

Abstract
The title compound, C18H18O4, is a triclinic polymorph of the previously reported monoclinic polymorph [Han & Zhen (2005 ▶). Acta Cryst. E61, o4358–o4359]. In the crystal of the triclinic polymorph, molecules are linked by two pairs of C—H⋯O hydrogen bonds, forming a two-dimensional network parallel to (102), and enclosing loops with graph set motifs of R 2 2(8) and R 2 2(6).
Related literature
For the monoclinic polymorph, see: Han & Zhen (2005 ▶). For related structures and the synthesis of similar compounds, see: Balić et al. (2012 ▶); Ma & Cao (2011 ▶); Dehno Khalaji et al. (2011 ▶); Narasimha Moorthy et al. (2005 ▶); Ilhan et al. (2007 ▶). For graph-set analysis of hydrogen bonds, see Bernstein et al. (1995 ▶).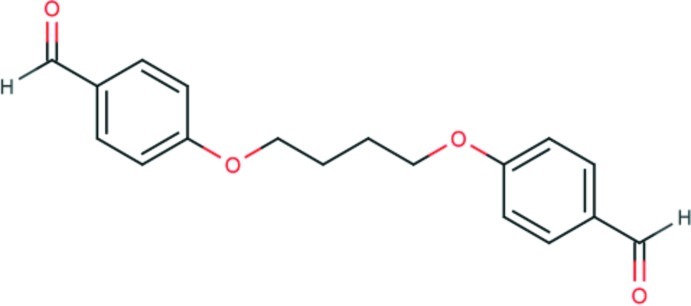
Experimental
Crystal data
C18H18O4
M r = 298.32
Triclinic,
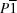
a = 4.4969 (2) Å
b = 7.9507 (6) Å
c = 11.0679 (8) Å
α = 73.854 (6)°
β = 84.788 (5)°
γ = 80.903 (5)°
V = 374.86 (4) Å3
Z = 1
Mo Kα radiation
μ = 0.09 mm−1
T = 190 K
0.59 × 0.35 × 0.21 mm
Data collection
Oxford Diffraction Xcalibur Sapphire3 diffractometer
Absorption correction: multi-scan (CrysAlis PRO; Oxford Diffraction, 2009 ▶) T min = 0.683, T max = 1.000
2235 measured reflections
1473 independent reflections
1272 reflections with I > 2σ(I)
R int = 0.010
Refinement
R[F 2 > 2σ(F 2)] = 0.043
wR(F 2) = 0.123
S = 1.04
1473 reflections
100 parameters
H-atom parameters constrained
Δρmax = 0.29 e Å−3
Δρmin = −0.17 e Å−3
Data collection: CrysAlis PRO (Oxford Diffraction, 2009 ▶); cell refinement: CrysAlis PRO; data reduction: CrysAlis PRO; program(s) used to solve structure: SIR2004 (Burla et al., 2005 ▶); program(s) used to refine structure: SHELXL97 (Sheldrick, 2008 ▶); molecular graphics: ORTEP-3 (Farrugia, 2012 ▶); software used to prepare material for publication: WinGX (Farrugia, 2012 ▶), PARST97 (Nardelli, 1995 ▶) and Mercury (Macrae et al., 2006 ▶).
Supplementary Material
Crystal structure: contains datablock(s) I, global. DOI: 10.1107/S1600536812050994/ng5308sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S1600536812050994/ng5308Isup2.hkl
Supplementary material file. DOI: 10.1107/S1600536812050994/ng5308Isup3.cml
Additional supplementary materials: crystallographic information; 3D view; checkCIF report
Table 1. Hydrogen-bond geometry (Å, °).
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| C6—H6⋯O2i | 0.95 | 2.58 | 3.4985 (16) | 162 |
| C1—H1⋯O1ii | 0.95 | 2.59 | 3.3953 (18) | 143 |
Symmetry codes: (i) 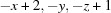 ; (ii)
; (ii) 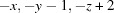 .
.
Acknowledgments
This work was supported by the Ministry of Science, Education and Sports of the Republic of Croatia (grant No. 119–1193079-1084). The authors wish to thank Professor Dubravka Matković-Čalogović for help with the crystallography.
supplementary crystallographic information
Comment
Reacent structural studies of dialdehydes (Balić et al. 2012; Narasimha Moorthy et al. 2005), or the so called two-arm aldehydes have proposed them as potential precursors for condensation reactions with primary amines (Ilhan et al. 2007; Ma & Cao 2011; Dehno Khalaji et al. 2011). In a relation to this structural studies a new triclinic polymorph of title compound was found. Previously reported monoclinic polymorph (Han & Zhen 2005) was reported in P21/c space group with Z=2. The new polymorph was found in P1 space group (Z=1), with different intermolecular interactions (Figure 1.).
The original polymorph crystallize in monoclinic space group P21/c, with a = 7.988 (2), b = 6.6635 (16), c= 14.260 (4) Å, β= 96.354 (4)° and Z = 2 (Han & Zhen 2005). The title compound crystallizes in the space group P1 with a = 4.5749 (7), b= 7.9467 (10), c = 14.260 (4) Å, α= 73.597 (11)°, β = 83.154 (11)°, γ = 80.533 (12)° and Z = 1. In the reported structure crystallographic inversion centre lies in the center of the molecule, so the asymmetric unit comprises only one half of the molecule. The molecular structure of the title compound is shown in Figure 2. In the triclinic polymorph the molecules are linked in centrosymetric dimers via weak C1— H1···O1 intermolecular interactions, as previously reported by Narasimha Moorthy et al. (2005) and Balić et al. (2012). Additional stabilization of crystal structure is accomplished by weak C4— H4···O2 (Figure 1.). In the previously reported monoclinic polymorph the dihedral angle between benzaldehyde group and four central carbon atoms is 62.82°, while in triclinic polymorph this angle is 42.07°. However, the largest difference between these two polymorphs is manifested by the presence of R22(6) and R22(8) (Bernstein et al. 1995) supramolecular motifs in the triclinic polymorph.
Experimental
The title compound was prepared by folowing procedure: p-hydroxybenzaldehyde (50 mmol) and K2CO3 (50 mmol) were mixed in DMF and the mixture was brought to brisk reflux. 25 mmol of butane-1,4-dibrom dissolved in DMF was then added and the reaction mixture was refluxed for 5 h. After the reaction was complete, 100 ml of water was added and the resulting percipitate was filtered and washed with water. Single crystals suitable for X-ray diffraction were grown via slow evaporation from ethanol solution of the title compound.
Refinement
All H atoms, were positioned geometrically and refined using a riding model with C—H = 0.93 - 0.97 Å and with Uiso(H) = 1.2 times Ueq(C).
Figures
Fig. 1.

Crystal packing of title compound viewed down the a axis with dased lines representing weak C— H···O [graph set R22(6), R22(8)] intermolecular interactions.
Fig. 2.
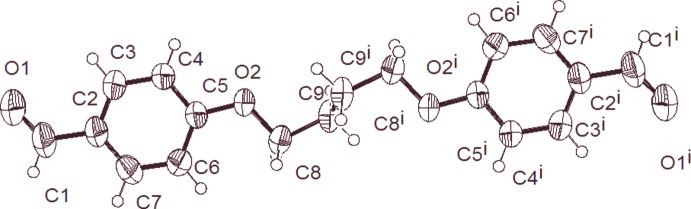
The molecular structure of the title compound with the atom numbering scheme. Displacement ellipsoids are drawn at the 50% probability level.
Crystal data
| C18H18O4 | Z = 1 |
| Mr = 298.32 | F(000) = 158 |
| Triclinic, P1 | Dx = 1.321 Mg m−3 |
| a = 4.4969 (2) Å | Mo Kα radiation, λ = 0.7107 Å |
| b = 7.9507 (6) Å | Cell parameters from 1657 reflections |
| c = 11.0679 (8) Å | θ = 4.6–28.5° |
| α = 73.854 (6)° | µ = 0.09 mm−1 |
| β = 84.788 (5)° | T = 190 K |
| γ = 80.903 (5)° | Block, colourless |
| V = 374.86 (4) Å3 | 0.59 × 0.35 × 0.21 mm |
Data collection
| Oxford Diffraction Xcalibur Sapphire3 diffractometer | 1473 independent reflections |
| Radiation source: Enhance (Mo) X-ray Source | 1272 reflections with I > 2σ(I) |
| Graphite monochromator | Rint = 0.010 |
| Detector resolution: 16.3426 pixels mm-1 | θmax = 26.0°, θmin = 4.6° |
| ω scans | h = −5→4 |
| Absorption correction: multi-scan (CrysAlis PRO; Oxford Diffraction, 2009) | k = −9→9 |
| Tmin = 0.683, Tmax = 1.000 | l = −13→13 |
| 2235 measured reflections |
Refinement
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.043 | Hydrogen site location: inferred from neighbouring sites |
| wR(F2) = 0.123 | H-atom parameters constrained |
| S = 1.04 | w = 1/[σ2(Fo2) + (0.0676P)2 + 0.0807P] where P = (Fo2 + 2Fc2)/3 |
| 1473 reflections | (Δ/σ)max < 0.001 |
| 100 parameters | Δρmax = 0.29 e Å−3 |
| 0 restraints | Δρmin = −0.17 e Å−3 |
Special details
| Experimental. (CrysAlis PRO RED; Oxford Diffraction, 2009) |
| Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes. |
| Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| O1 | 0.2410 (3) | −0.49759 (14) | 0.85962 (11) | 0.0490 (4) | |
| O2 | 0.8584 (2) | 0.19238 (11) | 0.61242 (8) | 0.0289 (3) | |
| C1 | 0.2367 (3) | −0.3608 (2) | 0.88964 (14) | 0.0377 (4) | |
| H1 | 0.1248 | −0.3496 | 0.9647 | 0.045* | |
| C2 | 0.3920 (3) | −0.21240 (17) | 0.81809 (13) | 0.0283 (3) | |
| C3 | 0.3932 (3) | −0.06701 (18) | 0.86379 (12) | 0.0308 (3) | |
| H3 | 0.2870 | −0.0627 | 0.9413 | 0.037* | |
| C4 | 0.5460 (3) | 0.07238 (17) | 0.79893 (12) | 0.0280 (3) | |
| H4 | 0.5453 | 0.1711 | 0.8315 | 0.034* | |
| C5 | 0.7004 (3) | 0.06505 (16) | 0.68513 (12) | 0.0242 (3) | |
| C6 | 0.6963 (3) | −0.07927 (18) | 0.63690 (13) | 0.0292 (3) | |
| H6 | 0.7991 | −0.0831 | 0.5586 | 0.035* | |
| C7 | 0.5437 (3) | −0.21533 (17) | 0.70273 (13) | 0.0305 (3) | |
| H7 | 0.5410 | −0.3130 | 0.6694 | 0.037* | |
| C8 | 0.8898 (3) | 0.33927 (16) | 0.65981 (12) | 0.0268 (3) | |
| H8A | 0.9914 | 0.2970 | 0.7406 | 0.032* | |
| H8B | 0.6892 | 0.4045 | 0.6742 | 0.032* | |
| C9 | 1.0762 (3) | 0.45809 (17) | 0.56216 (12) | 0.0276 (3) | |
| H9A | 1.2708 | 0.3883 | 0.5457 | 0.033* | |
| H9B | 1.1206 | 0.5532 | 0.5967 | 0.033* |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| O1 | 0.0659 (8) | 0.0366 (7) | 0.0488 (7) | −0.0282 (5) | 0.0138 (6) | −0.0119 (5) |
| O2 | 0.0378 (5) | 0.0208 (5) | 0.0297 (5) | −0.0128 (4) | 0.0082 (4) | −0.0076 (4) |
| C1 | 0.0431 (8) | 0.0373 (8) | 0.0326 (8) | −0.0182 (6) | 0.0064 (6) | −0.0048 (6) |
| C2 | 0.0292 (7) | 0.0262 (7) | 0.0282 (7) | −0.0087 (5) | −0.0004 (5) | −0.0026 (5) |
| C3 | 0.0335 (7) | 0.0356 (8) | 0.0233 (6) | −0.0095 (6) | 0.0045 (5) | −0.0072 (6) |
| C4 | 0.0337 (7) | 0.0255 (7) | 0.0271 (7) | −0.0079 (5) | 0.0007 (5) | −0.0094 (5) |
| C5 | 0.0245 (6) | 0.0211 (6) | 0.0257 (6) | −0.0057 (5) | 0.0005 (5) | −0.0032 (5) |
| C6 | 0.0342 (7) | 0.0254 (7) | 0.0291 (7) | −0.0087 (5) | 0.0073 (5) | −0.0095 (6) |
| C7 | 0.0352 (7) | 0.0235 (7) | 0.0346 (7) | −0.0101 (5) | 0.0047 (6) | −0.0094 (6) |
| C8 | 0.0309 (7) | 0.0215 (7) | 0.0301 (7) | −0.0083 (5) | −0.0009 (5) | −0.0080 (5) |
| C9 | 0.0275 (6) | 0.0230 (7) | 0.0328 (7) | −0.0086 (5) | −0.0023 (5) | −0.0051 (6) |
Geometric parameters (Å, º)
| O1—C1 | 1.2196 (18) | C5—C6 | 1.3969 (18) |
| O2—C5 | 1.3593 (15) | C6—C7 | 1.3704 (18) |
| O2—C8 | 1.4372 (15) | C6—H6 | 0.9500 |
| C1—C2 | 1.4652 (18) | C7—H7 | 0.9500 |
| C1—H1 | 0.9500 | C8—C9 | 1.5105 (17) |
| C2—C3 | 1.3857 (19) | C8—H8A | 0.9900 |
| C2—C7 | 1.396 (2) | C8—H8B | 0.9900 |
| C3—C4 | 1.3871 (18) | C9—C9i | 1.525 (3) |
| C3—H3 | 0.9500 | C9—H9A | 0.9900 |
| C4—C5 | 1.3933 (18) | C9—H9B | 0.9900 |
| C4—H4 | 0.9500 | ||
| C5—O2—C8 | 118.23 (10) | C7—C6—H6 | 120.1 |
| O1—C1—C2 | 124.62 (14) | C5—C6—H6 | 120.1 |
| O1—C1—H1 | 117.7 | C6—C7—C2 | 120.93 (13) |
| C2—C1—H1 | 117.7 | C6—C7—H7 | 119.5 |
| C3—C2—C7 | 118.64 (12) | C2—C7—H7 | 119.5 |
| C3—C2—C1 | 120.77 (13) | O2—C8—C9 | 107.25 (10) |
| C7—C2—C1 | 120.58 (13) | O2—C8—H8A | 110.3 |
| C2—C3—C4 | 121.52 (12) | C9—C8—H8A | 110.3 |
| C2—C3—H3 | 119.2 | O2—C8—H8B | 110.3 |
| C4—C3—H3 | 119.2 | C9—C8—H8B | 110.3 |
| C3—C4—C5 | 118.82 (12) | H8A—C8—H8B | 108.5 |
| C3—C4—H4 | 120.6 | C8—C9—C9i | 113.78 (13) |
| C5—C4—H4 | 120.6 | C8—C9—H9A | 108.8 |
| O2—C5—C4 | 124.74 (12) | C9i—C9—H9A | 108.8 |
| O2—C5—C6 | 115.04 (11) | C8—C9—H9B | 108.8 |
| C4—C5—C6 | 120.22 (12) | C9i—C9—H9B | 108.8 |
| C7—C6—C5 | 119.85 (12) | H9A—C9—H9B | 107.7 |
| O1—C1—C2—C3 | −175.28 (14) | C3—C4—C5—C6 | 1.0 (2) |
| O1—C1—C2—C7 | 4.3 (2) | O2—C5—C6—C7 | 179.96 (11) |
| C7—C2—C3—C4 | −1.3 (2) | C4—C5—C6—C7 | −1.0 (2) |
| C1—C2—C3—C4 | 178.22 (12) | C5—C6—C7—C2 | −0.2 (2) |
| C2—C3—C4—C5 | 0.2 (2) | C3—C2—C7—C6 | 1.4 (2) |
| C8—O2—C5—C4 | 5.11 (18) | C1—C2—C7—C6 | −178.21 (12) |
| C8—O2—C5—C6 | −175.90 (10) | C5—O2—C8—C9 | 179.31 (10) |
| C3—C4—C5—O2 | 179.95 (11) | O2—C8—C9—C9i | 64.84 (16) |
Symmetry code: (i) −x+2, −y+1, −z+1.
Hydrogen-bond geometry (Å, º)
| D—H···A | D—H | H···A | D···A | D—H···A |
| C6—H6···O2ii | 0.95 | 2.58 | 3.4985 (16) | 162 |
| C1—H1···O1iii | 0.95 | 2.59 | 3.3953 (18) | 143 |
Symmetry codes: (ii) −x+2, −y, −z+1; (iii) −x, −y−1, −z+2.
Footnotes
Supplementary data and figures for this paper are available from the IUCr electronic archives (Reference: NG5308).
References
- Balić, T., Marković, B. & Balić, I. (2012). Acta Cryst. E68, o2664. [DOI] [PMC free article] [PubMed]
- Bernstein, J., Davis, R. E., Shimoni, L. & Chang, N.-L. (1995). Angew. Chem. Int. Ed. Engl. 34, 1555–1573.
- Burla, M. C., Caliandro, R., Camalli, M., Carrozzini, B., Cascarano, G. L., De Caro, L., Giacovazzo, C., Polidori, G. & Spagna, R. (2005). J. Appl. Cryst. 38, 381–388.
- Dehno Khalaji, A., Hafez Ghoran, S., Gotoh, K. & Ishida, H. (2011). Acta Cryst. E67, o2484. [DOI] [PMC free article] [PubMed]
- Farrugia, L. J. (2012). J. Appl. Cryst. 45, 849–854.
- Han, J.-R. & Zhen, X.-L. (2005). Acta Cryst. E61, o4358–o4359.
- Ilhan, S., Temel, H., Yilmaz, I. & Kilic, A. (2007). Transition Met. Chem. 32, 344–349.
- Ma, Z. & Cao, Y. (2011). Acta Cryst. E67, o1503. [DOI] [PMC free article] [PubMed]
- Macrae, C. F., Edgington, P. R., McCabe, P., Pidcock, E., Shields, G. P., Taylor, R., Towler, M. & van de Streek, J. (2006). J. Appl. Cryst. 39, 453–457.
- Narasimha Moorthy, J., Natarajan, R. & Venugopalan, P. (2005). J. Mol. Struct. 741, 107–114.
- Nardelli, M. (1995). J. Appl. Cryst. 28, 659.
- Oxford Diffraction (2009). CrysAlis PRO Oxford Diffraction Ltd, Yarnton, England.
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablock(s) I, global. DOI: 10.1107/S1600536812050994/ng5308sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S1600536812050994/ng5308Isup2.hkl
Supplementary material file. DOI: 10.1107/S1600536812050994/ng5308Isup3.cml
Additional supplementary materials: crystallographic information; 3D view; checkCIF report