

Abstract
In the title compound, C15H14N2O, the fused five- and six-membered ring system is essentially planar, the maximum deviation from the mean plane being 0.009 (1) Å. The benzimidazol-2(3H)-one residue is nearly perpendicular to the benzyl ring, forming a dihedral angle of 77.41 (6)°. In the crystal, inversion dimers are formed by pairs of N—H⋯O hydrogen bonds; these dimers are linked by weak C—H⋯O interactions into a two-dimensional array in the (102) plane.
Related literature
For pharmacological and biochemical properties of benzimidazole derivatives, see: Lee et al. (2004 ▶); Deligeorgiev et al. (2011 ▶); Scott et al. (2002 ▶); Gothelf et al. (1998 ▶). For related structures, see: Belaziz et al. (2012 ▶); Ouzidan et al. (2011 ▶).
Experimental
Crystal data
C15H14N2O
M r = 238.28
Monoclinic,
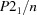
a = 12.5585 (5) Å
b = 5.7181 (2) Å
c = 17.4153 (7) Å
β = 95.277 (2)°
V = 1245.31 (8) Å3
Z = 4
Mo Kα radiation
μ = 0.08 mm−1
T = 296 K
0.51 × 0.42 × 0.15 mm
Data collection
Bruker X8 APEXII diffractometer
16486 measured reflections
3211 independent reflections
2157 reflections with I > 2σ(I)
R int = 0.029
Refinement
R[F 2 > 2σ(F 2)] = 0.044
wR(F 2) = 0.122
S = 1.02
3211 reflections
164 parameters
H-atom parameters constrained
Δρmax = 0.17 e Å−3
Δρmin = −0.15 e Å−3
Data collection: APEX2 (Bruker, 2009 ▶); cell refinement: SAINT (Bruker, 2009 ▶); data reduction: SAINT; program(s) used to solve structure: SHELXS97 (Sheldrick, 2008 ▶); program(s) used to refine structure: SHELXL97 (Sheldrick, 2008 ▶); molecular graphics: ORTEP-3 for Windows (Farrugia, 2012 ▶); software used to prepare material for publication: PLATON (Spek, 2009 ▶) and publCIF (Westrip,2010 ▶).
Supplementary Material
Crystal structure: contains datablock(s) I, global. DOI: 10.1107/S1600536812050726/tk5182sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S1600536812050726/tk5182Isup2.hkl
Supplementary material file. DOI: 10.1107/S1600536812050726/tk5182Isup3.cml
Additional supplementary materials: crystallographic information; 3D view; checkCIF report
Table 1. Hydrogen-bond geometry (Å, °).
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| N1—H1N1⋯O1i | 0.94 | 1.91 | 2.8317 (15) | 166 |
| C15—H15C⋯O1ii | 0.96 | 2.58 | 3.514 (2) | 165 |
| C8—H8A⋯O1iii | 0.97 | 2.61 | 3.5504 (18) | 164 |
Symmetry codes: (i) 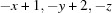 ; (ii)
; (ii) 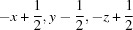 ; (iii)
; (iii)  .
.
Acknowledgments
The authors thank the Unit of Support for Technical and Scientific Research (UATRS, CNRST) for the X-ray measurements.
supplementary crystallographic information
Comment
The development of benzimidazole derivatives has experienced in recent years, a considerable expansion following reports of biological activities presented by this type of compound. Benzimidazole derivatives are endowed with anti-viral, anti-ulcer, anti-hypertensive and anti-cancer activities (Lee et al., 2004; Deligeorgiev et al., 2011; Scott et al., 2002). Heterocycles containing the benzimidazole nucleus are also antagonists of a number of biological receptors, namely angiotensin II and prostaglandin D2 (Gothelf et al., 1998).
In a previous study, we reacted benzimidazol-2-one with dodecyl bromide in the presence of a catalytic quantity of tetra-n-butylammonium bromide under mild conditions to form 1-dodecyl-1H-benzimidazol-2(3H)-one (Belaziz et al., 2012; Ouzidan et al., 2011). The study is extended to the synthesis of new benzimidazol-2-one derivative by action of methylbenzyl bromide with 1H-benzimidazol-2(3H)-one to form the title compound (Scheme 1).
The crystal structure of the title compound, C15H14N2O, is built up from two fused five- and six-membered rings (C1-C7,N1,N2,O1) linked to (C8-C15) the p-methyl-benzyl residue as shown in Fig. 1. The fused-ring system is essentially planar, with the maximum deviation of 0.009 (1) Å for the N2 atom. The dihedral angle between the benzimidazol-2(3H-one system and the (C9 to C14) benzyl ring is 77.41 (6)°.
In the crystal, inversion dimers are linked by N1—H1N···O1 hydrogen bonds. These are linked by weak C8–H8A···O1 and C15–H15C···O1 non-classic hydrogen bonds to form a layer parallel to (1 0 2); see Fig. 2 and Table 1.
Experimental
To 1H-benzimidazol-2(3H)-one (0.2 g, 1.49 mmol), potassium carbonate (0.41 g, 3 mmol) and tetra-n-butylammonium bromide (0.05 g, 0.15 mmol) in DMF (15 ml) was added methyl benzyl bromide (0.33 g, 1.78 mmol). Stirring was continued at room temperature for 6 h. The salt was removed by filtration and the filtrate concentrated under reduced pressure. The residue was separated by chromatography on a column of silica gel with ethyl acetate/hexane (1/2) as eluent. The compound was recrystallized from hexane to give colorless crystals.
Refinement
H atoms were located in a difference map and treated as riding with N—H = 0.94 Å, C—H = 0.93 Å (aromatic), C—H = 0.97 Å (methylene) and C—H = 0.96 Å (methyl), and with Uiso(H) = 1.2 Ueq (N, aromatic-C, methylene-C) and Uiso(H) = 1.5 Ueq(methyl-C). Two reflections, i.e. (-1 0 1) and (1 0 1), were omitted owing to poor agreement.
Figures
Fig. 1.
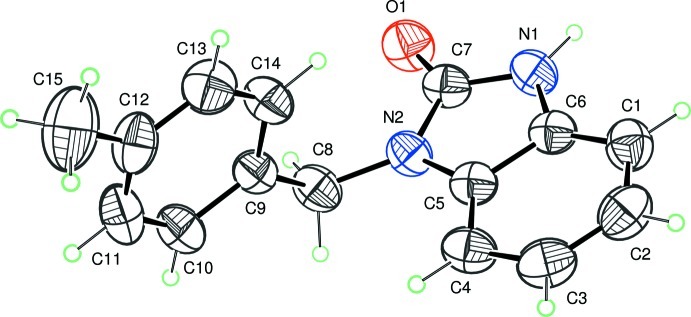
Molecular structure of the title compound with the atom-labelling scheme. Displacement ellipsoids are drawn at the 50% probability level. H atoms are represented as small circles.
Fig. 2.

Portion of the unit cell showing intermolecular interactions (dashed lines) as detailed in Table 1.
Crystal data
| C15H14N2O | F(000) = 504 |
| Mr = 238.28 | Dx = 1.271 Mg m−3 |
| Monoclinic, P21/n | Melting point: 456 K |
| Hall symbol: -P 2yn | Mo Kα radiation, λ = 0.71073 Å |
| a = 12.5585 (5) Å | Cell parameters from 3211 reflections |
| b = 5.7181 (2) Å | θ = 3.3–28.7° |
| c = 17.4153 (7) Å | µ = 0.08 mm−1 |
| β = 95.277 (2)° | T = 296 K |
| V = 1245.31 (8) Å3 | Block, colourless |
| Z = 4 | 0.51 × 0.42 × 0.15 mm |
Data collection
| Bruker X8 APEXII diffractometer | 2157 reflections with I > 2σ(I) |
| Radiation source: fine-focus sealed tube | Rint = 0.029 |
| Graphite monochromator | θmax = 28.7°, θmin = 3.3° |
| φ and ω scans | h = −16→10 |
| 16486 measured reflections | k = −7→7 |
| 3211 independent reflections | l = −23→23 |
Refinement
| Refinement on F2 | Secondary atom site location: difference Fourier map |
| Least-squares matrix: full | Hydrogen site location: inferred from neighbouring sites |
| R[F2 > 2σ(F2)] = 0.044 | H-atom parameters constrained |
| wR(F2) = 0.122 | w = 1/[σ2(Fo2) + (0.0511P)2 + 0.2271P] where P = (Fo2 + 2Fc2)/3 |
| S = 1.02 | (Δ/σ)max < 0.001 |
| 3211 reflections | Δρmax = 0.17 e Å−3 |
| 164 parameters | Δρmin = −0.15 e Å−3 |
| 0 restraints | Extinction correction: SHELXL97 (Sheldrick, 2008), Fc*=kFc[1+0.001xFc2λ3/sin(2θ)]-1/4 |
| Primary atom site location: structure-invariant direct methods | Extinction coefficient: 0.0047 (19) |
Special details
| Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes. |
| Refinement. Refinement of F2 against all reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on all data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| O1 | 0.40817 (8) | 0.80403 (18) | 0.04185 (6) | 0.0541 (3) | |
| N1 | 0.59419 (10) | 0.8138 (2) | 0.06169 (7) | 0.0456 (3) | |
| H1N1 | 0.6036 | 0.9478 | 0.0317 | 0.055* | |
| C6 | 0.67241 (12) | 0.6686 (2) | 0.09809 (7) | 0.0416 (3) | |
| N2 | 0.50984 (9) | 0.51864 (19) | 0.10975 (6) | 0.0425 (3) | |
| C9 | 0.41188 (10) | 0.3448 (2) | 0.21353 (8) | 0.0399 (3) | |
| C5 | 0.61861 (11) | 0.4813 (2) | 0.12841 (7) | 0.0405 (3) | |
| C8 | 0.42295 (12) | 0.3671 (2) | 0.12812 (8) | 0.0475 (4) | |
| H8A | 0.4343 | 0.2127 | 0.1073 | 0.057* | |
| H8B | 0.3565 | 0.4273 | 0.1029 | 0.057* | |
| C4 | 0.67330 (13) | 0.3032 (3) | 0.16849 (8) | 0.0498 (4) | |
| H4 | 0.6375 | 0.1783 | 0.1886 | 0.060* | |
| C14 | 0.44588 (12) | 0.5170 (2) | 0.26587 (8) | 0.0498 (4) | |
| H14 | 0.4778 | 0.6517 | 0.2487 | 0.060* | |
| C10 | 0.36386 (12) | 0.1473 (3) | 0.24098 (9) | 0.0500 (4) | |
| H10 | 0.3403 | 0.0291 | 0.2069 | 0.060* | |
| C7 | 0.49498 (12) | 0.7221 (2) | 0.06784 (8) | 0.0425 (3) | |
| C1 | 0.78212 (12) | 0.6843 (3) | 0.10756 (9) | 0.0518 (4) | |
| H1 | 0.8181 | 0.8099 | 0.0879 | 0.062* | |
| C3 | 0.78367 (13) | 0.3187 (3) | 0.17738 (9) | 0.0568 (4) | |
| H3 | 0.8228 | 0.2012 | 0.2039 | 0.068* | |
| C12 | 0.38486 (12) | 0.2947 (3) | 0.37129 (9) | 0.0568 (4) | |
| C2 | 0.83703 (13) | 0.5043 (3) | 0.14782 (9) | 0.0581 (4) | |
| H2 | 0.9113 | 0.5093 | 0.1550 | 0.070* | |
| C11 | 0.35059 (12) | 0.1242 (3) | 0.31856 (9) | 0.0582 (4) | |
| H11 | 0.3178 | −0.0095 | 0.3356 | 0.070* | |
| C13 | 0.43294 (13) | 0.4911 (3) | 0.34360 (9) | 0.0580 (4) | |
| H13 | 0.4571 | 0.6084 | 0.3779 | 0.070* | |
| C15 | 0.37084 (17) | 0.2654 (5) | 0.45607 (11) | 0.0944 (7) | |
| H15A | 0.3991 | 0.4000 | 0.4839 | 0.142* | |
| H15B | 0.4084 | 0.1281 | 0.4753 | 0.142* | |
| H15C | 0.2962 | 0.2495 | 0.4628 | 0.142* |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| O1 | 0.0523 (7) | 0.0521 (6) | 0.0563 (6) | 0.0055 (5) | −0.0040 (5) | 0.0144 (5) |
| N1 | 0.0529 (8) | 0.0409 (6) | 0.0424 (7) | 0.0004 (5) | 0.0018 (5) | 0.0097 (5) |
| C6 | 0.0511 (9) | 0.0410 (7) | 0.0326 (7) | 0.0025 (6) | 0.0027 (6) | −0.0010 (5) |
| N2 | 0.0466 (7) | 0.0404 (6) | 0.0399 (6) | 0.0001 (5) | 0.0003 (5) | 0.0076 (5) |
| C9 | 0.0376 (8) | 0.0381 (7) | 0.0431 (8) | 0.0002 (5) | −0.0018 (6) | 0.0029 (5) |
| C5 | 0.0484 (9) | 0.0403 (7) | 0.0323 (7) | 0.0019 (6) | 0.0006 (6) | −0.0010 (5) |
| C8 | 0.0535 (9) | 0.0437 (7) | 0.0440 (8) | −0.0072 (6) | −0.0024 (7) | 0.0014 (6) |
| C4 | 0.0621 (10) | 0.0427 (8) | 0.0432 (8) | 0.0042 (6) | −0.0029 (7) | 0.0051 (6) |
| C14 | 0.0565 (10) | 0.0425 (8) | 0.0497 (9) | −0.0090 (6) | 0.0009 (7) | −0.0003 (6) |
| C10 | 0.0496 (9) | 0.0448 (8) | 0.0542 (9) | −0.0094 (6) | −0.0019 (7) | 0.0020 (6) |
| C7 | 0.0520 (9) | 0.0398 (7) | 0.0348 (7) | 0.0029 (6) | 0.0000 (6) | 0.0033 (5) |
| C1 | 0.0519 (10) | 0.0568 (9) | 0.0471 (9) | −0.0046 (7) | 0.0063 (7) | −0.0004 (6) |
| C3 | 0.0599 (11) | 0.0558 (9) | 0.0526 (9) | 0.0145 (7) | −0.0063 (8) | 0.0023 (7) |
| C12 | 0.0410 (9) | 0.0808 (12) | 0.0485 (9) | 0.0004 (8) | 0.0044 (7) | 0.0068 (8) |
| C2 | 0.0490 (10) | 0.0715 (11) | 0.0529 (9) | 0.0090 (8) | 0.0000 (7) | −0.0049 (8) |
| C11 | 0.0481 (10) | 0.0646 (10) | 0.0619 (11) | −0.0107 (7) | 0.0054 (8) | 0.0173 (8) |
| C13 | 0.0599 (11) | 0.0654 (10) | 0.0478 (9) | −0.0044 (8) | −0.0003 (7) | −0.0104 (7) |
| C15 | 0.0829 (15) | 0.148 (2) | 0.0539 (12) | −0.0091 (14) | 0.0142 (10) | 0.0111 (12) |
Geometric parameters (Å, º)
| O1—C7 | 1.2338 (16) | C14—C13 | 1.386 (2) |
| N1—C7 | 1.3649 (18) | C14—H14 | 0.9300 |
| N1—C6 | 1.3933 (17) | C10—C11 | 1.383 (2) |
| N1—H1N1 | 0.9411 | C10—H10 | 0.9300 |
| C6—C1 | 1.375 (2) | C1—C2 | 1.392 (2) |
| C6—C5 | 1.3959 (19) | C1—H1 | 0.9300 |
| N2—C7 | 1.3770 (16) | C3—C2 | 1.380 (2) |
| N2—C5 | 1.3913 (17) | C3—H3 | 0.9300 |
| N2—C8 | 1.4521 (17) | C12—C11 | 1.381 (2) |
| C9—C14 | 1.3822 (19) | C12—C13 | 1.383 (2) |
| C9—C10 | 1.3857 (19) | C12—C15 | 1.512 (2) |
| C9—C8 | 1.5120 (19) | C2—H2 | 0.9300 |
| C5—C4 | 1.3811 (18) | C11—H11 | 0.9300 |
| C8—H8A | 0.9700 | C13—H13 | 0.9300 |
| C8—H8B | 0.9700 | C15—H15A | 0.9600 |
| C4—C3 | 1.383 (2) | C15—H15B | 0.9600 |
| C4—H4 | 0.9300 | C15—H15C | 0.9600 |
| C7—N1—C6 | 110.24 (11) | C9—C10—H10 | 119.7 |
| C7—N1—H1N1 | 121.3 | O1—C7—N1 | 127.42 (13) |
| C6—N1—H1N1 | 128.2 | O1—C7—N2 | 125.97 (13) |
| C1—C6—N1 | 132.24 (13) | N1—C7—N2 | 106.61 (12) |
| C1—C6—C5 | 121.27 (13) | C6—C1—C2 | 117.16 (14) |
| N1—C6—C5 | 106.49 (12) | C6—C1—H1 | 121.4 |
| C7—N2—C5 | 109.63 (11) | C2—C1—H1 | 121.4 |
| C7—N2—C8 | 123.58 (12) | C2—C3—C4 | 121.57 (14) |
| C5—N2—C8 | 126.76 (11) | C2—C3—H3 | 119.2 |
| C14—C9—C10 | 118.08 (13) | C4—C3—H3 | 119.2 |
| C14—C9—C8 | 122.55 (12) | C11—C12—C13 | 117.44 (15) |
| C10—C9—C8 | 119.36 (12) | C11—C12—C15 | 120.94 (18) |
| C4—C5—N2 | 131.55 (13) | C13—C12—C15 | 121.61 (17) |
| C4—C5—C6 | 121.43 (14) | C3—C2—C1 | 121.44 (15) |
| N2—C5—C6 | 107.02 (11) | C3—C2—H2 | 119.3 |
| N2—C8—C9 | 113.95 (11) | C1—C2—H2 | 119.3 |
| N2—C8—H8A | 108.8 | C12—C11—C10 | 121.61 (15) |
| C9—C8—H8A | 108.8 | C12—C11—H11 | 119.2 |
| N2—C8—H8B | 108.8 | C10—C11—H11 | 119.2 |
| C9—C8—H8B | 108.8 | C12—C13—C14 | 121.43 (15) |
| H8A—C8—H8B | 107.7 | C12—C13—H13 | 119.3 |
| C5—C4—C3 | 117.12 (14) | C14—C13—H13 | 119.3 |
| C5—C4—H4 | 121.4 | C12—C15—H15A | 109.5 |
| C3—C4—H4 | 121.4 | C12—C15—H15B | 109.5 |
| C9—C14—C13 | 120.74 (14) | H15A—C15—H15B | 109.5 |
| C9—C14—H14 | 119.6 | C12—C15—H15C | 109.5 |
| C13—C14—H14 | 119.6 | H15A—C15—H15C | 109.5 |
| C11—C10—C9 | 120.69 (14) | H15B—C15—H15C | 109.5 |
| C11—C10—H10 | 119.7 | ||
| C7—N1—C6—C1 | −179.97 (14) | C8—C9—C10—C11 | −178.65 (14) |
| C7—N1—C6—C5 | −0.56 (15) | C6—N1—C7—O1 | −179.46 (13) |
| C7—N2—C5—C4 | −179.25 (14) | C6—N1—C7—N2 | 1.05 (15) |
| C8—N2—C5—C4 | −1.2 (2) | C5—N2—C7—O1 | 179.35 (13) |
| C7—N2—C5—C6 | 0.82 (14) | C8—N2—C7—O1 | 1.3 (2) |
| C8—N2—C5—C6 | 178.83 (12) | C5—N2—C7—N1 | −1.15 (15) |
| C1—C6—C5—C4 | −0.6 (2) | C8—N2—C7—N1 | −179.24 (12) |
| N1—C6—C5—C4 | 179.90 (12) | N1—C6—C1—C2 | −179.93 (14) |
| C1—C6—C5—N2 | 179.33 (12) | C5—C6—C1—C2 | 0.7 (2) |
| N1—C6—C5—N2 | −0.16 (14) | C5—C4—C3—C2 | 0.3 (2) |
| C7—N2—C8—C9 | −117.18 (14) | C4—C3—C2—C1 | −0.1 (2) |
| C5—N2—C8—C9 | 65.08 (17) | C6—C1—C2—C3 | −0.4 (2) |
| C14—C9—C8—N2 | 26.23 (19) | C13—C12—C11—C10 | 0.2 (2) |
| C10—C9—C8—N2 | −155.20 (13) | C15—C12—C11—C10 | −179.31 (17) |
| N2—C5—C4—C3 | −179.82 (14) | C9—C10—C11—C12 | −0.4 (2) |
| C6—C5—C4—C3 | 0.1 (2) | C11—C12—C13—C14 | 0.3 (2) |
| C10—C9—C14—C13 | 0.5 (2) | C15—C12—C13—C14 | 179.84 (16) |
| C8—C9—C14—C13 | 179.13 (14) | C9—C14—C13—C12 | −0.7 (2) |
| C14—C9—C10—C11 | 0.0 (2) |
Hydrogen-bond geometry (Å, º)
| D—H···A | D—H | H···A | D···A | D—H···A |
| N1—H1N1···O1i | 0.94 | 1.91 | 2.8317 (15) | 166 |
| C15—H15C···O1ii | 0.96 | 2.58 | 3.514 (2) | 165 |
| C8—H8A···O1iii | 0.97 | 2.61 | 3.5504 (18) | 164 |
Symmetry codes: (i) −x+1, −y+2, −z; (ii) −x+1/2, y−1/2, −z+1/2; (iii) x, y−1, z.
Footnotes
Supplementary data and figures for this paper are available from the IUCr electronic archives (Reference: TK5182).
References
- Belaziz, D., Kandri Rodi, Y., Ouazzani Chahdi, F., Essassi, E. M., Saadi, M. & El Ammari, L. (2012). Acta Cryst. E68, o3212. [DOI] [PMC free article] [PubMed]
- Bruker (2009). APEX2 and SAINT Bruker AXS Inc., Madison, Wisconsin, USA.
- Deligeorgiev, T., Kaloyanova, S. & Vasilev, S. (2011). Dyes Pigm. 90, 170–1276.
- Farrugia, L. J. (2012). J. Appl. Cryst. 45, 849–854.
- Gothelf, K. V. & Jørgensen, K. A. (1998). Chem. Rev. 98, 863–909. [DOI] [PubMed]
- Lee, Y. H. & Pavlostathis, S. G. (2004). Water Res. 38, 1838–1852. [DOI] [PubMed]
- Ouzidan, Y., Essassi, E. M., Luis, S. V., Bolte, M. & El Ammari, L. (2011). Acta Cryst. E67, o1822. [DOI] [PMC free article] [PubMed]
- Scott, L. J., Dunn, C. J., Mallarkey, G. & Sharpe, M. (2002). Drugs, 62, 1503–1538. [DOI] [PubMed]
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Spek, A. L. (2009). Acta Cryst. D65, 148–155. [DOI] [PMC free article] [PubMed]
- Westrip, S. P. (2010). J. Appl. Cryst. 43, 920–925.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablock(s) I, global. DOI: 10.1107/S1600536812050726/tk5182sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S1600536812050726/tk5182Isup2.hkl
Supplementary material file. DOI: 10.1107/S1600536812050726/tk5182Isup3.cml
Additional supplementary materials: crystallographic information; 3D view; checkCIF report