijer AJ, Codogno P, Ogier-Denis E. The tumor suppressor PTEN positively regulates macroautophagy by inhibiting the phosphatidylinositol 3-kinase/protein kinase B pathway. J Biol Chem. 2001;276:35243–35246. doi: 10.1074/jbc.C100319200. [DOI] [PubMed] [Google Scholar]
- 18.Mizushima N. The role of the Atg1/ULK1 complex in autophagy regulation. Current opinion in cell biology. 2010;22:132–139. doi: 10.1016/j.ceb.2009.12.004. [DOI] [PubMed] [Google Scholar]
- 19.Dou Z, Chattopadhyay M, Pan JA, Guerriero JL, Jiang YP, Ballou LM, Yue Z, Lin RZ, Zong WX. The class IA phosphatidylinositol 3-kinase p110-beta subunit is a positive regulator of autophagy. J Cell Biol. 2010;191:827–843. doi: 10.1083/jcb.201006056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Herman PK, Emr SD. Characterization of VPS34, a gene required for vacuolar protein sorting and vacuole segregation in Saccharomyces cerevisiae. Mol Cell Biol. 1990;10:6742–6754. doi: 10.1128/mcb.10.12.6742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schu PV, Takegawa K, Fry MJ, Stack JH, Waterfield MD, Emr SD. Phosphatidylinositol 3-kinase encoded by yeast VPS34 gene essential for protein sorting. Science. 1993;260:88–91. doi: 10.1126/science.8385367. [DOI] [PubMed] [Google Scholar]
- 22.Blommaart EF, Krause U, Schellens JP, Vreeling-Sindelarova H, Meijer AJ. The phosphatidylinositol 3-kinase inhibitors wortmannin and LY294002 inhibit autophagy in isolated rat hepatocytes. Eur J Biochem. 1997;243:240–246. doi: 10.1111/j.1432-1033.1997.0240a.x. [DOI] [PubMed] [Google Scholar]
- 23.Kiel JA, Rechinger KB, van der Klei IJ, Salomons FA, Titorenko VI, Veenhuis M. The Hansenula polymorpha PDD1 gene product, essential for the selective degradation of peroxisomes, is a homologue of Saccharomyces cerevisiae Vps34p. Yeast. 1999;15:741–754. doi: 10.1002/(SICI)1097-0061(19990630)15:9<741::AID-YEA416>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 24.Kihara A, Noda T, Ishihara N, Ohsumi Y. Two distinct Vps34 phosphatidylinositol 3-kinase complexes function in autophagy and carboxypeptidase Y sorting in Saccharomyces cerevisiae. J Cell Biol. 2001;152:519–530. doi: 10.1083/jcb.152.3.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Petiot A, Ogier-Denis E, Blommaart EF, Meijer AJ, Codogno P. Distinct classes of phosphatidylinositol 3’-kinases are involved in signaling pathways that control macroautophagy in HT-29 cells. J Biol Chem. 2000;275:992–998. doi: 10.1074/jbc.275.2.992. [DOI] [PubMed] [Google Scholar]
- 26.Obara K, Sekito T, Ohsumi Y. Assortment of phosphatidylinositol 3-kinase complexes--Atg14p directs association of complex I to the pre-autophagosomal structure in Saccharomyces cerevisiae. Mol Biol Cell. 2006;17:1527–1539. doi: 10.1091/mbc.E05-09-0841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Obara K, Ohsumi Y. PtdIns 3-Kinase Orchestrates Autophagosome Formation in Yeast. J Lipids. 2011;2011:498768. doi: 10.1155/2011/498768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Axe EL, Walker SA, Manifava M, Chandra P, Roderick HL, Habermann A, Griffiths G, Ktistakis NT. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J Cell Biol. 2008;182:685–701. doi: 10.1083/jcb.200803137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Matsunaga K, Saitoh T, Tabata K, Omori H, Satoh T, Kurotori N, Maejima I, Shirahama-Noda K, Ichimura T, Isobe T, et al. Two Beclin 1-binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nature cell biology. 2009;11:385–396. doi: 10.1038/ncb1846. [DOI] [PubMed] [Google Scholar]
- 30.Zhong Y, Wang QJ, Li X, Yan Y, Backer JM, Chait BT, Heintz N, Yue Z. Distinct regulation of autophagic activity by Atg14L and Rubicon associated with Beclin 1-phosphatidylinositol-3-kinase complex. Nature cell biology. 2009;11:468–476. doi: 10.1038/ncb1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Funderburk SF, Wang QJ, Yue Z. The Beclin 1-VPS34 complex--at the crossroads of autophagy and beyond. Trends in cell biology. 2010;20:355–362. doi: 10.1016/j.tcb.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Matsunaga K, Morita E, Saitoh T, Akira S, Ktistakis NT, Izumi T, Noda T, Yoshimori T. Autophagy requires endoplasmic reticulum targeting of the PI3-kinase complex via Atg14L. J Cell Biol. 2010;190:511–521. doi: 10.1083/jcb.200911141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Itakura E, Kishi C, Inoue K, Mizushima N. Beclin 1 forms two distinct phosphatidylinositol 3-kinase complexes with mammalian Atg14 and UVRAG. Mol Biol Cell. 2008;19:5360–5372. doi: 10.1091/mbc.E08-01-0080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Takahashi Y, Coppola D, Matsushita N, Cualing HD, Sun M, Sato Y, Liang C, Jung JU, Cheng JQ, Mule JJ, et al. Bif-1 interacts with Beclin 1 through UVRAG and regulates autophagy and tumorigenesis. Nature cell biology. 2007;9:1142–1151. doi: 10.1038/ncb1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liang C, Lee JS, Inn KS, Gack MU, Li Q, Roberts EA, Vergne I, Deretic V, Feng P, Akazawa C, et al. Beclin1-binding UVRAG targets the class C Vps complex to coordinate autophagosome maturation and endocytic trafficking. Nature cell biology. 2008;10:776–787. doi: 10.1038/ncb1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fimia GM, Stoykova A, Romagnoli A, Giunta L, Di Bartolomeo S, Nardacci R, Corazzari M, Fuoco C, Ucar A, Schwartz P, et al. Ambra1 regulates autophagy and development of the nervous system. Nature. 2007;447:1121–1125. doi: 10.1038/nature05925. [DOI] [PubMed] [Google Scholar]
- 37.Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, Packer M, Schneider MD, Levine B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. 2005;122:927–939. doi: 10.1016/j.cell.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 38.Sinha S, Levine B. The autophagy effector Beclin 1: a novel BH3-only protein. Oncogene. 2008;27(Suppl 1):S137–148. doi: 10.1038/onc.2009.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wei Y, Pattingre S, Sinha S, Bassik M, Levine B. JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol Cell. 2008;30:678–688. doi: 10.1016/j.molcel.2008.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Backer JM. The regulation and function of Class III PI3Ks: novel roles for Vps34. Biochem J. 2008;410:1–17. doi: 10.1042/BJ20071427. [DOI] [PubMed] [Google Scholar]
- 41.Furuya T, Kim M, Lipinski M, Li J, Kim D, Lu T, Shen Y, Rameh L, Yankner B, Tsai LH, et al. Negative regulation of Vps34 by Cdk mediated phosphorylation. Mol Cell. 2010;38:500–511. doi: 10.1016/j.molcel.2010.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu J, Xia H, Kim M, Xu L, Li Y, Zhang L, Cai Y, Norberg HV, Zhang T, Furuya T, et al. Beclin1 controls the levels of p53 by regulating the deubiquitination activity of USP10 and USP13. Cell. 2011;147:223–234. doi: 10.1016/j.cell.2011.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Obara K, Noda T, Niimi K, Ohsumi Y. Transport of phosphatidylinositol 3-phosphate into the vacuole via autophagic membranes in Saccharomyces cerevisiae. Genes Cells. 2008;13:537–547. doi: 10.1111/j.1365-2443.2008.01188.x. [DOI] [PubMed] [Google Scholar]
- 44.Ktistakis N, Chandra P, Walker S, Manifava M, Musiwaro P, Axe E. Early events regulating autophagosome formation. Autophagy. 2009;5:897–897. [Google Scholar]
- 45.Dove SK, Piper RC, McEwen RK, Yu JW, King MC, Hughes DC, Thuring J, Holmes AB, Cooke FT, Michell RH, et al. Svp1p defines a family of phosphatidylinositol 3,5-bisphosphate effectors. Embo J. 2004;23:1922–1933. doi: 10.1038/sj.emboj.7600203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Krick R, Busse RA, Scacioc A, Stephan M, Janshoff A, Thumm M, Kuhnel K. Structural and functional characterization of the two phosphoinositide binding sites of PROPPINs, a beta-propeller protein family. Proc Natl Acad Sci U S A. 2012;109:E2042–2049. doi: 10.1073/pnas.1205128109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Watanabe Y, Kobayashi T, Yamamoto H, Hoshida H, Akada R, Inagaki F, Ohsumi Y, Noda NN. Structure-based Analyses Reveal Distinct Binding Sites for Atg2 and Phosphoinositides in Atg18. J Biol Chem. 2012;287:31681–31690. doi: 10.1074/jbc.M112.397570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Baskaran S, Ragusa MJ, Boura E, Hurley JH. Two-Site Recognition of Phosphatidylinositol 3-Phosphate by PROPPINs in Autophagy. Mol Cell. 2012 doi: 10.1016/j.molcel.2012.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Proikas-Cezanne T, Waddell S, Gaugel A, Frickey T, Lupas A, Nordheim A. WIPI-1alpha (WIPI49), a member of the novel 7-bladed WIPI protein family, is aberrantly expressed in human cancer and is linked to starvation-induced autophagy. Oncogene. 2004;23:9314–9325. doi: 10.1038/sj.onc.1208331. [DOI] [PubMed] [Google Scholar]
- 50.Polson HE, de Lartigue J, Rigden DJ, Reedijk M, Urbe S, Clague MJ, Tooze SA. Mammalian Atg18 (WIPI2) localizes to omegasome-anchored phagophores and positively regulates LC3 lipidation. Autophagy. 2010;6 doi: 10.4161/auto.6.4.11863. [DOI] [PubMed] [Google Scholar]
- 51.Krick R, Tolstrup J, Appelles A, Henke S, Thumm M. The relevance of the phosphatidylinositolphosphat-binding motif FRRGT of Atg18 and Atg21 for the Cvt pathway and autophagy. FEBS letters. 2006;580:4632–4638. doi: 10.1016/j.febslet.2006.07.041. [DOI] [PubMed] [Google Scholar]
- 52.Obara K, Sekito T, Niimi K, Ohsumi Y. The Atg18-Atg2 complex is recruited to autophagic membranes via phosphatidylinositol 3-phosphate and exerts an essential function. J Biol Chem. 2008;283:23972–23980. doi: 10.1074/jbc.M803180200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nair U, Cao Y, Xie Z, Klionsky DJ. Roles of the lipid-binding motifs of Atg18 and Atg21 in the cytoplasm to vacuole targeting pathway and autophagy. J Biol Chem. 2010;285:11476–11488. doi: 10.1074/jbc.M109.080374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lu Q, Yang P, Huang X, Hu W, Guo B, Wu F, Lin L, Kovacs AL, Yu L, Zhang H. The WD40 repeat PtdIns(3)P-binding protein EPG-6 regulates progression of omegasomes to autophagosomes. Dev Cell. 2011;21:343–357. doi: 10.1016/j.devcel.2011.06.024. [DOI] [PubMed] [Google Scholar]
- 55.Krick R, Henke S, Tolstrup J, Thumm M. Dissecting the localization and function of Atg18, Atg21 and Ygr223c. Autophagy. 2008;4:896–910. doi: 10.4161/auto.6801. [DOI] [PubMed] [Google Scholar]
- 56.Simonsen A, Birkeland HC, Gillooly DJ, Mizushima N, Kuma A, Yoshimori T, Slagsvold T, Brech A, Stenmark H. Alfy, a novel FYVE-domain-containing protein associated with protein granules and autophagic membranes. J Cell Sci. 2004;117:4239–4251. doi: 10.1242/jcs.01287. [DOI] [PubMed] [Google Scholar]
- 57.Clausen TH, Lamark T, Isakson P, Finley K, Larsen KB, Brech A, Overvatn A, Stenmark H, Bjorkoy G, Simonsen A, et al. p62/SQSTM1 and ALFY interact to facilitate the formation of p62 bodies/ALIS and their degradation by autophagy. Autophagy. 2010;6:330–344. doi: 10.4161/auto.6.3.11226. [DOI] [PubMed] [Google Scholar]
- 58.Filimonenko M, Isakson P, Finley KD, Anderson M, Jeong H, Melia TJ, Bartlett BJ, Myers KM, Birkeland HC, Lamark T, et al. The selective macroautophagic degradation of aggregated proteins requires the PI3P-binding protein Alfy. Mol Cell. 2010;38:265–279. doi: 10.1016/j.molcel.2010.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Longatti A, Tooze SA. Vesicular trafficking and autophagosome formation. Cell death and differentiation. 2009;16:956–965. doi: 10.1038/cdd.2009.39. [DOI] [PubMed] [Google Scholar]
- 60.Noda T, Fujita N, Yoshimori T. The late stages of autophagy: how does the end begin? Cell death and differentiation. 2009;16:984–990. doi: 10.1038/cdd.2009.54. [DOI] [PubMed] [Google Scholar]
- 61.Weidberg H, Shvets E, Elazar Z. Biogenesis and cargo selectivity of autophagosomes. Annu Rev Biochem. 2011;80:125–156. doi: 10.1146/annurev-biochem-052709-094552. [DOI] [PubMed] [Google Scholar]
- 62.Ogawa M, Yoshikawa Y, Kobayashi T, Mimuro H, Fukumatsu M, Kiga K, Piao Z, Ashida H, Yoshida M, Kakuta S, et al. A Tecpr1-dependent selective autophagy pathway targets bacterial pathogens. Cell host & microbe. 2011;9:376–389. doi: 10.1016/j.chom.2011.04.010. [DOI] [PubMed] [Google Scholar]
- 63.Chen D, Fan W, Lu Y, Ding X, Chen S, Zhong Q. A mammalian autophagosome maturation mechanism mediated by TECPR1 and the Atg12-Atg5 conjugate. Mol Cell. 2012;45:629–641. doi: 10.1016/j.molcel.2011.12.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pankiv S, Alemu EA, Brech A, Bruun JA, Lamark T, Overvatn A, Bjorkoy G, Johansen T. FYCO1 is a Rab7 effector that binds to LC3 and PI3P to mediate microtubule plus end-directed vesicle transport. J Cell Biol. 2010;188:253–269. doi: 10.1083/jcb.200907015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jager S, Bucci C, Tanida I, Ueno T, Kominami E, Saftig P, Eskelinen EL. Role for Rab7 in maturation of late autophagic vacuoles. J Cell Sci. 2004;117:4837–4848. doi: 10.1242/jcs.01370. [DOI] [PubMed] [Google Scholar]
- 66.Gutierrez MG, Munafo DB, Beron W, Colombo MI. Rab7 is required for the normal progression of the autophagic pathway in mammalian cells. J Cell Sci. 2004;117:2687–2697. doi: 10.1242/jcs.01114. [DOI] [PubMed] [Google Scholar]
- 67.Juhasz G, Hill JH, Yan Y, Sass M, Baehrecke EH, Backer JM, Neufeld TP. The class III PI(3)K Vps34 promotes autophagy and endocytosis but not TOR signaling in Drosophila. J Cell Biol. 2008;181:655–666. doi: 10.1083/jcb.200712051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhou X, Takatoh J, Wang F. The mammalian class 3 PI3K (PIK3C3) is required for early embryogenesis and cell proliferation. PLoS One. 2011;6:e16358. doi: 10.1371/journal.pone.0016358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhou X, Wang L, Hasegawa H, Amin P, Han BX, Kaneko S, He Y, Wang F. Deletion of PIK3C3/Vps34 in sensory neurons causes rapid neurodegeneration by disrupting the endosomal but not the autophagic pathway. Proc Natl Acad Sci U S A. 2010;107:9424–9429. doi: 10.1073/pnas.0914725107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.McLeod IX, Zhou X, Li QJ, Wang F, He YW. The class III kinase Vps34 promotes T lymphocyte survival through regulating IL-7Ralpha surface expression. J Immunol. 2011;187:5051–5061. doi: 10.4049/jimmunol.1100710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jaber N, Dou Z, Chen JS, Catanzaro J, Jiang YP, Ballou LM, Selinger E, Ouyang X, Lin RZ, Zhang J, et al. Class III PI3K Vps34 plays an essential role in autophagy and in heart and liver function. Proc Natl Acad Sci U S A. 2012;109:2003–2008. doi: 10.1073/pnas.1112848109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Willinger T, Flavell RA. Canonical autophagy dependent on the class III phosphoinositide-3 kinase Vps34 is required for naive T-cell homeostasis. Proc Natl Acad Sci U S A. 2012;109:8670–8675. doi: 10.1073/pnas.1205305109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Falasca M, Maffucci T. Regulation and cellular functions of class II phosphoinositide 3-kinases. The Biochemical journal. 2012;443:587–601. doi: 10.1042/BJ20120008. [DOI] [PubMed] [Google Scholar]
- 74.Shin HW, Hayashi M, Christoforidis S, Lacas-Gervais S, Hoepfner S, Wenk MR, Modregger J, Uttenweiler-Joseph S, Wilm M, Nystuen A, et al. An enzymatic cascade of Rab5 effectors regulates phosphoinositide turnover in the endocytic pathway. J Cell Biol. 2005;170:607–618. doi: 10.1083/jcb.200505128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Codogno P, Mehrpour M, Proikas-Cezanne T. Canonical and non-canonical autophagy: variations on a common theme of self-eating? Nat Rev Mol Cell Biol. 2012;13:7–12. doi: 10.1038/nrm3249. [DOI] [PubMed] [Google Scholar]
- 76.Nishida Y, Arakawa S, Fujitani K, Yamaguchi H, Mizuta T, Kanaseki T, Komatsu M, Otsu K, Tsujimoto Y, Shimizu S. Discovery of Atg5/Atg7-independent alternative macroautophagy. Nature. 2009;461:654–658. doi: 10.1038/nature08455. [DOI] [PubMed] [Google Scholar]
- 77.Mauthe M, Jacob A, Freiberger S, Hentschel K, Stierhof YD, Codogno P, Proikas-Cezanne T. Resveratrol-mediated autophagy requires WIPI-1-regulated LC3 lipidation in the absence of induced phagophore formation. Autophagy. 2011;7:1448–1461. doi: 10.4161/auto.7.12.17802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Vergne I, Roberts E, Elmaoued RA, Tosch V, Delgado MA, Proikas-Cezanne T, Laporte J, Deretic V. Control of autophagy initiation by phosphoinositide 3-phosphatase Jumpy. Embo J. 2009;28:2244–2258. doi: 10.1038/emboj.2009.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Dowling JJ, Low SE, Busta AS, Feldman EL. Zebrafish MTMR14 is required for excitation-contraction coupling, developmental motor function and the regulation of autophagy. Hum Mol Genet. 2010;19:2668–2681. doi: 10.1093/hmg/ddq153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Taguchi-Atarashi N, Hamasaki M, Matsunaga K, Omori H, Ktistakis NT, Yoshimori T, Noda T. Modulation of local PtdIns3P levels by the PI phosphatase MTMR3 regulates constitutive autophagy. Traffic. 2010;11:468–478. doi: 10.1111/j.1600-0854.2010.01034.x. [DOI] [PubMed] [Google Scholar]
- 81.Martin KR, Xu Y, Looyenga BD, Davis RJ, Wu CL, Tremblay ML, Xu HE, MacKeigan JP. Identification of PTPsigma as an autophagic phosphatase. J Cell Sci. 2011;124:812–819. doi: 10.1242/jcs.080341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Cebollero E, van der Vaart A, Zhao M, Rieter E, Klionsky DJ, Helms JB, Reggiori F. Phosphatidylinositol-3-phosphate clearance plays a key role in autophagosome completion. Curr Biol. 2012;22:1545–1553. doi: 10.1016/j.cub.2012.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.de Lartigue J, Polson H, Feldman M, Shokat K, Tooze SA, Urbe S, Clague MJ. PIKfyve regulation of endosome-linked pathways. Traffic. 2009;10:883–893. doi: 10.1111/j.1600-0854.2009.00915.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ho CY, Alghamdi TA, Botelho RJ. Phosphatidylinositol-3,5-bisphosphate: no longer the poor PIP2. Traffic. 2012;13:1–8. doi: 10.1111/j.1600-0854.2011.01246.x. [DOI] [PubMed] [Google Scholar]
- 85.Efe JA, Botelho RJ, Emr SD. The Fab1 phosphatidylinositol kinase pathway in the regulation of vacuole morphology. Curr Opin Cell Biol. 2005;17:402–408. doi: 10.1016/j.ceb.2005.06.002. [DOI] [PubMed] [Google Scholar]
- 86.Dove SK, Dong K, Kobayashi T, Williams FK, Michell RH. Phosphatidylinositol 3,5-bisphosphate and Fab1p/PIKfyve underPPIn endo-lysosome function. The Biochemical journal. 2009;419:1–13. doi: 10.1042/BJ20081950. [DOI] [PubMed] [Google Scholar]
- 87.Rusten TE, Vaccari T, Lindmo K, Rodahl LM, Nezis IP, Sem-Jacobsen C, Wendler F, Vincent JP, Brech A, Bilder D, et al. ESCRTs and Fab1 regulate distinct steps of autophagy. Curr Biol. 2007;17:1817–1825. doi: 10.1016/j.cub.2007.09.032. [DOI] [PubMed] [Google Scholar]
- 88.Ferguson CJ, Lenk GM, Meisler MH. Defective autophagy in neurons and astrocytes from mice deficient in PI(3,5)P2. Hum Mol Genet. 2009;18:4868–4878. doi: 10.1093/hmg/ddp460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ferguson CJ, Lenk GM, Meisler MH. PtdIns(3,5)P2 and autophagy in mouse models of neurodegeneration. Autophagy. 2010;6:170–171. doi: 10.4161/auto.6.1.10626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ferguson CJ, Lenk GM, Jones JM, Grant AE, Winters JJ, Dowling JJ, Giger RJ, Meisler MH. Neuronal expression of Fig4 is both necessary and sufficient to prevent spongiform neurodegeneration. Hum Mol Genet. 2012;21:3525–3534. doi: 10.1093/hmg/dds179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Yamashita S, Oku M, Wasada Y, Ano Y, Sakai Y. PI4P-signaling pathway for the synthesis of a nascent membrane structure in selective autophagy. J Cell Biol. 2006;173:709–717. doi: 10.1083/jcb.200512142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wang K, Yang Z, Nair U, Mao K, Liu X, Klionsky DJ. Phosphatatidylinositol 4-kinases are required for autophagic membrane trafficking. J Biol Chem. 2012 doi: 10.1074/jbc.M112.371591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Villasmil ML, Bankaitis VA, Mousley CJ. The oxysterol-binding protein superfamily: new concepts and old proteins. Biochemical Society transactions. 2012;40:469–473. doi: 10.1042/BST20120012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.LeBlanc MA, McMaster CR. Lipid binding requirements for oxysterol-binding protein Kes1 inhibition of autophagy and endosome-trans-Golgi trafficking pathways. J Biol Chem. 2010;285:33875–33884. doi: 10.1074/jbc.M110.147264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Yu L, McPhee CK, Zheng L, Mardones GA, Rong Y, Peng J, Mi N, Zhao Y, Liu Z, Wan F, et al. Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature. 2010;465:942–946. doi: 10.1038/nature09076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Rong Y, Liu M, Ma L, Du W, Zhang H, Tian Y, Cao Z, Li Y, Ren H, Zhang C, et al. Clathrin and phosphatidylinositol-4,5-bisphosphate regulate autophagic lysosome reformation. Nature cell biology. 2012;14:924–934. doi: 10.1038/ncb2557. [DOI] [PubMed] [Google Scholar]
- 97.Jenkins GM, Frohman MA. Phospholipase D: a lipid centric review. Cell Mol Life Sci. 2005;62:2305–2316. doi: 10.1007/s00018-005-5195-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Wiczer BM, Thomas G. Phospholipase D and mTORC1: nutrients are what bring them together. Sci Signal. 2012;5:pe13. doi: 10.1126/scisignal.2003019. [DOI] [PubMed] [Google Scholar]
- 99.Dall’Armi C, Hurtado-Lorenzo A, Tian H, Morel E, Nezu A, Chan RB, Yu WH, Robinson KS, Yeku O, Small SA, et al. The phospholipase D1 pathway modulates macroautophagy. Nat Commun. 2010;1:142. doi: 10.1038/ncomms1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Yoon MS, Du G, Backer JM, Frohman MA, Chen J. Class III PI-3-kinase activates phospholipase D in an amino acid-sensing mTORC1 pathway. J Cell Biol. 2011;195:435–447. doi: 10.1083/jcb.201107033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Shahnazari S, Yen WL, Birmingham CL, Shiu J, Namolovan A, Zheng YT, Nakayama K, Klionsky DJ, Brumell JH. A diacylglycerol-dependent signaling pathway contributes to regulation of antibacterial autophagy. Cell host & microbe. 2010;8:137–146. doi: 10.1016/j.chom.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Cemma M, Brumell JH. Interactions of pathogenic bacteria with autophagy systems. Curr Biol. 2012;22:R540–545. doi: 10.1016/j.cub.2012.06.001. [DOI] [PubMed] [Google Scholar]
- 103.Zheng YT, Shahnazari S, Brech A, Lamark T, Johansen T, Brumell JH. The adaptor protein p62/SQSTM1 targets invading bacteria to the autophagy pathway. J Immunol. 2009;183:5909–5916. doi: 10.4049/jimmunol.0900441. [DOI] [PubMed] [Google Scholar]
- 104.Tan SH, Shui G, Zhou J, Li JJ, Bay BH, Wenk MR, Shen HM. Induction of autophagy by palmitic acid via protein kinase C-mediated signaling pathway independent of mTOR (mammalian target of rapamycin) J Biol Chem. 2012;287:14364–14376. doi: 10.1074/jbc.M111.294157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Sakaki K, Wu J, Kaufman RJ. Protein kinase Ctheta is required for autophagy in response to stress in the endoplasmic reticulum. J Biol Chem. 2008;283:15370–15380. doi: 10.1074/jbc.M710209200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Manjithaya R, Anjard C, Loomis WF, Subramani S. Unconventional secretion of Pichia pastoris Acb1 is dependent on GRASP protein, peroxisomal functions, and autophagosome formation. J Cell Biol. 2010;188:537–546. doi: 10.1083/jcb.200911149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Moreau K, Ravikumar B, Puri C, Rubinsztein DC. Arf6 promotes autophagosome formation via effects on phosphatidylinositol 4,5-bisphosphate and phospholipase D. J Cell Biol. 2012;196:483–496. doi: 10.1083/jcb.201110114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Ravikumar B, Moreau K, Jahreiss L, Puri C, Rubinsztein DC. Plasma membrane contributes to the formation of pre-autophagosomal structures. Nature cell biology. 2010;12:747–757. doi: 10.1038/ncb2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Schweitzer JK, Sedgwick AE, D’Souza-Schorey C. ARF6-mediated endocytic recycling impacts cell movement, cell division and lipid homeostasis. Seminars in cell & developmental biology. 2011;22:39–47. doi: 10.1016/j.semcdb.2010.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Jovanovic OA, Brown FD, Donaldson JG. An effector domain mutant of Arf6 implicates phospholipase D in endosomal membrane recycling. Mol Biol Cell. 2006;17:327–335. doi: 10.1091/mbc.E05-06-0523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Henage LG, Exton JH, Brown HA. Kinetic analysis of a mammalian phospholipase D: allosteric modulation by monomeric GTPases, protein kinase C, and polyphosphoinositides. J Biol Chem. 2006;281:3408–3417. doi: 10.1074/jbc.M508800200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kim JH, Park JM, Yea K, Kim HW, Suh PG, Ryu SH. Phospholipase D1 mediates AMP-activated protein kinase signaling for glucose uptake. PLoS One. 2010;5:e9600. doi: 10.1371/journal.pone.0009600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Bodemann BO, Orvedahl A, Cheng T, Ram RR, Ou YH, Formstecher E, Maiti M, Hazelett CC, Wauson EM, Balakireva M, et al. RalB and the exocyst mediate the cellular starvation response by direct activation of autophagosome assembly. Cell. 2011;144:253–267. doi: 10.1016/j.cell.2010.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Mihaylova MM, Shaw RJ. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nature cell biology. 2011;13:1016–1023. doi: 10.1038/ncb2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Sun Y, Chen J. mTOR signaling: PLD takes center stage. Cell Cycle. 2008;7:3118–3123. doi: 10.4161/cc.7.20.6881. [DOI] [PubMed] [Google Scholar]
- 116.Xu L, Salloum D, Medlin PS, Saqcena M, Yellen P, Perrella B, Foster DA. Phospholipase D mediates nutrient input to mammalian target of rapamycin complex 1 (mTORC1) J Biol Chem. 2011;286:25477–25486. doi: 10.1074/jbc.M111.249631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Bridges D, Ma JT, Park S, Inoki K, Weisman LS, Saltiel AR. Phosphatidylinositol 3,5-bisphosphate plays a role in the activation and subcellular localization of mechanistic target of rapamycin 1. Mol Biol Cell. 2012;23:2955–2962. doi: 10.1091/mbc.E11-12-1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Bedia C, Levade T, Codogno P. Regulation of autophagy by sphingolipids. Anticancer Agents Med Chem. 2011;11:844–853. doi: 10.2174/187152011797655131. [DOI] [PubMed] [Google Scholar]
- 119.Scarlatti F, Bauvy C, Ventruti A, Sala G, Cluzeaud F, Vandewalle A, Ghidoni R, Codogno P. Ceramide-mediated macroautophagy involves inhibition of protein kinase B and up-regulation of beclin 1. J Biol Chem. 2004;279:18384–18391. doi: 10.1074/jbc.M313561200. [DOI] [PubMed] [Google Scholar]
- 120.Pattingre S, Bauvy C, Carpentier S, Levade T, Levine B, Codogno P. Role of JNK1-dependent Bcl-2 phosphorylation in ceramide-induced macroautophagy. J Biol Chem. 2009;284:2719–2728. doi: 10.1074/jbc.M805920200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Guenther GG, Peralta ER, Rosales KR, Wong SY, Siskind LJ, Edinger AL. Ceramide starves cells to death by downregulating nutrient transporter proteins. Proc Natl Acad Sci U S A. 2008;105:17402–17407. doi: 10.1073/pnas.0802781105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Lavieu G, Scarlatti F, Sala G, Carpentier S, Levade T, Ghidoni R, Botti J, Codogno P. Regulation of autophagy by sphingosine kinase 1 and its role in cell survival during nutrient starvation. J Biol Chem. 2006;281:8518–8527. doi: 10.1074/jbc.M506182200. [DOI] [PubMed] [Google Scholar]
- 123.Lepine S, Allegood JC, Park M, Dent P, Milstien S, Spiegel S. Sphingosine-1-phosphate phosphohydrolase-1 regulates ER stress-induced autophagy. Cell death and differentiation. 2011;18:350–361. doi: 10.1038/cdd.2010.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Hayashi-Nishino M, Fujita N, Noda T, Yamaguchi A, Yoshimori T, Yamamoto A. A subdomain of the endoplasmic reticulum forms a cradle for autophagosome formation. Nat Cell Biol. 2009;11:1433–1437. doi: 10.1038/ncb1991. [DOI] [PubMed] [Google Scholar]
- 125.Yla-Anttila P, Vihinen H, Jokitalo E, Eskelinen EL. 3D tomography reveals connections between the phagophore and endoplasmic reticulum. Autophagy. 2009;5:1180–1185. doi: 10.4161/auto.5.8.10274. [DOI] [PubMed] [Google Scholar]
- 126.Hailey DW, Rambold AS, Satpute-Krishnan P, Mitra K, Sougrat R, Kim PK, Lippincott-Schwartz J. Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell. 2010;141:656–667. doi: 10.1016/j.cell.2010.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Koga H, Kaushik S, Cuervo AM. Altered lipid content inhibits autophagic vesicular fusion. Faseb J. 2010;24:3052–3065. doi: 10.1096/fj.09-144519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Rodriguez-Navarro JA, Kaushik S, Koga H, Dall’Armi C, Shui G, Wenk MR, Di Paolo G, Cuervo AM. Inhibitory effect of dietary lipids on chaperone-mediated autophagy. Proc Natl Acad Sci U S A. 2012;109:E705–714. doi: 10.1073/pnas.1113036109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Wenk MR. Lipidomics: new tools and applications. Cell. 2010;143:888–895. doi: 10.1016/j.cell.2010.11.033. [DOI] [PubMed] [Google Scholar]