

Summary
Plasmacytoid dendritic cells (pDCs) mediate type I interferon (IFN-I) responses to viruses that are recognized through the Toll-like receptor 7 (TLR7) or TLR9 signaling pathway. However, it is unclear how pDCs regulate the antiviral responses via innate and adaptive immune cells. We generated diphtheria toxin receptor transgenic mice to selectively deplete pDCs by administration of diphtheria toxin. pDC-depleted mice were challenged with viruses known to activate pDCs. In murine cytomegalovirus (MCMV) infection, pDC depletion reduced early IFN-I production and augmented viral burden facilitating the expansion of natural killer (NK) cells expressing the MCMV-specific receptor Ly49H. During vesicular stomatitis virus (VSV) infection, pDC depletion enhanced early viral replication and impaired the survival and accumulation of virus-specific cytotoxic T lymphocytes. We conclude that pDCs mediate early antiviral IFN-I responses and influence the accrual of virus-specific NK or CD8+ T cells in a virus-dependent manner.
Introduction
Plasmacytoid dendritic cells (pDCs) are bone marrow-derived leukocytes that secrete large amounts of type I interferons (IFN-I), i.e., IFN-α and -β, in response to a variety of viruses in vitro and in vivo (Gilliet et al., 2008). IFN-I confer resistance to viral infections and promote apoptosis of virally infected cells (García-Sastre and Biron, 2006; Honda et al., 2005c; Pestka et al., 2004). Moreover, IFN-I promote natural killer (NK) cell, dendritic cell (DC), T cell, and B cell functions (Banchereau and Pascual, 2006; García-Sastre and Biron, 2006; Kolumam et al., 2005; Le Bon and Tough, 2008). Thus, pDCs have been implicated in the control of both innate and adaptive host antiviral responses.
Besides producing IFN-I, pDCs may contribute to antiviral defense through additional mechanisms. pDCs express major histocompatibility complex (MHC) molecules and costimulatory molecules, and therefore may directly present viral antigens to CD4+ T cells and cross-present viral antigens to CD8+ T cells (Villadangos and Young, 2008). In addition, pDCs are a source of proinflammatory chemokines, including CCL3, CCL4, CCL5, CXCL9, and CXCL10, which can attract activated CD4+ and CD8+ T cells to sites of infection (Sozzani et al., 2010). pDCs also secrete interleukin-12 (IL-12), contributing to T helper 1 (Th1) cell polarization of CD4+ T cells (Asselin-Paturel et al., 2001; Cella et al., 2000). Additionally, pDCs can directly kill virus-infected cells through FasL- and tumor necrosis factor-related apoptosis inducing ligand (TRAIL)-dependent mechanisms (Chaperot et al., 2006; Hardy et al., 2007).
pDCs detect RNA and DNA viruses through two endosomal sensors, Toll-like receptors (TLR) 7 and TLR9, which induce secretion of IFN-I through the MyD88-interferon regulatory factor 7 (IRF7) signaling pathway (Blasius and Beutler, 2010; Honda et al., 2005a; Pichlmair and Reis e Sousa, 2007; Takeuchi and Akira, 2009; Thompson and Iwasaki, 2008). Viruses reach TLR7 and TLR9 via receptor-mediated endocytosis or transport of cytosolic replication intermediates into endosomes by autophagy (Thompson and Iwasaki, 2008; Wang et al., 2007). Various DNA and RNA viruses activate pDCs (Borrow and Bhardwaj, 2008; Cervantes-Barragan et al., 2007; Delale et al., 2005; Diebold et al., 2004; Jung et al., 2008; Krug et al., 2004a, 2004b; Steinberg et al., 2009; Thompson and Iwasaki, 2008; Yoneyama et al., 2005; Zucchini et al., 2008) without the need for viral replication (Kumagai et al., 2009). Moreover, TLR7-, TLR9-, MyD88-, and IRF7-deficient mice fail to secrete sufficient IFN-I after certain viral infections, resulting in increased viral replication and mortality in comparison to wild-type mice (Delale et al., 2005; Honda et al., 2005b; Steinberg et al., 2009; Thompson and Iwasaki, 2008; Zucchini et al., 2008). However, it is not clear whether pDCs are primarily responsible for TLR7- or TLR9-MyD88-IRF7-mediated antiviral responses in vivo. Moreover, whether pDCs impact the control of viral infection via mechanisms other than IFN-I in vivo is poorly understood.
One approach to assessing pDC functions in vivo is to analyze antiviral host responses in mice lacking pDCs. To this end, pDCs have been depleted by the administration of monoclonal antibodies specific for pDC surface antigens such as Gr-1 (Asselin-Paturel et al., 2001) or bone marrow stromal antigen 2 (BST-2) (Asselin-Paturel et al., 2003; Blasius et al., 2006b; Krug et al., 2004a). Although informative, one limitation of antibody (Ab) depletion studies is that the Gr-1 antigen (Ag) is expressed by pDCs, plasma cells (Wrammert et al., 2002), inflammatory monocytes (Barbalat et al., 2009), subsets of T cells (Walunas et al., 1995), and granulocytes. Moreover, the BST-2 Ag is expressed on pDCs and plasma cells in naive mice but is induced on most cell types after stimulation with IFN-I or IFN-γ (Blasius et al., 2006b). Therefore, pDC-depleting Abs can deplete additional cell types during viral infection and the subsequent immune response, thus confounding the interpretation of these studies.
An alternative approach to Ab depletion is to evaluate mutant mice that are deficient for pDCs, specifically mice lacking the transcription factor E2-2 (Cisse et al., 2008) or with a hypomorphic mutation of Ikaros (Ikzf1L/L) (Allman et al., 2006). Because Ikaros is also expressed in non-pDC subsets and pDCs are not completely eliminated in Ikzf1L/L mice (Allman et al., 2006), this mouse model presents similar limitations as pDC-depleting antibodies. E2-2-deficient mice have a more specific pDC defect, but have not yet been evaluated for susceptibility to viral infections.
To precisely address the impact of pDCs in innate and adaptive antiviral immune responses, we generated transgenic (Tg) mice that express the diphtheria toxin receptor (DTR) under the control of the highly specific human pDC gene promoter, BDCA-2. Administration of diphtheria toxin (DT) to these mice resulted in an almost complete and selective depletion of pDCs. pDC-depleted mice were challenged with representative DNA and RNA viruses, murine cytomegalovirus (MCMV), and vesicular stomatitis virus (VSV), respectively. Our results demonstrated that pDCs provide an immediate but limited source of IFN-I that restricts viral burden only in the very early phase of infection. Lack of pDC-mediated containment of MCMV resulted in the increased expansion of NK cells expressing the Ly49H receptor. In contrast, during VSV infection, pDC depletion reduced the amplitude of primary CD8+ T cell responses resulting from impaired survival of virus-specific cytotoxic T cells (CTLs). Thus, pDCs impact virus-specific NK cell or CD8+ T cell responses in a fashion that is dependent on the infecting agent.
Results
Specific pDC Depletion in BDCA2-DTR Transgenic Mice
Evaluating the role of DCs in orchestrating immune responses has been greatly facilitated by the generation of Tg mice that express DTR under the control of the DC-specific CD11c promoter (Jung et al., 2002). Administration of DT to CD11c-DTR Tg mice selectively eliminates CD11chi classical DCs. Emulating this approach, we generated Tg mice that express DTR under the blood dendritic cell antigen 2 (BDCA-2) promoter. BDCA-2 is a C-type lectin that is exclusively expressed by human pDCs (Gilliet et al., 2008). Although BDCA-2 is not encoded in mouse, we assumed that its promoter contains target sequences for pDC-specific transcription factors that might be conserved across species. Indeed, this assumption was correct; the transcription factor E2-2, which is a specific regulator of pDC development in mouse and human, was subsequently shown to interact with elements in the BDCA-2 promoter (Cisse et al., 2008). Therefore, we generated a BDCA2-DTR Tg mouse on a C57BL/6 background in which pDCs were systemically depleted with high efficiency and specificity 24 hr after DT administration (Figure 1). The DT-induced depletion persisted for 2 to 3 days; however, pDCs could be depleted for longer periods with repeated DT administration. The depletion efficiency in blood, spleen, liver, and lymph nodes was approximately 90% 24–48 hr after DT treatment (data not shown). To further demonstrate that DT specifically depletes pDCs, BDCA2-DTR Tg mice were bred to SiglecH-eGFP (enhanced green fluorescent protein) gene-targeted mice (Figure S1A available online). In these mice, pDCs expressed high amounts of eGFP, whereas classical DCs expressed low amounts of eGFP (Figure S1B). DT administration to BDCA2-DTR Tg × SiglecH-eGFP gene-targeted mice resulted in the selective depletion of pDCs whereas classical DCs were left intact (Figure S1C). Thus, the BDCA2-DTR Tg mouse is a unique tool to specifically investigate the function of pDCs in a variety of models.
Figure 1. Generation of BDCA2-DTR Tg Mice and Selective Depletion of pDCs.
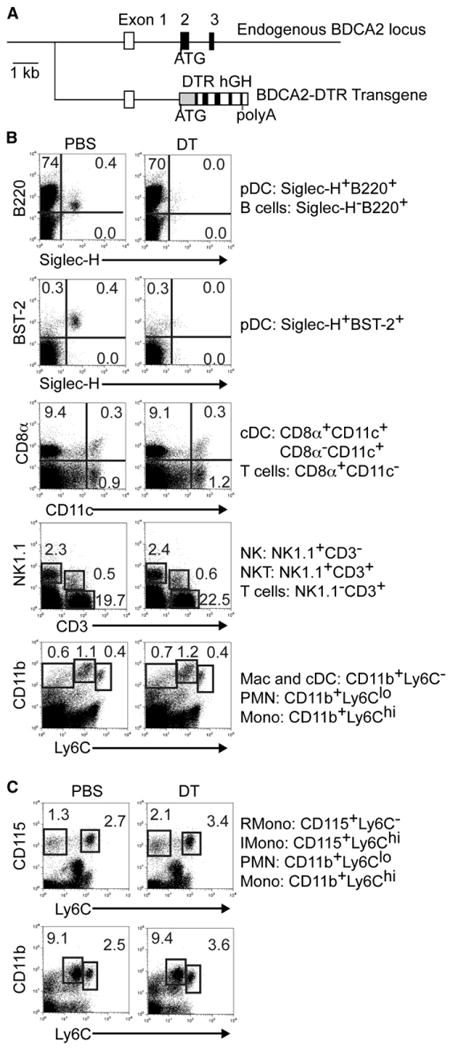
(A) The construct for BDCA2-DTR Tg mice includes a 5 kb fragment upstream of the ATG of the BDCA-2 gene, a cDNA fragment encoding DTR, and the human growth hormone (hGH) gene polyadenylation signal.
(B and C) DT administration specifically depletes pDCs in BDCA2-DTR Tg mice. Cell subsets in spleen (B) and blood (C) were analyzed by flow cytometry 24 hr after DT injection. Data shown are representative of at least three experiments. Classical DCs (cDC), macrophages (Mac), resident (R), inflammatory (I), monocytes (Mono), and granulocytes (PMN).
pDCs Limit MCMV Replication Independently of Ly49H+ NK Cells
NK cells play a critical role in controlling MCMV infection by killing infected cells in the spleen and producing IFN-γ in the liver (Biron et al., 1999). In C57BL/6 mice, NK cells recognize MCMV-infected cells when the MCMV-encoded m157 protein expressed on the surface of infected cells binds to the Ly49H activating receptor on a subset of NK cells (Lanier, 2008). NK cells are also activated by IFN-I and other cytokines (Biron et al., 1999; García-Sastre and Biron, 2006), which are released by MCMV-activated pDCs. Thus, it has been thought that pDCs may contribute to anti-MCMV defense primarily by activating NK cell-mediated surveillance.
To address the role of pDCs in controlling viral burden, we infected control and DT-treated mice with different doses of MCMV intraperitoneally (i.p.) and examined viral titers in multiple tissues. In control mice, MCMV systemically activated pDCs in vivo, as assessed by expression of the activation marker PDC-TREM (Figure S2A; Watarai et al., 2008). In DT-treated mice, pDCs were effectively depleted in spleen and liver without having major effects on the total numbers of cells (Figure S2B–S2D). Furthermore, we confirmed that DT treatment did not eliminate cells critical for antiviral responses such as virus-specific NK cells and CD8+ T cells during MCMV infection (Figures S2E and S2F). On day 3 postinfection (p.i.), viral titers in the spleen were significantly higher in DT-treated mice when a dose of 5 × 104 plaque-forming units (pfu) per mouse was administered whereas no differences were observed at a higher (1 × 105 pfu) dose (Figure 2A). When we quantified viral titers in the livers on day 3 p.i., we found that pDC-depleted mice had significantly higher viral titers than control mice only at a low dose (1 × 104 pfu) (Figure 2B).
Figure 2. pDC-Mediated Inhibition of MCMV Replication Is Dependent on Viral Inoculum.
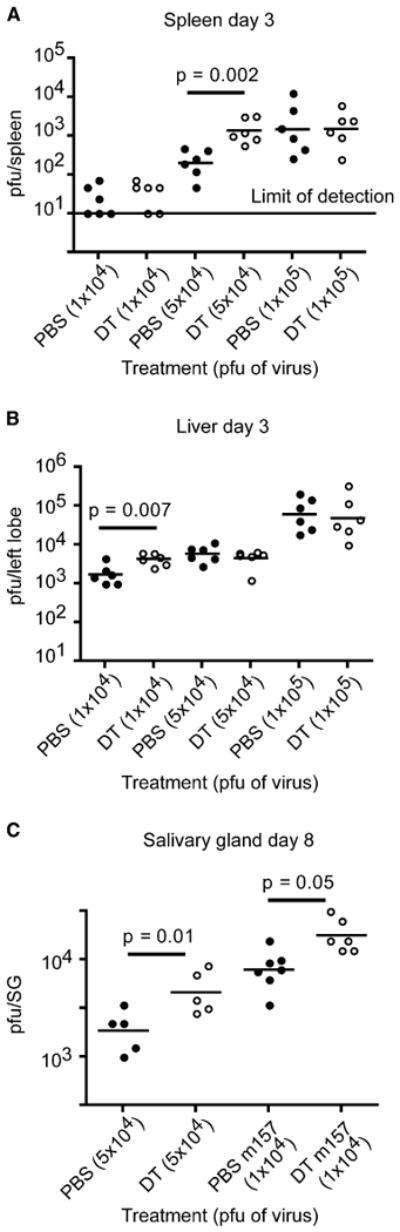
(A and B) PBS- or DT-treated mice were infected i.p. with different doses of MCMV and viral titers in spleen (A) and liver (B) were enumerated on day 3 p.i. by plaque assay.
(C) PBS- or DT-treated mice were infected i.p. with MCMV or Δm157 MCMV and salivary gland (SG) viral titers were calculated on day 8 p.i. p value, unpaired, two-tailed Student's t test. Data shown are from two experiments.
Because MCMV has tropism for the salivary glands and replicates there during the later stages of infection (Krmpotic et al., 2003), we next evaluated salivary gland viral titers in control and pDC-depleted mice on day 8 p.i. In these experiments, wild-type (WT) MCMV (5 × 104 pfu) as well as MCMV lacking m157 (Δm157 MCMV) (1 × 104 pfu) were used to infect mice. Our rationale for using Δm157 MCMV is that this virus escapes surveillance by Ly49H+ NK cells in C57BL/6 mice (Lanier, 2008), allowing the evaluation of pDC impact on viral control independently of Ly49H+ NK cells. Salivary glands from pDC-depleted mice infected with either WT or Δm157 MCMV had significantly elevated viral loads (Figure 2C). We also examined the salivary glands from control and pDC-depleted mice infected with 1 × 105 pfu of WT MCMV for 8 days and found no differences in viral titers (data not shown). These results demonstrate that at lower doses of MCMV, the presence of pDCs can limit viral burden in the spleen, liver, and salivary glands. However, at higher inoculi of MCMV, pDCs are not sufficient to control viral loads. The results obtained with Δm157 MCMV infection also indicate that pDCs can control MCMV infection independently of Ly49H+ NK cells.
pDC Depletion Has Distinct Effects on Nonspecific and Specific NK Cell Activation
It has been established that NKcell surveillance of MCMV infection develops through distinct phases (Dokun et al., 2001). In the early phase of infection, NKcell activation and secretion of IFN-γ is independent of Ly49H engagement but depends on cytokines. During the later phase of infection, Ly49H+ NK cells are selectively activated and proliferate in response to MCMV. After viral clearance, Ly49H+ NK cells undergo contraction and generate a population of memory cells (Sun et al., 2009). To assess the effect of pDCs on NK cell activity in the early phase of infection, we infected pDC-depleted and control mice with MCMV and measured spleen NK cell frequencies, activation, ability to kill target cells, and intracellular IFN-γ content. The overall frequencies of NK cells in the spleen declined overtime in both groups of mice (Figure 3A). At 36 hr p.i., NK cells from pDC-depleted mice had reduced expression of CD69 compared to control mice whereas mice from both groups at 48 hr p.i. expressed similar amounts of CD69 (Figure 3B). At 24 hr p.i., NK cells from pDC-depleted mice were less efficient at killing RMA-S target cells (Figure 3C). At 36 and 48 hr, this difference in specific lysis was no longer observed between control and pDC-depleted mice. Thus, in the absence of pDCs, activation and cytotoxicity of nonspecific NK cells is impaired very early on during infection.
Figure 3. pDC Impact on Nonspecific and Specific NK Cell Activation during MCMV Infection.
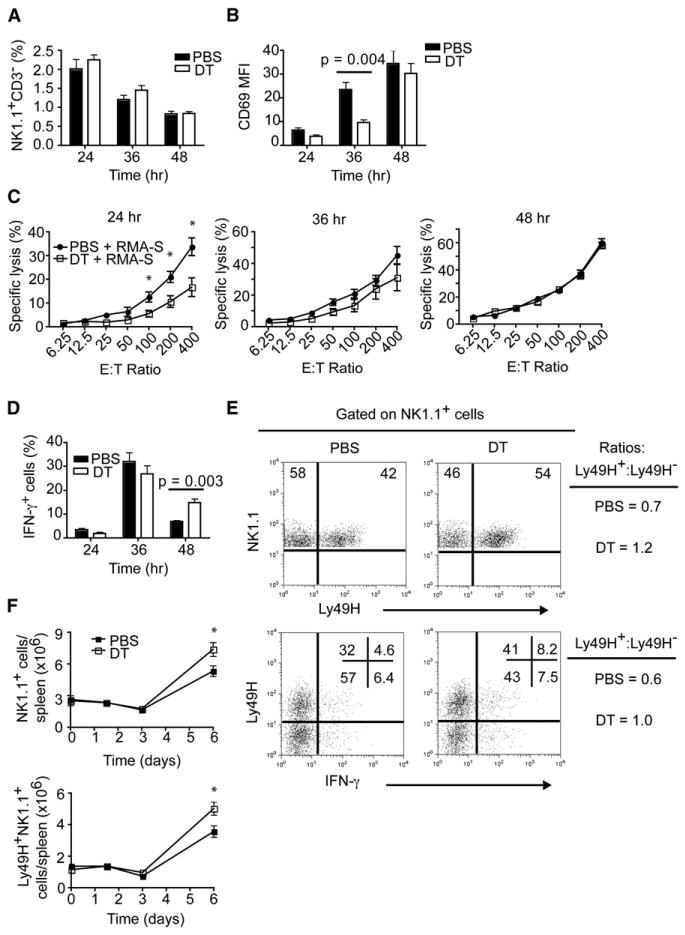
Mice were infected i.p. with MCMV (5 × 104 pfu).
(A) Frequencies of NK cells (NK1.1+CD3−) in spleens.
(B) Expression of CD69 on splenic NK cells.
(C) Cytotoxic capacity of splenic NK cells from control and pDC-depleted mice with RMA-S target cells in standard 4 hr 51Cr release assays.
(D) Frequencies of IFN-γ+ NK cells in spleens of MCMV-infected mice.
(E) Frequencies of Ly49H+ and IFN-γ+-producing NK cells on day 3 p.i. Data are representative of six to eight mice from two experiments.
(F) Total numbers of NK1.1+ and Ly49H+NK1.1+ cells during MCMV infection.
(B and D) p value, unpaired, two-tailed Student's t test.
(C and F) Asterisks denote p values ≤ 0.03 as measured by unpaired, two-tailed Student's t test. (A–D, F) Data are from two experiments (mean ± SEM, n = 5–7).
The effect of pDC depletion on NK cell secretion of IFN-γ was opposite. Frequencies of IFN-γ-producing NK cells were similar in control and pDC-depleted mice at 24 and 36 hr p.i. At 48 hr, IFN-γ-producing NK cells declined in both groups of mice; however, there were significantly more IFN-γ-producing NK cells (∼2.5-fold) in pDC-depleted mice (Figure 3D). Additionally, we observed increased frequencies of IFN-γ-producing NKT cells in the spleens and livers of pDC-depleted mice compared to controls (Figure S3). Thus, in the absence of pDCs, IFN-γ secretion by NK cells and NKT cells is facilitated.
To assess the effect of pDCs on the MCMV-specific NK cell response, we infected pDC-depleted and control mice with MCMV and measured frequencies of Ly49H+ and Ly49H− NK cells and their intracellular IFN-γ content 3 days p.i. pDC-depleted mice showed increased frequencies of Ly49H+ NK cells as well as more IFN-γ-producing Ly49H+ NK cells than control mice (Figure 3E). These results prompted us to examine NK1.1+ and Ly49H+ NK cell numbers at different time points p.i. As anticipated, pDC-depleted mice had a greater accumulation of total NK1.1+ and Ly49H+NKcells during the later phase of infection (Figure 3F). Taken together, these results demonstrate that pDCs promote transient NK cell activation and cytotoxicity in the early phase of MCMV infection, but limit NK cell and NKT cell secretion of IFN-γ. At later time points, pDC depletion does not affect the selective expansion and function of Ly49H+ NK cells, which are in fact more abundant, most probably as a response to increased viral replication and subsequent engagement of Ly49H by m157.
pDCs Are a Transient Source of IFN-I that Modulates IL-12 and IFN-γ Production
To investigate the mechanisms by which pDCs control low-intermediate inoculi of MCMV, induce activation of NK cells, and modulate NK cell and NKT cell secretion of IFN-γ, we examined serum cytokine concentrations in pDC-depleted mice at different time points. pDC-depleted mice produced significantly less IFN-α at 36 hr p.i. whereas no differences in serum IFN-α were observed at 48 hr (Figure 4A). Thus, pDCs are the major source of IFN-I during the initial stages of MCMV infection whereas non-pDCs are responsible for the IFN-I produced at later time points (Asselin-Paturel et al., 2001; Dalod et al., 2002, 2003; Delale et al., 2005; Scheu et al., 2008). In addition, pDC-depleted mice had 3- to 4-fold more serum IFN-γ than nondepleted mice 48 hr p.i. (Figure 4B), reflecting the increased IFN-γ production by NK cells and NKT cells. The impact of pDC depletion on IFN-γ serum concentrations was dependent on the viral inoculum. DT-treated mice infected with low or intermediate doses of WT MCMV had higher concentrations of serum IFN-γ compared to control mice (Figure 4C). DT-treated mice infected with Δm157 MCMV also had elevated amounts of serum IFN-γ. In contrast, no differences were apparent when DT-treated mice were infected with 1 × 105 pfu of WT virus. These results corroborate that the role of pDCs in early cytokine responses to MCMV infection is prominent only at low viral inoculi.
Figure 4. Impact of pDCs on Cytokine Production, DC Activation, and Early Anti-MCMV CD8+T Cell Responses.
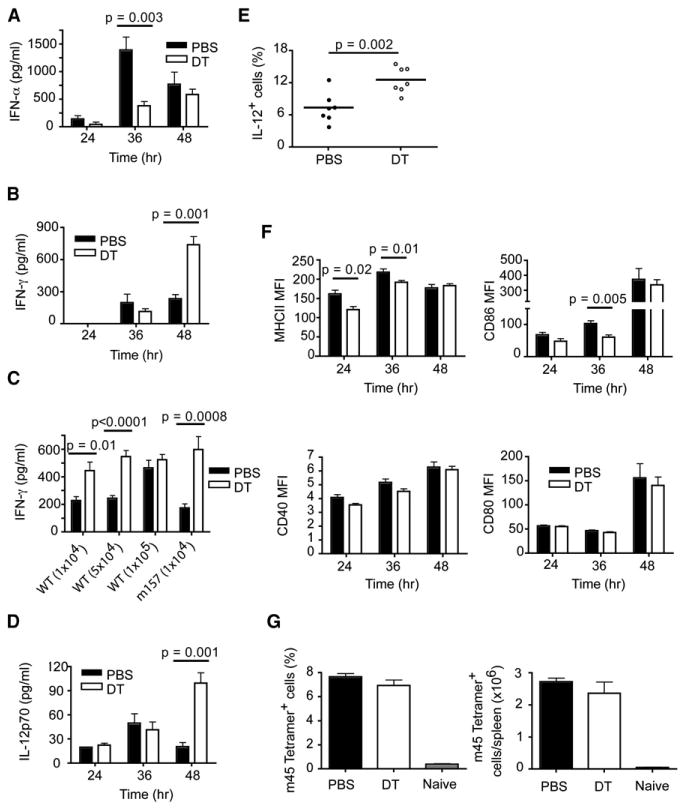
(A, B, D–G) PBS- and DT-treated mice were infected i.p. with 5 × 104 pfu of MCMV and serum samples were analyzed for IFN-α (A), IFN-γ (B), and IL-12p70 (D).
(C) PBS- and DT-treated mice were infected i.p. with different doses of WT MCMV or Δm157 MCMV and serum IFN-γ concentrations were quantified 48 hr p.i.
(E) Frequencies of IL-12+CD11chi cells were determined 48 hr p.i. by intracellular staining.
(F) Expression based on mean fluorescence intensity (MFI) of costimulatory (CD86, CD40, CD80) and MHC class II molecules on CD11chi DCs.
(G) Frequencies and absolute numbers of Ag-specific CD8+ T cells on day 6 p.i.
p value, unpaired, two-tailed Student's t test. Data are from two (A, B, D–G) or three (C) experiments (mean ± SEM, n = 5–25).
The increase of serum IFN-γ in pDC-depleted mice was mirrored by a parallel increase of serum IL-12 (Figure 4D), which probably triggered IFN-γ production. It was previously shown that CD11b+ DCs from MCMV-infected mice depleted of pDCs with anti-Gr-1 produce more IL-12 (Dalod et al., 2002). Consistent with these results, we found more IL-12-producing CD11chi cells in DT-treated mice than in the control group (Figure 4E). Most probably this reflects an inhibitory effect of the IFN-α released by pDCs on IL-12 secretion by classical DCs (Dalod et al., 2002, 2003). Taken together, these results demonstrate that pDCs provide a transient source of IFN-I that limits the IL-12-IFN-γ axis in response to low viral inoculi.
Impact of pDCs on DC Activation and Early MCMV-Specific CD8+ T Cell Responses
We observed that CD11chi DCs from MCMV-infected pDC-depleted mice expressed lower amounts of MHC class II at 24 and 36 hr p.i. and CD86 at 36 hr p.i.; however, these differences ceased at 48 hr (Figure 4F). pDC depletion had little effect on CD40 or CD80 expression by CD11chi DCs. Thus, the absence of pDCs affects the activation of DCs during the initial phase of MCMV infection. However, this transient delay of DC activation was insufficient to affect subsequent CD8+ T cell responses in the C57BL/6 background during the first week of infection (Figure 4G).
pDCs Mediate Early IFN-I Responses during VSV Infection
To obtain a broader view of pDC function in viral infections, we assessed the contribution of pDCs in an infection model by using an RNA virus. Because the TLR7-MyD88-IRF7 pathway has been shown to be important for the control of VSV infection and for pDC responses to VSV (Honda etal., 2005b; Thompson and Iwasaki, 2008), we evaluated the role of pDCs during an infection with recombinant VSV expressing ovalbumin (VSV-OVA). To this end, control and pDC-depleted mice were infected intravenously (i.v.) with VSV-OVA. In control mice, B220+Siglec-H+ spleen pDCs were activated and expressed PDC-TREM as early as 6 hr p.i. PDC-TREM could also be detected on pDCs in the blood, bone marrow, and liver 24 hr p.i. (Figure S4). We also measured serum IFN-α concentrations and virus titers in the spleen at different time points p.i. In the absence of pDCs, IFN-α was reduced at 6 hr p.i. but not at 12 or 24 hr (Figure 5A). We also found a significant difference in viral loads between control and pDC-depleted mice at 6 hr p.i., whereas it was difficult to detect VSV-OVA in the periphery after 24 hr (Figure 5B and data not shown). Thus, pDCs contribute to IFN-I production and control viral burden very early during VSV-OVA infection.
Figure 5. pDCs Augment Ag-Specific CD8+T Cell Responses during VSV-OVA Infection.
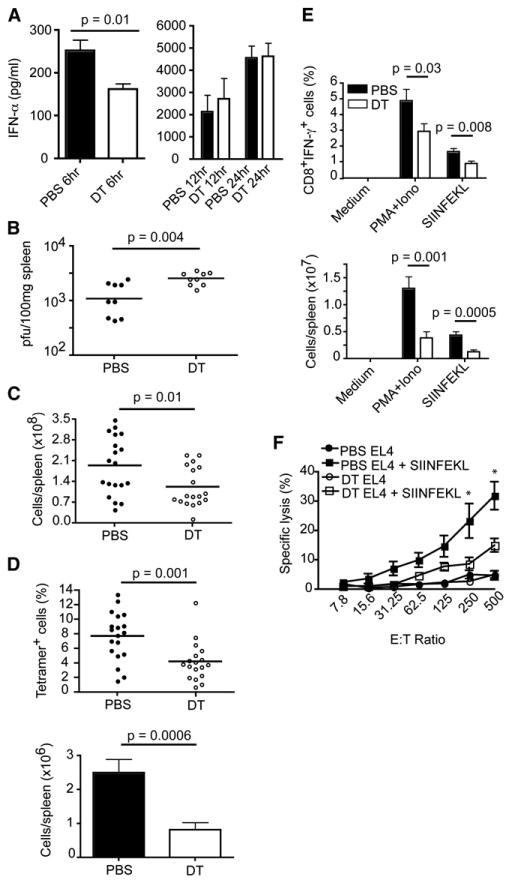
PBS- or DT-treated mice were infected i.v. with 5 × 106 pfu (A, C–F) or 5 × 105 pfu (B) of VSV-OVA.
(A) IFN-α concentrations in serum were determined at 6, 12, or 24 hr p.i.
(B) Viral titers in spleens were measured at 6 hr p.i.
(C–F) Spleens from VSV-OVA-infected mice were analyzed on day 7 p.i.
(C) Spleen cellularity.
(D) Spleen cells from infected mice were stained with H-2Kb OVA257-264 peptide tetramers and anti-CD8. Frequencies and totalnumbers ofAg-specific CD8+ T cells were determined by flow cytometry.
(E) Frequencies and numbers of CD8+ T cells producing IFN-γ after restimulation with SIINFEKL were determined by flow cytometry.
(A–E) p value, unpaired, two-tailed Student's t test. Data are from two to five experiments (mean ± SEM, n = 6–19).
(F) Antigen-specific cytotoxicity was assessed in standard 4 hr 51Cr release assays. Asterisks denote pvalues ≤0.03 as measured by unpaired, two-tailed Student's t test. Data are from two experiments (mean ± SEM, n = 6–7).
pDCs Influence Anti-VSV CD8+ T Cell Responses In Vivo
Next we analyzed spleens from VSV-OVA-infected mice on day 7 p.i. Spleens from pDC-depleted mice contained fewer cells and were slightly smaller than controls (Figure 5C and data not shown). Closer examination with H-2Kb OVA257-264 peptide tetramers revealed reduced frequencies and numbers of Ag-specific CD8+ T cells in pDC-depleted mice (Figure 5D). We also noted a reduction in the frequencies and numbers of Ag-specific CD8+ T cells from pDC-depleted mice that produced IFN-γ after stimulation with PMA+Ionomycin or OVA257-264 (SIINFEKL) peptide (Figure 5E). To test whether Ag-specific CD8+ T cells from both groups of mice were capable of killing target cells, we incubated splenocytes with either untreated or SIINFEKL-pulsed EL4 cells in standard chromium release assays. In accordance with the reduced frequencies of tetramer+ and IFN-γ+ cells in pDC-depleted mice, less specific lysis was observed compared to controls (Figure 5F). These data show that pDCs are important for the establishment of viral-specific CD8+ T cell responses in this model of viral infection.
pDCs Enhance the Accumulation of Ag-Specific CD8+ T Cells during VSV Infection
To corroborate that pDCs indeed facilitate the accumulation of Ag-specific CD8+ T cells during VSV infection, we set up an adoptive transfer system in which CD8+ T cells from OT-I TCR Tg mice were labeled with the cytoplasmic dye CFSE and injected into BDCA2-DTR Tg mice. The following day, mice were administered either phosphate-buffered saline (PBS) or DT and then infected with VSV-OVA. When spleens were examined 66 hr p.i., we found that the transferred CD8+ T cells in both groups of mice were proliferating based on CFSE dilutions (Figure 6A); however, the frequencies as well as the absolute numbers of CD8+Vα2+CFSE+ cells were 3-fold higher in nondepleted mice (Figure 6B) (Vα2 is the T cell receptor [TCR] α chain used by OT-I OVA-specific CD8+ T cells). Taken together, these data demonstrate that pDCs enhance the accumulation of Ag-specific CD8+ T cells during VSV infection.
Figure 6. pDCs Enhance Accumulation of Ag-Specific CD8+T Cells during VSV-OVA Infection.
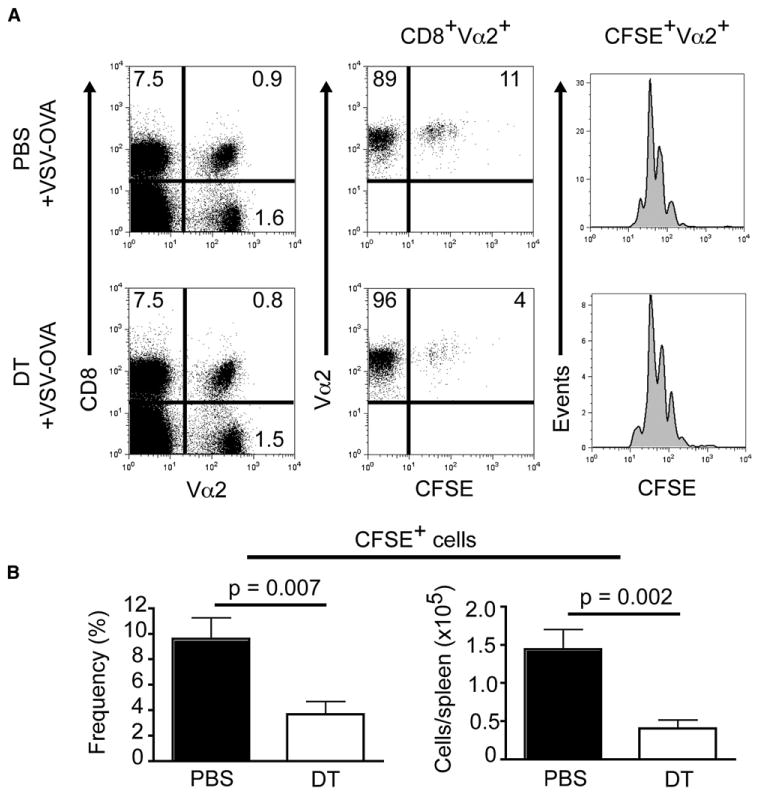
Purified CD8+ T cells from OT-I TCR Tg mice were CFSE labeled and adoptively transferred into BDCA2-DTR Tg mice. Mice were injected the following day with PBS or DT, then infected i.v. with VSV-OVA (5 × 105 pfu) 24 hr later. Splenocytes isolated 66 hr p.i. were stained with anti-CD8 and -Vα2 then analyzed by flow cytometry. Data are from two experiments (mean ± SEM, n = 8).
(A) Dot plots show the frequencies of CFSE+ cells among CD8+Vα2+ T cells. Histograms show the number of divisions based on CFSE dilution.
(B) Frequencies and absolute numbers of CFSE+CD8+Vα2+ T cells. p value, unpaired, two-tailed Student's t test.
pDCs Promote the Survival of VSV-Specific CD8+ T Cells
We next asked how pDCs contribute to the accumulation of Ag-specific CD8+ T cells. pDCs may elicit the expansion of CD8+ T cells by activating bystander DCs (Yoneyama et al., 2005). Therefore, we examined DC numbers, activation state, and Ag presentation in control and pDC-depleted VSV-OVA-infected mice, but found no differences in DC numbers (Figure S5A) or the upregulation of costimulatory or MHC class II molecules on CD11chi DCs (data not shown). We also observed no differences in the ability of CD11c+ DCs enriched from VSV-OVA-infected control or pDC-depleted mice to present Ag to CD8+ T or CD4+ T cells purified from OT-I or OT-II TCR Tg mice, respectively (Figure S5B). We also sorted DC subsets from VSV-OVA-infected control and pDC-depleted mice and found (1) that CD8α+ DCs and CD8α− DCs from both groups of mice were equally capable of presenting Ag to OT-I and OT-II cells and (2) that pDCs do not present Ag to OT-I or OT-II cells (data not shown).
pDCs preferentially secrete chemokines such as CCL3 and CCL4 (Sozzani et al., 2010), which have been shown to recruit naive CD8+ T cells into priming sites. Thus, depleting pDCs may affect the recruitment of naive CD8+ T cells to the spleens of VSV-OVA-infected mice. Quantification of these two chemokines in the serum of VSV-OVA-infected mice revealed that both groups of mice produced CCL3 and CCL4 (data not shown and Figure 7A); however, there was a significant reduction in serum CCL4 in pDC-depleted mice 24 hr p.i. To assess whether the reduction in CCL4 affected the recruitment of Ag-specific CD8+ T cells, we examined the frequencies of adoptively transferred OT-I cells in spleens at early time points p.i. in control and pDC-depleted mice and compared them to that of naive mice. At 6 hr p.i., the frequencies of OT-I cells in control and pDC-depleted mice were comparable to uninfected mice, indicating that recruitment of Ag-specific CD8+ T cells was not impaired. At 22 hr p.i., the frequencies of OT-I began to decline and this reduction was more pronounced in pDC-depleted mice compared to PBS controls (Figure 7B), suggesting that pDCs may impact the survival of Ag-specific CD8+ T cells.
Figure 7. Recruitment and Survival of Ag-Specific CD8+T Cells during VSV-OVA Infection.
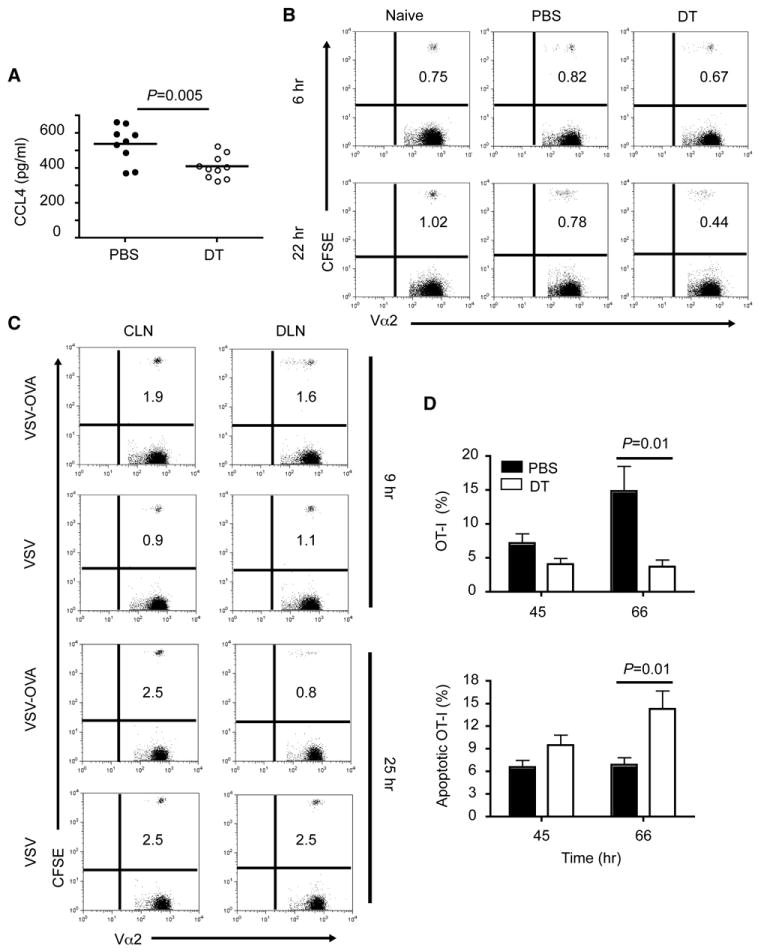
(A) Serum concentrations of CCL4 in VSV-OVA-infected mice 24 hr p.i. Data are from three experiments.
(B) Purified CD8+ T cells from OT-I TCR Tg mice were CFSE labeled and adoptively transferred into BDCA2-DTR Tg mice. Mice were depleted and infected as described in Figure 6. Splenocytes isolated at6or22 hr p.i. were stained with anti-Vα2then analyzed by flow cytometry. Dot plots show the frequencies of CFSE+ cells among Vα2+ cells. Data are representative of three to eight mice per group.
(C) Mice were injected with 2 × 106 CFSE-labeled CD8+ T cells from OT-I mice then infected in the footpad with VSV or VSV-OVA (1 × 106 pfu) 24 hr later. Controlateral (CLN) and draining (DLN) lymph nodes were stained with anti-Vα2 and analyzed by flow cytometry at 9 or 25 hr p.i. Data are representative of three mice per group.
(D) Mice were injected with CFSE- or eFluor 670-labeled OT-I cells, then depleted or not and infected as described in Figure 6. Splenocytes were stained with anti-CD8, anti-Vα2, and Annexin V or CaspACE FITC-VAD-FMK 45 or 66 hr p.i. Bar graphs show the frequencies of OT-I cells among CD8+Va2+ cells (top) and frequencies of apoptotic OT-I cells (bottom). Data are from two experiments (mean ± SEM, n = 4–8).
p value, unpaired, two-tailed Student's t test.
To address whether the initial decline in OT-I frequencies was due to Ag-induced apoptosis, we infected mice in the footpads with VSV-OVA or VSV and compared the frequencies of OT-I cells in the contralateral lymph nodes (CLN) to those in the draining lymph nodes (DLN) (Figure 7C). At 9 hr p.i., the frequencies of OT-I begin to decline in the DLN of VSV-OVA-infected mice but not in the DLN of VSV-infected mice. At 25 hr p.i., the reduction in frequencies was even more dramatic in VSV-OVA-infected mice, suggesting that OT-I cells do in fact undergo Ag-induced apoptosis. To corroborate this finding, we evaluated the apoptosis of adoptively transferred OT-I cells in spleens of control or pDC-depleted mice infected i.v. with VSV-OVA by using Annexin V and CaspACE FITC-VAD-FMK (which binds to activated caspase). As anticipated, more apoptotic Ag-specific CD8+ T cells were evident in pDC-depleted mice, especially at 66 hr p.i. (Figure 7D). Taken together, these results demonstrate that pDCs promote the survival of Ag-specific CD8+ T cells during VSV-OVA infection.
Discussion
In this study, we have addressed the contribution of pDCs to antiviral responses by using a BDCA2-DTR Tg mouse that we have generated in our lab. We first showed that pDCs can be almost entirely and specifically depleted in these mice, providing an optimal experimental system to evaluate pDC functions in vivo. This experimental model appears to be superior to depleting antibodies, because the available antibodies also recognize non-pDCs, potentially depleting other cells involved in immune responses. Cross-reactivity is particularly problematic when BST-2 antibodies are used during viral infections, because BST-2 is upregulated on a wide variety of cells in response to IFNs (Blasius et al., 2006b), resulting in their depletion. Although Siglec-H is predominately expressed on pDCs (Blasius et al., 2006a; Zhang et al., 2006), SiglecH-eGFP gene-targeted mice exhibited eGFP expression not only in pDCs but also in classical DCs, suggesting that the promoter of BDCA-2 is more suitable than that of Siglec-H to selectively drive gene expression in pDCs.
The effectiveness of the BDCA2-DTR Tg mouse model in evaluating antiviral pDC functions was confirmed by our results in the MCMV and VSV infections. We demonstrated that pDCs are essential for the production of IFN-I only during the initial stages of MCMV infection, which is in agreement with a study by Delale et al. (2005). Additionally, we found that the impact of pDC depletion on anti-MCMV responses is highly dependent on initial viral dose, demonstrating that the antiviral capacity of pDCs is limited. Because defects in IFN-I or MyD88 signaling (Dalod et al., 2002, 2003; Delale et al., 2005; Krug et al., 2004a; Steinberg et al., 2009; Zucchini et al., 2008) have more profound impacts on anti-MCMV control than does pDC depletion, we conclude that pDCs account only in part for the antiviral responses mediated by IFN-I and MyD88 signaling and other cells must be involved. CD11b+ DCs are also capable of producing IFN-I and activating NK cells in a TLR-independent manner after infection with MCMV (Andoniou et al., 2005). In addition, both TLR3 and TLR2 pathways, which are not operating in pDCs, have been implicated in the secretion of IFN-I during MCMV infection (Barbalat et al., 2009; Szomolanyi-Tsuda et al., 2006; Tabeta et al., 2004).
Although in the MCMV model pDCs efficiently contained viral replication at low-to-intermediate viral loads but were insufficient at restraining high viral loads, we must emphasize that most natural infections in vivo occur by transmission of low numbers of virions. Therefore, pDCs might play a critical role in containing naturally spreading viral infections in physiological settings. Furthermore, the inability of pDCs to augment antiviral responses when higher MCMV doses are administered may be due to their limited numbers in infected organs, such as spleen, liver, and salivary glands. However, pDCs may be able to control higher viral loads during human infections such as varicella zoster virus, hepatitisCvirus, and molluscum contagiosum virus, which cause considerable accumulation of pDCs at sites of infection (Sozzani et al., 2010).
Previous studies have shown that pDCs activate NK cells, suggesting that pDCs control viral spreading and replication indirectly through NK cell-mediated killing of MCMV-infected cells. In our study we evaluated the impact of pDC depletion on the control of Δm157 MCMV, which escapes surveillance by Ly49H+ NK cells (Lanier, 2008), and found that pDCs contribute to the control of MCMV independently of Ly49H+ NK cells. Thus, IFN-I released by pDCs controls MCMV replication directly by inducing an antiviral state in other cells. As previously reported, we confirmed that pDCs also induce NK cell activation, but only during the early nonspecific phase of the NK cell response. In contrast, pDCs were not required for activation of MCMV-specific Ly49H+ NK cells, which in fact were more frequent in pDC-depleted mice. This expansion of Ly49H+ cells is most probably a consequence of increased viral titers and thus may compensate for the lack of pDCs and their ability to control viral replication. Accordingly, mice lacking IFN-I signaling exhibit preferential expansion of Ly49H+ cells (Geurs et al., 2009).
pDC depletion resulted in an increased frequency of IFN-γ-producing NK cells and NKT cells in the spleen and liver, as wellas increased serum concentrations of IL-12p70 and production of IL-12 by classical DCs. Previous studies in interferon alpha receptor (IFNAR) gene-targeted mice and mice depleted of pDCs with antibodies have demonstrated that IFN-I signaling in classical DCs reduces IL-12 production during MCMV infection (Dalod et al., 2002, 2003). Our data corroborate a cross-talk between pDCs and DCs such that pDC-released IFN-I limits IL-12 production by DCs and consequently IFN-γ production by NK cells and NKT cells. However, the impact of pDC depletion on systemic IL-12 was not as strong as that induced by blockade of IFN-I signaling, indicating that pDCs are only one of the sources of IFN-I that contribute to the control of the IL-12-IFN-γ axis.
Infection of BDCA2-DTR mice with VSV indicated a role for pDCs in the very early production of IFN-I and control of viral burden. Moreover, pDCs promoted the accumulation of Ag-specific CD8+ T cells, unveiling an important function of pDCs in adaptive immune responses. This pDC function may explain the delayed accumulation of T cells in the bronchoalveolar space of Ikzf1L/L mice infected with influenza (Wolf et al., 2009) and the defective CTL responses against HSV-1 infection in pDC-depleted mice (Yoneyama et al., 2005). We explored several mechanisms by which pDCs may promote CTL accumulation. We noticed that pDC depletion resulted in a significant reduction in serum concentrations of CCL4, which attracts CTLs to priming sites. However, pDC depletion had no clear impact on the recruitment of adoptively transferred Ag-specific CTLs. Although pDCs have been implicated in Ag presentation in several models (Villadangos and Young, 2008), pDCs did not present Ag in VSV-OVA infection nor increase the Ag-presenting capacity of classical DCs. Instead we found that pDCs promoted the survival of Ag-specific CTLs. Because pDCs limit viral replication early in VSV-OVA infection, CTL survival may be explained by reduced activation-induced apoptosis. Additionally, pDCs may promote CTL survival through soluble factors such as IFN-I (Kolumam et al., 2005; Marrack and Kappler, 2004).
Not surprisingly, the defect of CTL accumulation in pDC-depleted mice infected with VSV-OVA had no obvious impact on viral loads in the brain at later time points (data not shown), because VSV clearance is mainly dependent on Ab responses (Steinhoff et al., 1995), which did not vary between control and pDC-depleted mice (data not shown). However, pDC-mediated accumulation of CTLs may be essential in the control of other experimental infections, such as murine hepatitis virus (MHV), herpes simplex virus 2 (HSV-2), and respiratory syncytial virus (RSV), in which pDC depletion impairs host antiviral responses (Cervantes-Barragan et al., 2007; Smit et al., 2006; Thompson and Iwasaki, 2008; Wang et al., 2006). In conclusion, analysis of MCMV and VSV infections in our newly generated BDCA2-DTR Tg mice demonstrates that pDCs provide an early and transient source of IFN-I that partially controls viral replication. This pDC-mediated control of viral burden impacts the accrual of virus-specific NK cells or CD8+ T cells in a virus-dependent manner.
Experimental Procedures
Mice and Treatments
All animal studies were approved by the Washington University Animal Studies Committee. BDCA2-DTR Tg and SiglecH-eGFP gene-targeted mice were maintained as heterozygotes and used at 7–12 weeks of age. Diphtheria toxin (DT, Sigma-Aldrich) was injected intraperitoneally (i.p.) at 100–120 ng/mouse. pDCs were depleted on days −1,1, and 3 in virus-infected mice. Mice were administered PBS or DT only on day −1 in adoptive transfer experiments with VSV-OVA. OT-I and OT-II TCR Tg mice were used between 8 and 12 weeks of age.
Viruses and Infections
Smith strains MCMV and AT1.5 (Δm157) were generous gifts of W. Yokoyama and A. French (Washington University, St. Louis, MO), respectively. MCMV tissue culture (TC) stocks were prepared by propagation in BALB/c NIH3T12 fibroblasts (3T12, ATCC). Salivary gland (SG) MCMV stocks were prepared from BALB/c mice that were infected i.p. with 1 × 106 pfu of TC stock. Indiana strain VSV-OVA and VSV were provided by L. Lefrancois (University of Connecticut, Farmington, CT) and D. Lenschow (Washington University), respectively. Mice were infected i.p. with different doses (specified in figures and text) of MCMV SG stocks. VSV-OVA was administered intravenously (i.v.) at doses of 5 × 105 or 5 × 106 pfu/mouse. For footpad (f.p.) infections, mice were injected with 1 × 106 pfu of VSV or VSV-OVA.
Virus Plaque Assays
MCMV and VSV titers were determined by standard plaque assays. A detailed description of methods can be found in the Supplemental Information.
Cell Preparations
Spleens were minced and digested for 45 min at 37°C in RPMI 1640 with collagenase D (Sigma-Aldrich). Single-cell suspensions of spleens and lymph nodes were prepared by passage through nylon mesh cell strainers (BD Biosciences). Red blood cells (RBC) were lysed with RBC lysis buffer (Sigma-Aldrich). Liver cells were isolated by digesting minced lobes for 1 hr at 37°C in RPMI 1640 containing DNase I (Sigma-Aldrich) and collagenase D. Leukocytes were isolated over a 40%–70% Percoll gradient. Whole blood was collected by cardiac puncture and bone marrow cells were harvested from tibias and femurs.
Antibodies and Flow Cytometry
A detailed list of antibodies, reagents, and staining methods can be found in the Supplemental Information. All flow cytometry was conducted on a dual laser FACSCalibur flow cytometer (BD Biosciences) and analyzed with FlowJo software (Tree Star, Inc.).
ELISA and Cytometric Bead Array
Serum samples from infected mice were collected at various time points p.i. IFN-α concentrations were determined by ELISA (PBL Interferon Source). IL-12p70and IFN-γ were measured by flow cytometry with the Mouse Inflammation CBA kit (BD Biosciences) and CCL3 and CCL4 were quantified by flow cytometry with Mouse CBA flex sets (BD Biosciences).
Cell Lines and Tissue Culture
EL4 and RMA-S cells were grown in complete RPMI: RPMI 1640 with 10% bovine calf serum (BCS), 1% glutamax, 1% nonessential amino acids, 1% sodium pyruvate, and 1% kanamycin sulfate (GIBCO-Invitrogen). 3T12 and Vero cells were cultured in complete DMEM: high-glucose DMEM, 10% BCS, 1% glutamax, 1% HEPES, and 1% penicillin plus streptomycin (GIBCO-Invitrogen). Primary cells were cultured in complete RPMI with 10% fetal calf serum (FCS, Hyclone).
Cytotoxicity Assays
For NK cell cytotoxicity assays, splenocytes from MCMV-infected mice were resuspended in complete RPMI and serially diluted in 96-well round bottom plates. RMA-S cells were labeled with 1 mCi/ml 51Crfor2 hrthen incubated with effector cells at 37°C for 4 hr. 51Cr release in supernatants was measured with a γ-counter. For Ag-specific lysis assays, splenocytes from mice infected with VSV-OVA were resuspended in complete RPMI and serially diluted. EL4 cells were pulsed or not pulsed with H-2Kb OVA257-264 peptide (SIINFEKL, 10 ng/ml) and labeled with 51Cr as described above.
T Cell Restimulation Assays
Splenocytes from VSV-OVA-infected mice were incubated at 37°C in complete RPMI alone or with PMA+Ionomycin or SIINFEKL (10 μg/ml) in the presence of brefeldin A. After 6 hr cells were intracellularly stained for IFN-γ.
Antigen Presentation Assays
DCs were enriched from VSV-OVA-infected mice 24 hr p.i. by positive selection with anti-CD11c beads (Miltenyi Biotec). DCs were incubated with CD8+ or CD4+ T cells purified from OT-I or OT-II TCR Tg mice, respectively, for 48 hr and IFN-γ was measured in culture supernatants.
T Cell Purification, CFSE Labeling, and Adoptive Transfer
Naive CD8+ or CD4+ T cells were obtained from OT-I or OT-II TCR Tg mice by negative selection with CD8+ or CD4+ T cell isolation kits (Miltenyi Biotec) according to the manufacturer's instructions. Purity was greater than 90% as determined by flow cytometry. Purified CD8+ T cells were labeled for 10 min at room temperature with 1–5 μM CFSE (Invitrogen-Molecular Probes) or cell proliferation dye eFluor 670 (eBioscience) and 1 × 106 labeled CD8+ T cells were injected i.v. into DTR mice. For f.p. infections, 2 × 106 CFSE-labeled CD8+ T cells were injected i.v. 24 hr before VSV or VSV-OVA.
Apoptosis Assessment
Spleens were harvested from VSV-OVA-infected mice and single-cell suspensions were prepared as described above. After surface staining, cells were incubated with Annexin V (BD Biosciences) or CaspACE FITC-VAD-FMK (Promega) as recommended by the manufacturers.
Statistical Analysis
The statistical significance of differences in mean values was analyzed with unpaired, two-tailed Student's t test. p values less than 0.05 were considered statistically significant.
Supplementary Material
Acknowledgments
We would like to thank W. Yokoyama, T.Cheng, A.French, R. Presti (Washington University, St. Louis, MO), L. Lefrançois (University of Connecticut, Farmington, CT), and M. Brown (University of Virginia, Charlottesville, VA) for reagents and advice on virus infections; S. Jung for the DTR plasmid (Weizmann Institute of Science, Rehovot, Israel); S. McCartney and K. Otero (Washington University) for helping with injections; S. Schloemann and O. Malkova for cell sorting; M.White for injection of the BDCA2-DTR transgene and the SiglecH-eGFP ES cells; and M. Cella, M. Diamond, A. French, T. Hannan, and P. Allen for helpful discussions and critically reading the manuscript. M.S. is supported by the NRSA training grant 5T32DK007296-30. Y.W. is supported by the Pulmonary and Critical Care training grant 2T32HL007317-31 from the National Heart, Lung, and Blood Institute. This project was supported by Juvenile Diabetes Research Foundation grant 24-2007-420 and National Institutes of Health grant CA109673 to M.C.
Footnotes
Supplemental Information: Supplemental Information includes Supplemental Experimental Procedures and five figures and can be found with this article online at doi:10.1016/j.immuni.2010.11.020.
References
- Allman D, Dalod M, Asselin-Paturel C, Delale T, Robbins SH, Trinchieri G, Biron CA, Kastner P, Chan S. Ikaros is required for plasmacytoid dendritic cell differentiation. Blood. 2006;108:4025–4034. doi: 10.1182/blood-2006-03-007757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andoniou CE, van Dommelen SL, Voigt V, Andrews DM, Brizard G, Asselin-Paturel C, Delale T, Stacey KJ, Trinchieri G, Degli-Esposti MA. Interaction between conventional dendritic cells and natural killer cells is integral to the activation of effective antiviral immunity. Nat Immunol. 2005;6:1011–1019. doi: 10.1038/ni1244. [DOI] [PubMed] [Google Scholar]
- Asselin-Paturel C, Boonstra A, Dalod M, Durand I, Yessaad N, Dezutter-Dambuyant C, Vicari A, O'Garra A, Biron C, Brière F, Trinchieri G. Mouse type I IFN-producing cells are immature APCs with plasmacytoid morphology. Nat Immunol. 2001;2:1144–1150. doi: 10.1038/ni736. [DOI] [PubMed] [Google Scholar]
- Asselin-Paturel C, Brizard G, Pin JJ, Brière F, Trinchieri G. Mouse strain differences in plasmacytoid dendritic cell frequency and function revealed by a novel monoclonal antibody. J Immunol. 2003;171:6466–6477. doi: 10.4049/jimmunol.171.12.6466. [DOI] [PubMed] [Google Scholar]
- Banchereau J, Pascual V. Type I interferon in systemic lupus erythematosus and other autoimmune diseases. Immunity. 2006;25:383–392. doi: 10.1016/j.immuni.2006.08.010. [DOI] [PubMed] [Google Scholar]
- Barbalat R, Lau L, Locksley RM, Barton GM. Toll-like receptor 2 on inflammatory monocytes induces type I interferon in response to viral but not bacterial ligands. Nat Immunol. 2009;10:1200–1207. doi: 10.1038/ni.1792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biron CA, Nguyen KB, Pien GC, Cousens LP, Salazar-Mather TP. Natural killer cells in antiviral defense: Function and regulation by innate cytokines. Annu Rev Immunol. 1999;17:189–220. doi: 10.1146/annurev.immunol.17.1.189. [DOI] [PubMed] [Google Scholar]
- Blasius AL, Beutler B. Intracellular toll-like receptors. Immunity. 2010;32:305–315. doi: 10.1016/j.immuni.2010.03.012. [DOI] [PubMed] [Google Scholar]
- Blasius AL, Cella M, Maldonado J, Takai T, Colonna M. Siglec-H is an IPC-specific receptor that modulates type I IFN secretion through DAP12. Blood. 2006a;107:2474–2476. doi: 10.1182/blood-2005-09-3746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blasius AL, Giurisato E, Cella M, Schreiber RD, Shaw AS, Colonna M. Bone marrow stromal cell antigen 2 is a specific marker of type I IFN-producing cells in the naive mouse, but a promiscuous cell surface antigen following IFN stimulation. J Immunol. 2006b;177:3260–3265. doi: 10.4049/jimmunol.177.5.3260. [DOI] [PubMed] [Google Scholar]
- Borrow P, Bhardwaj N. Innate immune responses in primary HIV-1 infection. Curr Opin HIV AIDS. 2008;3:36–44. doi: 10.1097/COH.0b013e3282f2bce7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cella M, Facchetti F, Lanzavecchia A, Colonna M. Plasmacytoid dendritic cells activated by influenza virus and CD40L drive a potent TH1 polarization. Nat Immunol. 2000;1:305–310. doi: 10.1038/79747. [DOI] [PubMed] [Google Scholar]
- Cervantes-Barragan L, Züst R, Weber F, Spiegel M, Lang KS, Akira S, Thiel V, Ludewig B. Control of coronavirus infection through plasmacytoid dendritic-cell-derived type I interferon. Blood. 2007;109:1131–1137. doi: 10.1182/blood-2006-05-023770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaperot L, Blum A, Manches O, Lui G, Angel J, Molens JP, Plumas J. Virus or TLR agonists induce TRAIL-mediated cytotoxic activity of plasmacytoid dendritic cells. J Immunol. 2006;176:248–255. doi: 10.4049/jimmunol.176.1.248. [DOI] [PubMed] [Google Scholar]
- Cisse B, Caton ML, Lehner M, Maeda T, Scheu S, Locksley R, Holmberg D, Zweier C, den Hollander NS, Kant SG, et al. Transcription factor E2-2 is an essential and specific regulator of plasmacytoid dendritic cell development. Cell. 2008;135:37–48. doi: 10.1016/j.cell.2008.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalod M, Salazar-Mather TP, Malmgaard L, Lewis C, Asselin-Paturel C, Brière F, Trinchieri G, Biron CA. Interferon alpha/beta and interleukin 12 responses to viral infections: Pathways regulating dendritic cell cytokine expression in vivo. J Exp Med. 2002;195:517–528. doi: 10.1084/jem.20011672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalod M, Hamilton T, Salomon R, Salazar-Mather TP, Henry SC, Hamilton JD, Biron CA. Dendritic cell responses to early murine cytomegalovirus infection: Subset functional specialization and differential regulation by interferon alpha/beta. J Exp Med. 2003;197:885–898. doi: 10.1084/jem.20021522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delale T, Paquin A, Asselin-Paturel C, Dalod M, Brizard G, Bates EE, Kastner P, Chan S, Akira S, Vicari A, et al. MyD88-dependent and -independent murine cytomegalovirus sensing for IFN-alpha release and initiation of immune responses in vivo. J Immunol. 2005;175:6723–6732. doi: 10.4049/jimmunol.175.10.6723. [DOI] [PubMed] [Google Scholar]
- Diebold SS, Kaisho T, Hemmi H, Akira S, Reis e Sousa C. Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science. 2004;303:1529–1531. doi: 10.1126/science.1093616. [DOI] [PubMed] [Google Scholar]
- Dokun AO, Kim S, Smith HR, Kang HS, Chu DT, Yokoyama WM. Specific and nonspecific NK cell activation during virus infection. Nat Immunol. 2001;2:951–956. doi: 10.1038/ni714. [DOI] [PubMed] [Google Scholar]
- García-Sastre A, Biron CA. Type 1 interferons and the virus-host relationship: A lesson in détente. Science. 2006;312:879–882. doi: 10.1126/science.1125676. [DOI] [PubMed] [Google Scholar]
- Geurs TL, Zhao YM, Hill EB, French AR. Ly49H engagement compensates for the absence of type I interferon signaling in stimulating NK cell proliferation during murine cytomegalovirus infection. J Immunol. 2009;183:5830–5836. doi: 10.4049/jimmunol.0901520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilliet M, Cao W, Liu YJ. Plasmacytoid dendritic cells: Sensing nucleic acids in viral infection and autoimmune diseases. Nat Rev Immunol. 2008;8:594–606. doi: 10.1038/nri2358. [DOI] [PubMed] [Google Scholar]
- Hardy AW, Graham DR, Shearer GM, Herbeuval JP. HIV turns plasmacytoid dendritic cells (pDC) into TRAIL-expressing killer pDC and down-regulates HIV coreceptors by Toll-like receptor 7-induced IFN-alpha. Proc Natl Acad Sci USA. 2007;104:17453–17458. doi: 10.1073/pnas.0707244104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda K, Ohba Y, Yanai H, Negishi H, Mizutani T, Takaoka A, Taya C, Taniguchi T. Spatiotemporal regulation of MyD88-IRF-7 signalling for robust type-I interferon induction. Nature. 2005a;434:1035–1040. doi: 10.1038/nature03547. [DOI] [PubMed] [Google Scholar]
- Honda K, Yanai H, Negishi H, Asagiri M, Sato M, Mizutani T, Shimada N, Ohba Y, Takaoka A, Yoshida N, Taniguchi T. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature. 2005b;434:772–777. doi: 10.1038/nature03464. [DOI] [PubMed] [Google Scholar]
- Honda K, Yanai H, Takaoka A, Taniguchi T. Regulation of the type I IFN induction: A current view. Int Immunol. 2005c;17:1367–1378. doi: 10.1093/intimm/dxh318. [DOI] [PubMed] [Google Scholar]
- Jung S, Unutmaz D, Wong P, Sano G, De los Santos K, Sparwasser T, Wu S, Vuthoori S, Ko K, Zavala F, et al. In vivo depletion of CD11c+ dendritic cells abrogates priming of CD8+ T cells by exogenous cell-associated antigens. Immunity. 2002;17:211–220. doi: 10.1016/s1074-7613(02)00365-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung A, Kato H, Kumagai Y, Kumar H, Kawai T, Takeuchi O, Akira S. Lymphocytoid choriomeningitis virus activates plasmacytoid dendritic cells and induces a cytotoxic T-cell response via MyD88. J Virol. 2008;82:196–206. doi: 10.1128/JVI.01640-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolumam GA, Thomas S, Thompson LJ, Sprent J, Murali-Krishna K. Type I interferons act directly on CD8 T cells to allow clonal expansion and memory formation in response to viral infection. J Exp Med. 2005;202:637–650. doi: 10.1084/jem.20050821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krmpotic A, Bubic I, Polic B, Lucin P, Jonjic S. Pathogenesis of murine cytomegalovirus infection. Microbes Infect. 2003;5:1263–1277. doi: 10.1016/j.micinf.2003.09.007. [DOI] [PubMed] [Google Scholar]
- Krug A, French AR, Barchet W, Fischer JA, Dzionek A, Pingel JT, Orihuela MM, Akira S, Yokoyama WM, Colonna M. TLR9-dependent recognition of MCMV by IPC and DC generates coordinated cytokine responses that activate antiviral NK cell function. Immunity. 2004a;21:107–119. doi: 10.1016/j.immuni.2004.06.007. [DOI] [PubMed] [Google Scholar]
- Krug A, Luker GD, Barchet W, Leib DA, Akira S, Colonna M. Herpes simplex virus type 1 activates murine natural interferon-producing cells through toll-like receptor 9. Blood. 2004b;103:1433–1437. doi: 10.1182/blood-2003-08-2674. [DOI] [PubMed] [Google Scholar]
- Kumagai Y, Kumar H, Koyama S, Kawai T, Takeuchi O, Akira S. Cutting Edge: TLR-dependent viral recognition along with type I IFN positive feedback signaling masks the requirement of viral replication for IFN-alpha production in plasmacytoid dendritic cells. J Immunol. 2009;182:3960–3964. doi: 10.4049/jimmunol.0804315. [DOI] [PubMed] [Google Scholar]
- Lanier LL. Evolutionary struggles between NK cells and viruses. Nat Rev Immunol. 2008;8:259–268. doi: 10.1038/nri2276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Bon A, Tough DF. Type I interferon as a stimulus for cross-priming. Cytokine Growth Factor Rev. 2008;19:33–40. doi: 10.1016/j.cytogfr.2007.10.007. [DOI] [PubMed] [Google Scholar]
- Marrack P, Kappler J. Control of T cell viability. Annu Rev Immunol. 2004;22:765–787. doi: 10.1146/annurev.immunol.22.012703.104554. [DOI] [PubMed] [Google Scholar]
- Pestka S, Krause CD, Walter MR. Interferons, interferon-like cytokines, and their receptors. Immunol Rev. 2004;202:8–32. doi: 10.1111/j.0105-2896.2004.00204.x. [DOI] [PubMed] [Google Scholar]
- Pichlmair A, Reis e Sousa C. Innate recognition of viruses. Immunity. 2007;27:370–383. doi: 10.1016/j.immuni.2007.08.012. [DOI] [PubMed] [Google Scholar]
- Scheu S, Dresing P, Locksley RM. Visualization of IFNbeta production by plasmacytoid versus conventional dendritic cells under specific stimulation conditions in vivo. Proc Natl Acad Sci USA. 2008;105:20416–20421. doi: 10.1073/pnas.0808537105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smit JJ, Rudd BD, Lukacs NW. Plasmacytoid dendritic cells inhibit pulmonary immunopathology and promote clearance of respiratory syncytial virus. J Exp Med. 2006;203:1153–1159. doi: 10.1084/jem.20052359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sozzani S, Vermi W, Del Prete A, Facchetti F. Trafficking properties of plasmacytoid dendritic cells in health and disease. Trends Immunol. 2010;31:270–277. doi: 10.1016/j.it.2010.05.004. [DOI] [PubMed] [Google Scholar]
- Steinberg C, Eisenächer K, Gross O, Reindl W, Schmitz F, Ruland J, Krug A. The IFN regulatory factor 7-dependent type I IFN response is not essential for early resistance against murine cytomegalovirus infection. Eur J Immunol. 2009;39:1007–1018. doi: 10.1002/eji.200838814. [DOI] [PubMed] [Google Scholar]
- Steinhoff U, Müller U, Schertler A, Hengartner H, Aguet M, Zinkernagel RM. Antiviral protection by vesicular stomatitis virus-specific antibodies in alpha/beta interferon receptor-deficient mice. J Virol. 1995;69:2153–2158. doi: 10.1128/jvi.69.4.2153-2158.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun JC, Beilke JN, Lanier LL. Adaptive immune features of natural killer cells. Nature. 2009;457:557–561. doi: 10.1038/nature07665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szomolanyi-Tsuda E, Liang X, Welsh RM, Kurt-Jones EA, Finberg RW. Role for TLR2 in NK cell-mediated control of murine cytomegalovirus in vivo. J Virol. 2006;80:4286–4291. doi: 10.1128/JVI.80.9.4286-4291.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabeta K, Georgel P, Janssen E, Du X, Hoebe K, Crozat K, Mudd S, Shamel L, Sovath S, Goode J, et al. Toll-like receptors 9 and 3 as essential components of innate immune defense against mouse cytomegalovirus infection. Proc Natl Acad Sci USA. 2004;101:3516–3521. doi: 10.1073/pnas.0400525101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi O, Akira S. Innate immunity to virus infection. Immunol Rev. 2009;227:75–86. doi: 10.1111/j.1600-065X.2008.00737.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson JM, Iwasaki A. Toll-like receptors regulation of viral infection and disease. Adv Drug Deliv Rev. 2008;60:786–794. doi: 10.1016/j.addr.2007.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villadangos JA, Young L. Antigen-presentation properties of plasmacytoid dendritic cells. Immunity. 2008;29:352–361. doi: 10.1016/j.immuni.2008.09.002. [DOI] [PubMed] [Google Scholar]
- Walunas TL, Bruce DS, Dustin L, Loh DY, Bluestone JA. Ly-6C is a marker of memory CD8+ T cells. J Immunol. 1995;155:1873–1883. [PubMed] [Google Scholar]
- Wang H, Peters N, Schwarze J. Plasmacytoid dendritic cells limit viral replication, pulmonary inflammation, and airway hyperresponsiveness in respiratory syncytial virus infection. J Immunol. 2006;177:6263–6270. doi: 10.4049/jimmunol.177.9.6263. [DOI] [PubMed] [Google Scholar]
- Wang JP, Asher DR, Chan M, Kurt-Jones EA, Finberg RW. Cutting edge: Antibody-mediated TLR7-dependent recognition of viral RNA. J Immunol. 2007;178:3363–3367. doi: 10.4049/jimmunol.178.6.3363. [DOI] [PubMed] [Google Scholar]
- Watarai H, Sekine E, Inoue S, Nakagawa R, Kaisho T, Taniguchi M. PDC-TREM, a plasmacytoid dendritic cell-specific receptor, is responsible for augmented production of type I interferon. Proc Natl Acad Sci USA. 2008;105:2993–2998. doi: 10.1073/pnas.0710351105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf AI, Buehler D, Hensley SE, Cavanagh LL, Wherry EJ, Kastner P, Chan S, Weninger W. Plasmacytoid dendritic cells are dispensable during primary influenza virus infection. J Immunol. 2009;182:871–879. doi: 10.4049/jimmunol.182.2.871. [DOI] [PubMed] [Google Scholar]
- Wrammert J, Källberg E, Agace WW, Leanderson T. Ly6C expression differentiates plasma cells from other B cell subsets in mice. Eur J Immunol. 2002;32:97–103. doi: 10.1002/1521-4141(200201)32:1<97::AID-IMMU97>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- Yoneyama H, Matsuno K, Toda E, Nishiwaki T, Matsuo N, Nakano A, Narumi S, Lu B, Gerard C, Ishikawa S, Matsushima K. Plasmacytoid DCs help lymph node DCs to induce anti-HSV CTLs. J Exp Med. 2005;202:425–435. doi: 10.1084/jem.20041961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Raper A, Sugita N, Hingorani R, Salio M, Palmowski MJ, Cerundolo V, Crocker PR. Characterization of Siglec-H as a novel endocytic receptor expressed on murine plasmacytoid dendritic cell precursors. Blood. 2006;107:3600–3608. doi: 10.1182/blood-2005-09-3842. [DOI] [PubMed] [Google Scholar]
- Zucchini N, Bessou G, Traub S, Robbins SH, Uematsu S, Akira S, Alexopoulou L, Dalod M. Cutting edge: Overlapping functions of TLR7 and TLR9 for innate defense against a herpesvirus infection. J Immunol. 2008;180:5799–5803. doi: 10.4049/jimmunol.180.9.5799. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.