

Abstract
Dysregulation of the PI3K/mTOR pathway, either through amplifications, deletions, or as a direct result of mutations, has been closely linked to the development and progression of a wide range of cancers. Moreover, this pathway activation is a poor prognostic marker for many tumor types and confers resistance to various cancer therapies. Here, we describe VS-5584, a novel, low-molecular weight compound with equivalent potent activity against mTOR (IC50 = 37 nmol/L) and all class I phosphoinositide 3-kinase (PI3K) isoforms IC50: PI3Kα = 16 nmol/L; PI3Kβ = 68 nmol/L; PI3Kγ = 25 nmol/L; PI3Kδ = 42 nmol/L, without relevant activity on 400 lipid and protein kinases. VS-5584 shows robust modulation of cellular PI3K/mTOR pathways, inhibiting phosphorylation of substrates downstream of PI3K and mTORC1/2. A large human cancer cell line panel screen (436 lines) revealed broad antiproliferative sensitivity and that cells harboring mutations in PI3KCA are generally more sensitive toward VS-5584 treatment. VS-5584 exhibits favorable pharmacokinetic properties after oral dosing in mice and is well tolerated. VS-5584 induces long-lasting and dose-dependent inhibition of PI3K/mTOR signaling in tumor tissue, leading to tumor growth inhibition in various rapalog-sensitive and -resistant human xenograft models. Furthermore, VS-5584 is synergistic with an EGF receptor inhibitor in a gastric tumor model. The unique selectivity profile and favorable pharmacologic and pharmaceutical properties of VS-5584 and its efficacy in a wide range of human tumor models supports further investigations of VS-5584 in clinical trials.
Introduction
The phosphoinositide 3-kinase (PI3K) signaling pathway is crucial to many aspects of cell growth and survival via its regulation of diverse physiologic processes that include cell-cycle progression, differentiation, transcription, translation, and apoptosis (1). The PI3K family of lipid kinases consists of 3 classes based on their substrate specificity and sequence homology. In class I PI3K, 4 isoforms of the catalytic subunit p110 have been identified, whereby the α and β isoforms are ubiquitously expressed and the γ and δ are mainly expressed in leukocytes (2). Dysregulation of the PI3K class I signaling pathway, either through gene amplification or as a direct result of mutations, has been closely linked to the development and progression of a wide range of cancers. Genetic alterations in proteins of this signaling pathway include p85 (regulatory subunit of PI3Kα), p110α, PDK1, PTEN, and Akt (3, 4). The dysregulated PI3K pathway induces a myriad of downstream effectors including mTOR. mTOR is a member of the PI3K-related kinase (PIKK) family, which includes PI3K, DNA-dependent protein kinase (DNA-PK), and ataxia telangiectasia mutated (ATM). Its catalytic kinase domain is highly homologous to the lipid kinase domain of PI3K. In mammals, mTOR is the catalytic subunit in 2 distinct complexes, mTORC1 and mTORC2. mTORC1 controls cellular growth by integrating signals from growth factor receptors and intracellular nutrient status. mTORC2 is less well understood but plays a role in the regulation of cellular survival and cell migration (5, 6). The mTOR signaling pathway has been suggested to be involved in multiple anticancer drug resistance mechanisms toward chemotherapeutics but also signal transduction inhibitors (small-molecule tyrosine kinase inhibitors and antibodies; ref. 4). Rapamycin and its analogs block mTORC1 activity and have shown single-agent activity in a small subsets of cancers (7). However, resistance has been shown to develop through activation of the PI3K signaling pathway including activation of mTORC2(8). To overcome this and also broaden applications, the rapalogs are now being evaluated in combination with other standard or targeted therapies. We have taken another approach, which is to directly block mTOR kinase with compounds that bind the ATP site, and therefore block both mTOR complexes. In addition, we wanted to generate compounds that also inhibit PI3K with equivalent potency to overcome one of the key resistance pathways activated by mTORC1 inhibition.
VS-5584 is a novel low-molecular weight compound with high and equivalent potency against mTOR and all PI3K class I isoforms but with no relevant activity for more than 400 lipid and protein kinases, and thereby having a differentiating profile compared with currently available clinical stage compounds. The present study characterizes the pharmacodynamic and pharmacokinetic relationship of VS-5584 in human tumor models and shows superior efficacy and broad antitumor efficacy of VS-5584 and tolerability across a range of cancer types.
Materials and Methods
Compounds and reagents
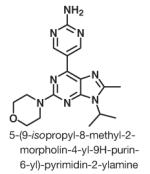
VS-5584 (formerly named SB2343), as depicted in Table 1: 5-(9-isopropyl-8-methyl-2-morpholin-4-yl-9H-purin-6-yl)-pyrimidin-2-ylamine was discovered and synthesized by S*BIO Pte Ltd. (Singapore). Synthesis of VS-5584 is described in a published patent application WO WO2010114484 (9). Gefitinib was purchased at LC Laboratories, 5-fluorouracil (5-FU) and everolimus were obtained from Sigma-Aldrich. For in vivo studies, VS-5584, everolimus, and gefitinib dosing solutions were prepared in 0.5% methylcellulose (w/v) and 0.1% Tween-80 in H2O (MC/Tween). 5-FU was dissolved in sterile saline. IFN-α, IFN-γ, interleukin (IL)-2, and IL-3 were purchased from i-DNA.
Table 1.
Summary table showing the chemical structure and lipid kinase in vitro enzyme profile of VS-5584
| Structure | Kinase | IC50 (nmol/L) |
|---|---|---|
|
mTOR | 37 (±7) |
| PI3Kα | 16 (±3) | |
| PI3Kβ | 68 (±9) | |
| PI3Kγ | 25 (±5) | |
| PI3Kδ | 42 (±8) | |
| ATM/ATR | >10,000 | |
| DNA-PK | 1,270 (±321) | |
| Vps34 | 7,470 (±1,300 |
NOTE: Values (mean ± SD) were obtained from S*BIO in-house assays (n > 3).
In vitro kinase assays
For details on the in vitro kinase assays for mTOR, class I PI3K α/β/γ/δ, ATM, ATR and for kinase profiling at Invitrogen, Millipore, Ambit, and ProQinase please see the Supplementary Information.
Cell culture and proliferation assay
SET-2 cells were obtained from Deutsche Sammlung von Mikroorganismen und Zellkulturen GmbH. SNU-478, SNU-1196, SNU-245, SNU-1079, SNU-308, and SNU-869 cells were purchased from Korean Cell Line Bank. MKN7 were obtained from the Japanese Collection of Research Bioresources. Experiments on multiple myeloma cells (H929, MM1.S, MM1.R, R8226, U266) and nasopharyngeal cells (CNE-1, CNE-2, HONE1, HK1) were conducted at Drs. Wee Joo Chng’s and Boon Cher Goh’s laboratory. All other cells were obtained from the American Type Culture Collection. All cells were cultivated according cultivated according to the vendor’s instructions, tested for mycoplasma contamination (Mycoplasma Plus PCR Primer Set, Stratagene; Agilent Technologies Inc), and verified by short tandem repeat (STR) profiling (John Hopkins University, Baltimore, MD). For proliferation assays in 96-well plates, cells were seeded at 30% to 50% confluency for adherent cells, or 2,000 to 6,000 cells for suspension cells and treated the following day with compounds (in triplicates) at concentrations up to 10 μmol/L for 48 hours. Cell viability was monitored using the CellTiter-Glo assay (Promega). Dose–response curves were plotted to determine IC50 values for the compounds using the XL-fit software (IDBS Ltd.). Detailed information on the drug sensitivity and genetic profiling cancer cell lines panel are provided in the Supplementary Information.
Western blot analysis
Cells were lysed and proteins immunoprecipitated as previously described (10). Western blot analyses were conducted according to standard methods. pAkt (S473l Cat #9271), pAkt (T308; Cat #2965), pS6 ribosomal protein (S240/244), pmTOR (S2481; Cat #2974), pErk1/2 (T202/y204; Cat# 4376), and anti-rabbit immunoglobulin G (IgG), horseradish peroxidase (HRP)-linked (Cat #7074) antibodies were purchased from Cell Signaling Technology. β-Actin (Cat #A2066) was purchased from Sigma. The images were captured digitally using the LAS-3000 Life Science Imager from Fujifilm. Densitometric analysis was conducted using the MultiGauge software (v3.1) from Fujifilm.
In vivo efficacy studies
Athymic BALB/c nude mice (BALB/cOlaHsd-Foxn1nu) were obtained from the Biological Resource Centre (BRC, Biopolis, Singapore). Fox-Chase severe combined immunodeficient (SCID) mice (CB17/Icr-Prkdcscid/CrlBltw) were obtained from Biolasco. Standard protocols were followed, in compliance with the guidelines of NIH and National Advisory Committee for Laboratory Animal Research guidelines [Institutional Animal Care and Use Committee (IACUC) approval #0800371].
Male (PC3 and COLO 205) or female (MV4-11 and HuH7) BALB/c nude mice or female SCID mice (NCI-N87) were implanted intradermally in the right flank with 5 × 106 (PC3, COLO205, HuH7, NCI-N87) or 1 × 107 (MV4-11) cells. Cells were resuspended in 70% (v/v; COLO205 and HuH7 only) or 50% (v/v) serum-free growth medium/Matrigel (Cat. No: 354248; BD Biosciences) and injected in a total volume of 100 μL, using a 27.5-gauge needle.
Dosing started 7 to 14 days after tumor implantation. VS-5584 was dosed daily orally. The reference compounds everolimus and gefitinib were dosed p.o. at 5 and 150 mg/kg, respectively, with everolimus dosed daily and gefitinib dosed for 5 day-on and 2 day-off in cycles. 5-FU was administered intraperitoneally, at 25 mg/kg, every Tuesday, Thursday, and Saturday. All statistics conducted were done using GraphPad Prism (v5), (GraphPad Software Inc.). For histology, please see the Supplementary Information.
Results
Kinase selectivity spectrum of VS-5584
VS-5584, 5-(9-isopropyl-8-methyl-2-morpholin-4-yl-9H-purin-6-yl)-pyrimidin-2-ylamine, a novel purine analog (11, 12; Table 1), was generated with the aid of computational chemistry to be a small-molecule ATP competitive inhibitor of PI3K and mTOR kinases with favorable pharmaceutical properties. The synthesis of the compound will be published in a separate article (13). To further explore its kinase selectivity spectrum, VS-5584 was profiled against 2 large kinase panels (>400 kinases) covering all major families of the human protein and lipid kinome.
VS-5584 is a potent inhibitor of mTOR (IC50 = 37 nmol/L) as well as class I PI3K isoforms (IC50: PI3Kα = 16 nmol/L; PI3Kβ = 68 nmol/L; PI3Kγ = 25 nmol/L; PI3Kδ = 42 nmol/L). The Ambit full panel screening revealed that besides mTOR and the PI3K family, only NEK2 and BTK showed potential binding (less than 5%) of VS-5584 (Supplementary Fig. S1). All other evaluated kinases showed negligible binding when tested up to 10 μmol/L VS-5584 (Table 1). Further analysis of 320 kinases (including NEK2 and BTK) in a radiometric kinase assay platform showed that no kinase showed an IC50 < 300 nmol/L except for the PIKK family (data not shown).
Modulation of PI3K/mTOR signaling pathways by VS-5584
To investigate whether the enzyme inhibitory properties of VS-5584 translate into modulation of the PI3K/mTOR signaling pathway, the phosphorylation status of downstream substrates were determined in human cancer cell lines with different genetic backgrounds.
First, the effects of VS-5584 on PC3, a prostate cancer cell line with PTEN deletion were examined. Treatment of the cells resulted in an equipotent inhibition of both the PI3K and the mTOR signaling cascade after 3 hours (Fig. 1A). The IC50 values for pS6(S240/244), pAkt(S473), and pAkt (T308) were 20, 23, and 15 nmol/L.
Figure 1.

VS-5584 effectively blocks PI3K/mTOR signaling in cancer cells with different genetic background. A, PC3 cells were treated with VS-5584 for 3 hours as indicated. After lysis, phosphorylation status of pS6 and pAkt were detected by immunoblotting. B, structure of everolimus. MV4-11 (C), NCI-N87 (D), COLO-205 (E), MDA-MB-231 (F), and HUH-7 (G) cells were treated with VS-5584 for 3 hours as indicated or everolimus (100 nmol/L for 3 hours). After lysis, phosphorylation status of pS6, pAkt, pmTOR, and pERK1/2 was detected by immunoblotting.
Next, the effect of VS-5584 on signaling of overexpressed or mutated receptor tyrosine kinases was studied. In the FLT3-ITD harboring MV4-11 cells, VS-5584 blocked pAkt(S473) and pAkt(T308) with an IC50 of 12 and 13 nmol/L (Fig. 1B). Everolimus (also known as RAD001; Fig. 1B), an inhibitor of mTORC1, but not mTORC2, was not able to inhibit phosphorylation of Akt (Fig. 1C). In the HER2 overexpressing gastric cancer cell line NCI-N87, VS-5584 potently blocked pS6. Interestingly, pAkt(S473) and pAkt(T308) showed a higher IC50 compared with pS6. mitogen-activated protein kinase (MAPK) activity was not blocked up to 1,000 nmol/L of VS-5584, showing specificity for the PI3K signaling pathway (Fig. 1D).
Furthermore, we investigated whether an activated Ras/MAPK pathway interferes in the inhibition of PI3K/mTOR by VS-5584. We treated Colo205 (BRAF V600E, mTOR P1193L) and MDA-MB-231 (BRAF G464V, KRAS G13D) with VS-5584 for 3 hours. Despite having an activated MAPK pathway, the IC50 of VS-5584 on pAkt and pS6 was not higher compared with PC3, MV4-11, or NCI-N87 (Fig. 1E and F).
In addition to the effects of VS-5584 on cell lines with known mutations in the PI3K/MAPK pathway, studies were conducted on HuH7 cells, which do not have any known genetic alterations in these signaling pathways. Similar to our earlier findings, VS-5584 blocked PI3K and mTOR signaling in the same range with IC50 of 20 nmol/L for pS6(S240/244) and 10 nmol/L for pAkt(S473; Fig. 1G).
Overall, these data show that VS-5584 effectively permeates cells to modulate signaling pathways downstream of PI3K/mTOR independent of the genetic background of the cells.
VS-5584 potently blocks proliferation in a broad spectrum of tumor cells
As the PI3K/mTOR signaling pathway regulates important functional responses including cell proliferation, the effects of VS-5584 on a panel of 51 cancer cell lines derived from both liquid and solid tumors of human origin were investigated.
Overall, VS-5584 showed high antiproliferative activity in a broad spectrum of cancer cells, with H929 (multiple myeloma) showing the highest sensitivity in our panel (IC50 = 48 nmol/L; Supplementary Fig. S2). Of note, VS-5584 was potent against many rapamycin-resistant cell lines.
Pharmacokinetic and pharmacodynamic properties of VS-5584
To investigate the efficacy of VS-5584 in disease models, the pharmacokinetic and pharmacodynamic profile of VS-5584 was determined to enable the selection of an optimal dosing schedule. A single oral dose of VS-5584 was rapidly absorbed with a tmax of 0.9 hours and an elimination half-life of 10 hours (Supplementary Fig. S3). To determine the pharmacokinetic and pharmacodynamic relationship in tumors, PC3-tumor–bearing mice were treated with a single dose of VS-5584 and plasma and tumors were harvested after 6 hours and analyzed for concentrations of VS-5584 and effects on target efficacy biomarkers. Plasma levels of VS-5584 increased dose-dependently (Fig. 2A). Plasma pharmacokinetic was not significantly different to tumor pharmacokinetic. Drug levels exceeded the IC50 for inhibition of the target kinases in the enzymatic and cell-based assays starting from 3.7 mg/kg. Dose-dependent inhibition of pAkt(S473) and pS6 (S240/244) was observed in tumor tissue with complete inhibition from 33 mg/kg (EC50 of 4.2 and 1.7 mg/kg; Fig. 2B). To study the time course of drug levels and inhibition of target kinase signaling in plasma and tumor, PC3-tumor–bearing mice were treated with a single oral dose of 33 mg/kg VS-5584 and the tissues harvested 1, 6, and 24 hours postdosing. The plasma concentration of VS-5584 following the 33 mg/kg dose of VS-5584 was highest 1 hour after dosing (1221 ng/mL or 3.55 μmol/L) and was still above concentrations required to block the targets in in vitro assays after 24 hours (15 ng/mL or 43 nmol/L; Fig. 2C). pAkt(S473) and pS6(S240/244) were blocked by 90% or more within 1 hour of VS-5584 treatment and remained inhibited by 60% to 70% after 24 hours (Fig. 2D).
Figure 2.

Pharmacokinetic (PK)/pharmacodynamic properties of VS-5584. A, PC3 tumor-bearing mice received various single doses of VS-5584 as indicated. Mice were sacrificed 6 hours postdosing and the concentration of VS-5584 was determined in blood plasma and tumor tissue. B, the phosphorylation status of pAKT(S473) and pS6(S240/244) in tumor lysates 6 hours postdosing was determined by immunoblotting. C, PC3 tumor-bearing mice received a single dose of 33 mg/kg VS-5584 as indicated. Mice were sacrificed 1, 6, and 24 hours postdosing and the concentration of VS-5584 was determined in blood plasma and tumor tissue. D, 1, 6, and 24 hours postdosing the phosphorylation status of pAKT(S473) and pS6(S240/244) in tumor lysates was determined by immunoblotting.
Having established the pharmacokinetic and pharmacodynamic relationships for VS-5584 after single dosing and showing that a single dose was well tolerated up to 100 mg/kg, the maximum-tolerated dose (MTD) after chronic dosing was determined. Male Balb/C nude mice were dosed once daily for 14 consecutive days. Doses of 25 and 35 mg/kg VS-5584 were well tolerated with maximum observed body weight losses of 3.1% and 13.9% (data not shown).
Peripheral blood cell counts remained within normal levels without significant changes throughout dosing after the 25 and 35 mg/kg doses of VS-5584 (data not shown).
In summary, VS-5584 shows good oral bioavailability with dose-linear pharmacokinetic and a profound and long-lasting pharmacodynamic response in tumor tissue following a single oral dose in tumor bearing mice.
VS-5584 is efficacious in a PTENnull human prostate PC3 xenograft model
For evaluation of efficacy in a rapamycin-sensitive PC3 engraftment model, tumor-bearing mice were treated with VS-5584 for 28 days in comparison with the rapalog everolimus. VS-5584 was well tolerated at both doses tested (11 and 25 mg/kg) with minimal weight loss (mean 4.7% on day 27; Supplementary Fig. S4). Treatment with VS-5584 led to significant tumor growth inhibition (TGI) of 79% and 113% for 11 and 25 mg/kg, respectively. Everolimus at 5 mg/kg showed a TGI of 133% (Fig. 3A).
Figure 3.

VS-5584 is efficacious in a PTENnull human prostate PC3 xenograft model and in a rapamycin-resistant human colorectal COLO-205 xenograft model. A, PC3 tumor-bearing mice (n = 7/group) were treated daily for 28 days as indicated and the TGI determined. ANOVA with Dunnett posttest was conducted; ***, P < 0.001. B and C, 6 hours after the last treatment on day 27 the phosphorylation status of pS6, pAkt(S473) in tumor tissue was analyzed. ANOVA with Dunnett posttest was conducted; **, P < 0.01. D, Colo-205 tumor-bearing nude mice (n = 13/group) were treated daily for 18 days and the tumor volume monitored. E, tumor weight for each group is shown. ANOVA with Dunnett posttest was conducted; ***, P < 0.001. F, 6 hours after the last treatment on day 17 the phosphorylation status of S6, Akt(S473) in tumor tissue were determined. G, on day 17, active vessels in the tumors were stained with FITC-conjugated Ricinus communis agglutinin I and the vessel score determined. t test was conducted; *, P < 0.05.
Having shown that acute dosing of VS-5584 led to significant inhibition of cellular biomarkers in tumor tissue, the pharmacodynamic marker modulation in the tumors was also determined after chronic dosing. VS-5584 induced a near-complete inhibition of pS6 and pAkt(S473) 6 hours after the last dose on day 27 (95% and 85%, respectively at 25 mg/kg). The lower dose of 11 mg/kg of VS-5584 also induced significant inhibition of pS6 and pAkt(S473; 79% and 38%, respectively; Fig. 3B and C).
These results further show the effective and long-lasting inhibition of PI3K and mTOR signaling by VS-5584 in tumor tissue and that this results in significant inhibition of tumor growth at well-tolerated doses.
Therapeutic effects of VS-5584 in a rapamycin-resistant human colorectal COLO-205 xenograft model
To investigate therapeutic effects of VS-5584 on a rapamycin-resistant model, we used a subcutaneous COLO-205 xenograft model. VS-5584 was very efficacious in this aggressive tumor model and showed a dose-dependent efficacy with TGIs of 45%, 85%, and 86% for 11, 25, or 35 mg/kg (Fig. 3D). Everolimus at 5 mg/kg led to 29% TGI, which was not statistically significant. VS-5584 also showed dose-dependent efficacy based on final tumor weights (Fig. 3E). Furthermore, analysis of pharmacodynamic markers showed that 1h after the last dose on day 17, tumors of the 25 mg/kg group showed a significant reduction of 95% and 85% of pS6 and pAkt (Fig. 3F).
Having shown that VS-5584 blocks tumor growth of established COLO-205 tumors, the effects of VS-5584 on tumor vascularization were investigated. The number of patent blood vessels in COLO 205 tumors was compared after 18 days of treatment with either vehicle, 25 mg/kg VS-5584 or everolimus. Fluorescein isothiocyanate (FITC)–conjugated Riccinus communis agglutinin 1 (RCA1), which only binds to functional blood vessels, was injected 30 minutes before harvesting the tumors. The overall vessel score of the whole tumor sections was significantly reduced in the 25 mg/kg treatment group, whereas everolimus treatment did not reduce the vessel score significantly (Fig. 3G and Supplementary Fig. S5).
In summary, we have shown that a well-tolerated dose of VS-5584 blocks mTOR and PI3K signaling in tumor tissue and reduces the number of functional blood vessels in the tumor and is efficacious in a rapalog-resistant COLO-205 xenograft model.
VS-5584 is efficacious in a FLT3-ITD AML xenograft model
MV4-11 xenografts were treated for 26 consecutive days with 3.7 and 11 mg/kg of VS-5584. VS-5584 treatment induced dose-dependent inhibition of tumor growth (28% for 3.7 mg/kg and 76% for 11 mg/kg; Supplementary Fig. S6A). All doses were well tolerated with no significant body weight loss (Supplementary Fig. S6B). To investigate target modulation, MV4-11 harboring mice were given a single dose of 3.7 and 11 mg/kg VS-5584 and tumor samples taken 4 hours and the tumor lysates were analyzed for pAkt(T308). VS-5584 treatment was able to block pAkt(T308) already at the lowest dose of 3.7 mg/kg (Supplementary Fig. S6C)
In summary, these data show the VS-5584 is also efficacious at low and well-tolerated dose in liquid tumor model, namely the FLT3-ITD harboring MV4-11 xenograft model.
VS-5584 is efficacious as a single agent and has synergistic effects in combination with an EGFRi in a gastric xenograft model
VS-5584 was compared with 5-FU in a HER2 overexpressing gastric xenograft model. 5-FU (25 mg/kg i.v.) or VS-5584 (25 mg/kg p.o.) inhibited NCI-N87 tumor growth by 32% and 121%, which was only statistically significant for VS-5584 (Fig. 4A). Structure of 5-FU is shown in Fig. 4B. Measurement of pharmacodynamic markers for PI3K and mTOR inhibition in tumor tissue 6 hours after the last dosing on day 16 showed significant inhibition by VS-5584 but not by 5-FU treatment (Fig. 4C and D).
Figure 4.

High dose of VS-5584 or a low dose in combination with an EGFRi is efficacious in a gastric xenograft model. A, NCI-N87 tumor-bearing mice (n = 12/group) were treated for 17 days with 25 mg/kg orally daily. VS-5584 or with 25 mg/kg 5-FU intraperitoneally every Tuesday, Thursday, and Saturday for 2 weeks, followed by 1 week break. TGI on volume is indicated. ANOVA with Dunnett posttest was conducted, ***, P < 0.001. B, structure of 5-FU. C and D, 6 hours after dosing on day 16 phosphorylation status of mTOR, S6, Akt, and ERK1/2 and total actin was determined in tumor tissue. E, NCI-N87 tumor-bearing mice (n = 10/group) were treated for 26 days with 11 mg/kg orally daily. VS-5584, 150 mg/kg gefitinib orally dosed for 5 day-on and 2 day-off in cycles and combined treatment of 11 mg/kg VS-5584 with 150 mg/kg gefitinib (dosing schedule was the same as monotherapy). ANOVA with Dunnett posttest was conducted, ***, P < 0.001. F, structure of gefitinib.
Having shown inhibition of tumor growth in this model using VS-5584 as monotherapy, it was of interest to determine whether VS-5584 could be safely and beneficially combined with an EGF receptor inhibitor (EGFRi), a targeted therapy that recently has been tested in phase II in gastric cancer. Monotreatment of NCI-N87 tumor-bearing mice with VS-5584 at 11 mg/kg or gefitinib at 150 mg/kg resulted in a TGI of 88% and 17% (P < 0.001; Fig. 4E; structure of gefitinib is shown in Fig. 4F), which was only statistically significant for VS-5584. Combination therapy at the same dose levels resulted in a TGI of 121% (P < 0.001). The Clarke’s combination index was −0.1 indicating synergism (14). The combination was very well tolerated with no significant body weight loss (data not shown). Our data show that this tumor is highly sensitive to VS-5584 as a single agent and that the drug can act synergistically with gefitinib.
Drug sensitivity profiling in the Genomics of Drug Sensitivity in Cancer cell line panel
To identify putative biomarkers of sensitivity, a large panel of genetically characterized cancer cell lines was screened with VS-5584 to identify genomic features associated with drug sensitivity (15, 16). The cell line collection includes many common and rare cancer subtypes, and encompasses much of the genomic diversity found in cancer and which seems to be important in influencing drug response (Supplementary Table S1). To identify genomic biomarkers of sensitivity and resistance in the 436 cancer cell lines treated, we used a multivariate ANOVA to correlate cell line IC50 values and the slope of the dose–response with mutations in 66 cancer genes (point mutations and/or gene amplification and homozygous deletions), 3 gene rearrangements, and microsatellite instability. Notably, consistent with the target of VS-5584, mutation of PIK3CA was the genetic event most significantly associated with sensitivity [P = 0.0018; n = 50 PIK3CA-mutated cell lines and 386 wild-type (WT) cell lines; Fig. 5A; Supplementary Table S2]. A wide range of sensitivities to VS-5584 was observed in PIK3CA-mutated cell lines and overall the effect is modest with a approximately 2-fold difference in mean IC50 (geometric mean for PIK3CA = 237 nmol/L vs. 394 nmol/L for WT; Fig. 5B).
Figure 5.

Cell line profiling identifies an association between VS-5584 sensitivity and PIK3CA mutational status. A, a volcano plot representation from a multivariate ANOVA for cancer gene mutations associated with sensitivity and resistance to VS-5584. Each circle represents the correlation between VS-5584 and a single cancer gene. The effect on drug response (x-axis) and significance of the association (y-axis; inverted scale) is shown, and circle size is proportional to the number of cell lines screened with the given mutation (range, 1–291 depending on the gene). B, a scatterplot of IC50 values plotted on a log scale for WT and PIK3CA-mutated cell lines. Each circle represents the IC50 from a single cell line and the gray bar indicates the geometric mean. C, VS-5584 IC50 values in PIK3CA-mutated cell lines categorized by tissue type. PIK3CA WT cell lines are shown for comparison. D, a comparison of IC50 values in WT and PIK3CA-mutated breast cancer cell lines. E, sensitivity to VS-5584 is dependent on both PIK3CA and ERBB2 mutational status. The + and − symbols indicate the presence or absence of the indicated mutation. Kruskal–Wallis nonparametric analysis of variance was used comparing WT cells to the indicated mutant cell line populations. Only significant associations are indicated.
Mutations of PIK3CA occur in a wide range of tissues and we observed variable sensitivity to VS-5584 across different tissue types. For example, bladder and colorectal cancer cell lines with PIK3CA mutations were relatively insensitive to the drug (geometric mean of IC50 values are 522 nmol/L (n=4) and 553 nmol/L (n=7) for bladder and colorectal cancer (large intestine), whereas breast and upper aerodigestive tract cell lines with a PIK3CA mutation seemed to have approximately 4-fold enhanced sensitivity to VS-5584 [geometric mean of IC50 are 162 nmol/L (n = 12) and 158 nmol/L (n = 5) for breast and upper aerodigestive tract; Fig. 5C]. Indeed, PIK3CA-mutated breast cancer cell lines were significantly associated with sensitivity to VS-5584 compared with PIK3CA WT cells (Kruskal–Wallis test; P < 0.05). Moreover, sensitivity was specifically correlated with PIK3CA mutation rather than tissue type because breast cancer cell lines are not associated with sensitivity to VS-5584 compared with other tissues (data not shown), and PIK3CA-mutated breast cancer cells lines were more sensitive to VS-5584 than breast cancer cell lines lacking this mutation (Mann–Whitney test; P = 0.002; n = 12 PIK3CA-mutated and n = 16 PIK3CA-WT breast cancer cell lines; Fig. 5D).
Amplification of ERBB2 (HER2) is frequently observed in breast cancer and cell lines with coincident ERBB2 and PIK3CA mutations were more sensitive to VS-5584 than cell lines with either mutation alone (Fig. 5E). Cell lines with coincident mutation (n = 7) had a mean IC50 of 147 nmol/L as compared with 394 nmol/L (n = 374) in cell lines with neither mutation. In contrast, a coincident mutation in KRAS, which is frequently mutated in lung and colorectal cancers together with PIK3CA, had the opposite effect and weakly suppressed the sensitivity of PI3K-mutated cells to VS-5584 (Supplementary Fig. S7A). This slight decrease in sensitivity was not statistically significant. Moreover, we did not observe increased sensitivity of ERBB2-amplified cells with coincident PIK3CA mutations to the EGFR/ERBB2 inhibitor, BIBW2992 (Supplementary Fig. S7B and Supplementary Table S1). Collectively, our data indicate that although sensitivity to VS-5584 is associated with mutation of PIK3CA, the greatest sensitivity is observed in PIK3CA-mutated breast cancer cell lines with coincident ERBB2 amplification.
Mutation of EZH2, encoding for a histone-lysine N-methyltransferase involved in regulation of chromatin structure and gene expression, was also significantly correlated with sensitivity (Fig. 5A and Supplementary Table S2). The mechanism of this sensitivity is currently unclear and warrants further investigation. Interestingly, mutations of APC, MYCL1, or MYCN were correlated with drug resistance (Fig. 5A).
Discussion
The PI3K/mTOR pathway is one of the most commonly activated signaling pathway in human cancer. Many players in the PI3K pathway are either amplified, have undergone LOH, or are targeted by somatic or germline alterations (4). These observations led to the development of rapamycin and rapalogs, which are allosteric, irreversible inhibitors of mTORC1, for cancer treatment. Temsirolimus was approved for metastatic renal cell carcinoma in 2007, serving to validate the PI3K/mTOR pathway as a therapeutic target in cancer (17). Despite some success in selected tumor types, rapalogs generally showed very limited anticancer efficacy as single agents and mostly lead to cytostatic effects (18). Negative feedback loops involving S6K have been described to have dramatic effects on drug responses for mTORC1 inhibitors (19). Activated mTORC1 initiates a negative feedback cascade via S6K to downregulate PI3K activity. Treating tumors with rapalogs can result in increased PI3K/Akt activity leading to an enhanced proliferation rate of the tumor (19). Some ATP-competitive mTOR TKI inhibitors that inhibit both mTORC1 and 2, such as OSI-027, AZD8055, and INK128 have been developed and are currently undergoing clinical trials. However, resistance to these selective mTORC inhibitors can still arise via the PI3K feedback mechanism, by increased Akt(T308) phosphorylation or activation of Akt-independent PI3K targets (20). We have shown herein that VS-5584, with its selective PI3K/mTOR kinase activity, can overcome these feedback signaling mechanisms. Furthermore, upregulation of the RAS-MAPK pathway that occurs after mTORC1 inhibition alone, was not detected with VS-5584 treatment as it simultaneously blocks mTORC2 and PI3K as well as mTORC1 (21).
Interestingly, the activity of VS-5584 on inhibition of target modulation biomarkers was independent of the genetic background of the cancer cells indicating that PTEN loss or mutation status of upstream receptor tyrosine kinases do not necessarily predict inhibition of these events.
To identify genomic alterations that affect tumor cell response toward pathway inhibition by VS-5584 treatment, we screened across a large numbers of cancer cell lines to identify genetic biomarkers of sensitivity. Using this unbiased approach, we identified that mutations of EZH2 or PIK3CA are correlated with drug sensitivity, whereas mutations of APC, MYCL1, or MYCN are correlated with drug resistance. Notably, the greatest sensitivity to VS-5584 was associated with breast cancer cells bearing coincident amplification of ERBB2 in addition to a PIK3CA mutation. The precise reasons for this sensitivity are currently unclear but may reflect an enhanced requirement for signaling through the PI3K/mTOR pathway in this specific genetic context. Indeed, mutations in PIK3CA in breast cancer are indicators of sensitivity to the antitumor effects of the PI3K inhibitor GDC-0941 (22). Collectively, these results indicate that coincident mutation of PIK3CA and amplification of ERBB2 may have potential use as biomarkers of sensitivity to VS-5584 and may prove useful for patient stratification during clinical testing.
In contrast, as mutations in APC, MYCL1, or MYCN were associated with drug resistance these may provide genetic markers for patient exclusion. Moreover, these data provide a rationale to explore combinations of VS-5584 with therapies that target pathways activated by these mutations.
The PI3K pathway has been shown to play an important role in tumor angiogenesis by regulating the production of VEGF (23, 24). We showed that treatment of highly vascularized COLO-205 xenografts with VS-5584 reduced the average number of functional blood vessels within the tumor. The therapeutic effect of VS-5584 may therefore be due to direct effects on tumor cells as well as an effect on tumor vasculariazation, as reported for other compounds blocking either PI3K or mTOR (25).
Activation of the PI3K pathway has been shown to induce resistance to chemotherapy as well as many targeted agents (26). Trastuzumab-resistant breast cancer cells have been shown to have upregulated PI3K/mTOR signaling and blockade of the pathway restored sensitivity toward trastuzumab (27). Patients who developed a resistance mechanism to EGFRi showed a continued activation of the PI3K/mTOR pathway (28). This has promoted trials of combinations of PI3K or mTOR pathway inhibitors with EGFRi. Here, we have shown that NCI-N87, a HER2 overexpressing cell line, is very sensitive to VS-5584 as a single agent and not to gefitinib. However, a low dose of VS-5584 was synergistic with gefitinib in a combination study and may provide a valid therapeutic strategy to be tested in the clinic.
Over the last 20 years, much research effort led to a great progress in the understanding the role of the PI3K/mTOR pathway in the initiation and development of cancer. Numerous PI3K-selective and dual PI3K/mTOR inhibitors with various inhibition profiles are currently under clinical investigation (29). Pan-class I PI3K or PI3K/mTOR inhibitors that are tested in clinical trials include NVP-BEZ235, GDC-0980, XL765, NVP-BKM120, XL147, SF1126, GSK2126458, and PF-04691502 (30–35). In contrast to other described ATP-competitive inhibitors, VS-5584 targets mTOR and class I PI3K in the same IC50 range but with no significant activity on other lipid and protein kinases tested (Table 1 and Supplementary Fig. S1). VS-5584 is efficacious against a broad spectrum of cell lines independently of rapalog-sensitivity and effectively blocks intracellular PI3K/mTOR signaling in these cells. VS-5584 has very good pharmacokinetic properties and effectively blocks mTORC1 and 2 as well as PI3K signaling in tumor tissue after once daily oral dosing. It is highly efficacious and well tolerated in all xenograft models tested so far, including models resistant to rapalogs and standard of care therapies. Furthermore, we have shown that VS-5584 is synergistic with an EGFRi in a gastric tumor model.
In summary, the favorable target selectivity profile, pharmacokinetic and pharmacodynamic properties of VS-5584 and as a consequence its efficacy in a range of tumors resistant to rapalogs and standard of care, provide a compelling rationale for the clinical evaluation of this drug in a range of liquid and solid tumor indications. The genetic markers of sensitivity and resistance identified in the large cell panel screen provide a rationale for patient selection for single-agent therapy as well as for drug combinations.
Footnotes
Disclosure of Potential Conflicts of Interest C. Benes has a commercial research grant from Astrazeneca and has honoraria from Speakers Bureau of EMD serono. No potential conflicts of interest were disclosed by the other authors.
Authors’ Contributions Conception and design: S. Hart, V. Novotny-Diermayr, K.C. Goh, M. Williams, H. Nagaraj, K. Ethirajulu, C. Benes, B. Dymock, J.M. Wood Development of methodology: S. Hart, V. Novotny-Diermayr, K.C. Goh, C. Amalini, B. Madan, U. McDermott
Acquisition of data (provided animals, acquired and managed patients, provided facilities, etc.): S. Hart, V. Novotny-Diermayr, K.C. Goh, Y.C. Tan, L.C. Ong, B.K. Ng, C. Amalini, K.M. Pasha, K. Ethirajulu, W.J. Chng, N. Mustafa, B.C. Goh, C. Benes, U. McDermott, M. Garnett
Analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis): S. Hart, V. Novotny-Diermayr, K.C. Goh, Y. C. Tan, L.C. Ong, A. Cheong, C. Amalini, R. Jayaraman, U. McDermott, M. Garnett
Writing, review, and/or revision of the manuscript: S. Hart, K.C. Goh, M. Williams, K. Ethirajulu, W.J. Chng, J.M. Wood
Administrative, technical, or material support (i.e., reporting or organizing data, constructing databases): S. Hart, M. Williams, Y.C. Tan, A. Cheong, N. Mustafa
Study supervision: S. Hart, V. Novotny-Diermayr, K.C. Goh, M. Williams
Note: Supplementary data for this article are available at Molecular Cancer Therapeutics Online (http://mct.aacrjournals.org/).
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
References
- 1.Vanhaesebroeck B, Guillermet-Guibert J, Graupera M, Bilanges B. The emerging mechanisms of isoform-specific PI3K signalling. Nat Rev Mol Cell Biol. 2010;11:329–41. doi: 10.1038/nrm2882. [DOI] [PubMed] [Google Scholar]
- 2.Engelman JA. Targeting PI3K signalling in cancer: opportunities, challenges and limitations. Nat Rev Cancer. 2009;9:550–62. doi: 10.1038/nrc2664. [DOI] [PubMed] [Google Scholar]
- 3.Vanhaesebroeck B, Vogt PK, Rommel C. PI3K: from the bench to the clinic and back. Curr Top Microbiol Immunol. 2010;347:1–19. doi: 10.1007/82_2010_65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu P, Cheng H, Roberts TM, Zhao JJ. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat Rev Drug Discov. 2009;8:627–44. doi: 10.1038/nrd2926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Huang S, Bjornsti MA, Houghton PJ. Rapamycins: mechanism of action and cellular resistance. Cancer Biol Ther. 2003;2:222–32. doi: 10.4161/cbt.2.3.360. [DOI] [PubMed] [Google Scholar]
- 6.Yea SS, Fruman DA. Cell signaling. New mTOR targets Grb attention. Science. 2011;332:1270–1. doi: 10.1126/science.1208071. [DOI] [PubMed] [Google Scholar]
- 7.Guertin DA, Sabatini DM. Defining the role of mTOR in cancer. Cancer Cell. 2007;12:9–22. doi: 10.1016/j.ccr.2007.05.008. [DOI] [PubMed] [Google Scholar]
- 8.Sarbassov DD, Ali SM, Sengupta S, Sheen JH, Hsu PP, Bagley AF, et al. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell. 2006;22:159–68. doi: 10.1016/j.molcel.2006.03.029. [DOI] [PubMed] [Google Scholar]
- 9.Chen D, Williams Minventors S *BIO PTE LTD., assignee Pyrimidine substituted purine compounds as kinase inhibitors. Oct 7, 2010. PCT application WO2010114484.
- 10.Borrell-Pages M, Rojo F, Albanell J, Baselga J, Arribas J. TACE is required for the activation of the EGFR by TGF-alpha in tumors. EMBO J. 2003;22:1114–24. doi: 10.1093/emboj/cdg111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Williams M, Hart S, Bonday Z, Goh KC, Diermayr-Novotny V, Loh YK, et al. A novel series of tetrasubstituted purines potently inhibit mTOR and PI3K, exhibit striking selectivity and demonstrate good pathway inhibition in vivo. Proceedings of the Special Conference of the American Association for Cancer Research – Targeting PI3K/mTOR Signaling in Cancer; San Francisco, CA. Philadelphia (PA). 2011 Feb 24–27; AACR; 2011. Abstract nr B26. [Google Scholar]
- 12.Hart S, Soh CK, Bonday Z, Goh KC, Diermayr-Novotny V, Loh YK, et al. SB2343, a novel tetra substituted purine, inhibits mTOR and PI3K equipotently, and has favorable pharmacokinetic and pharmacodynamic properties and is orally efficacious. Proceedings of the Special Conference of the American Association for Cancer Research – Targeting PI3K/mTOR Signaling in Cancer; San Francisco, CA. Philadelphia (PA). 2011 Feb 24–27; AACR; 2011. Abstract nr A25. [Google Scholar]
- 13.Williams M, Hart S, Chen D, Kumar H, Blanchard S, Poulsen A, et al. Discovery of 5-(2-morpholino-9H-purin-6-yl)pyrimidin-2-amine derivate (SB2343), a highly selective pan-PI3/mTOR kinase inhibitor. J Med Chem. 2013 In preparation. [Google Scholar]
- 14.Clarke R. Issues in experimental design and endpoint analysis in the study of experimental cytotoxic agents in vivo in breast cancer and other models. Breast Cancer Res Treat. 1997;46:255–78. doi: 10.1023/a:1005938428456. [DOI] [PubMed] [Google Scholar]
- 15.Garnett MJ, Edelman EJ, Heidorn SJ, Greenman CD, Dastur A, Lau KW, et al. Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature. 2012;483:570–5. doi: 10.1038/nature11005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Garnett MJ, McDermott U. Exploiting genetic complexity in cancer to improve therapeutic strategies. Drug Discov Today. 2012;17:188–93. doi: 10.1016/j.drudis.2012.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lamm W, Vogl UM, Bojic M, Zielinski C, Klingler C, Kramer G, et al. Safety and efficacy of temsirolimus in heavily pretreated patients with metastatic renal cell carcinoma. Acta Oncol. 2011;51:101–6. doi: 10.3109/0284186X.2011.589404. [DOI] [PubMed] [Google Scholar]
- 18.Willems L, Tamburini J, Chapuis N, Lacombe C, Mayeux P, Bouscary D. PI3K and mTOR signaling pathways in cancer: new data on targeted therapies. Curr Oncol Rep. 2012;14:129–38. doi: 10.1007/s11912-012-0227-y. [DOI] [PubMed] [Google Scholar]
- 19.O’Reilly KE, Rojo F, She QB, Solit D, Mills GB, Smith D, et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 2006;66:1500–8. doi: 10.1158/0008-5472.CAN-05-2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Feldman ME, Apsel B, Uotila A, Loewith R, Knight ZA, Ruggero D, et al. Active-site inhibitors of mTOR target rapamycin-resistant outputs of mTORC1 and mTORC2. PLoS Biol. 2009;7:e38. doi: 10.1371/journal.pbio.1000038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Carracedo A, Pandolfi PP. The PTEN-PI3K pathway: of feedbacks and cross-talks. Oncogene. 2008;27:5527–41. doi: 10.1038/onc.2008.247. [DOI] [PubMed] [Google Scholar]
- 22.O’Brien C, Wallin JJ, Sampath D, GuhaThakurta D, Savage H, Punnoose EA, et al. Predictive biomarkers of sensitivity to the phosphatidylinositol 3′ kinase inhibitor GDC-0941 in breast cancer preclinical models. Clin Cancer Res. 2010;16:3670–83. doi: 10.1158/1078-0432.CCR-09-2828. [DOI] [PubMed] [Google Scholar]
- 23.Graupera M, Guillermet-Guibert J, Foukas LC, Phng LK, Cain RJ, Salpekar A, et al. Angiogenesis selectively requires the p110alpha isoform of PI3K to control endothelial cell migration. Nature. 2008;453:662–6. doi: 10.1038/nature06892. [DOI] [PubMed] [Google Scholar]
- 24.Schnell CR, Stauffer F, Allegrini PR, O’Reilly T, McSheehy PM, Dartois C, et al. Effects of the dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor NVP-BEZ235 on the tumor vasculature: implications for clinical imaging. Cancer Res. 2008;68:6598–607. doi: 10.1158/0008-5472.CAN-08-1044. [DOI] [PubMed] [Google Scholar]
- 25.Cook KM, Figg WD. Angiogenesis inhibitors: current strategies and future prospects. CA Cancer J Clin. 2010;60:222–43. doi: 10.3322/caac.20075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McCubrey JA, Steelman LS, Kempf CR, Chappell WH, Abrams SL, Stivala F, et al. Therapeutic resistance resulting from mutations in Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR signaling pathways. J Cell Physiol. 2011;226:2762–81. doi: 10.1002/jcp.22647. [DOI] [PubMed] [Google Scholar]
- 27.Liu M, Howes A, Lesperance J, Stallcup WB, Hauser CA, Kadoya K, et al. Antitumor activity of rapamycin in a transgenic mouse model of ErbB2-dependent human breast cancer. Cancer Res. 2005;65:5325–36. doi: 10.1158/0008-5472.CAN-04-4589. [DOI] [PubMed] [Google Scholar]
- 28.Bianco R, Garofalo S, Rosa R, Damiano V, Gelardi T, Daniele G, et al. Inhibition of mTOR pathway by everolimus cooperates with EGFR inhibitors in human tumours sensitive and resistant to anti-EGFR drugs. Br J Cancer. 2008;98:923–30. doi: 10.1038/sj.bjc.6604269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ciraolo E, Morello F, Hirsch E. Present and future of PI3K pathway inhibition in cancer: perspectives and limitations. Curr Med Chem. 2011;18:2674–85. doi: 10.2174/092986711796011193. [DOI] [PubMed] [Google Scholar]
- 30.Bowles DW, Jimeno A. New phosphatidylinositol 3-kinase inhibitors for cancer. Expert Opin Investig Drugs. 2011;20:507–18. doi: 10.1517/13543784.2011.562192. [DOI] [PubMed] [Google Scholar]
- 31.Wallin JJ, Edgar KA, Guan J, Berry M, Prior WW, Lee L, et al. GDC-0980 is a novel class I PI3K/mTOR kinase inhibitor with robust activity in cancer models driven by the PI3K pathway. Mol Cancer Ther. 2011;10:2426–36. doi: 10.1158/1535-7163.MCT-11-0446. [DOI] [PubMed] [Google Scholar]
- 32.Salphati L, Wong H, Belvin M, Bradford D, Edgar KA, Prior WW, et al. Pharmacokinetic-pharmacodynamic modeling of tumor growth inhibition and biomarker modulation by the novel phosphatidylinositol 3-kinase inhibitor GDC-0941. Drug Metab Dispos. 2010;38:1436–42. doi: 10.1124/dmd.110.032912. [DOI] [PubMed] [Google Scholar]
- 33.Bendell JC, Rodon J, Burris HA, de Jonge M, Verweij J, Birle D, et al. Phase I, dose-escalation study of BKM120, an oral pan-class I PI3K inhibitor, in patients with advanced solid tumors. J Clin Oncol. 2012;30:282–90. doi: 10.1200/JCO.2011.36.1360. [DOI] [PubMed] [Google Scholar]
- 34.Kurtz JE, Ray-Coquard I. PI3 kinase inhibitors in the clinic: an update. Anticancer Res. 2012;32:2463–70. [PubMed] [Google Scholar]
- 35.Bartholomeusz C, Gonzalez-Angulo AM. Targeting the PI3K signaling pathway in cancer therapy. Expert Opin Ther Targets. 2012;16:121–30. doi: 10.1517/14728222.2011.644788. [DOI] [PubMed] [Google Scholar]