

SUMMARY
Histone modifications regulate chromatin-dependent processes, yet the mechanisms by which they contribute to specific outcomes remain unclear. H3K4me3 is a prominent histone mark that is associated with active genes and promotes transcription through interactions with effector proteins that include initiation factor TFIID. We demonstrate that H3K4me3-TAF3 interactions direct global TFIID recruitment to active genes, some of which are p53 targets. Further analyses show that (i) H3K4me3 enhances p53-dependent transcription by stimulating preinitiation complex (PIC) formation; (ii) H3K4me3, through TAF3 interactions, can act either independently or cooperatively with the TATA box to direct PIC formation and transcription; and (iii) H3K4me3-TAF3/TFIID interactions regulate gene-selective functions of p53 in response to genotoxic stress. Our findings indicate a mechanism by which H3K4me3 directs PIC assembly for the rapid induction of specific p53 target genes.
INTRODUCTION
Histone modifications contribute to gene regulation through direct effects on chromatin structure and through the recruitment of effector proteins (Bannister and Kouzarides, 2011; Campos and Reinberg, 2009; Taverna et al., 2007). H3K4me3 is a histone mark associated with promoters and early transcribed regions of active genes (Bernstein et al., 2005; Heintzman et al., 2007). Whereas studies in yeast indicated that H3K4me3 is a consequence of transcription, studies in metazoans have favored more causal roles in transcription (reviewed in Vermeulen and Timmers, 2010). Nonetheless, the functions of H3K4me3 in initiation versus early elongation are not well established. However, in support of a function at initiation, a seminal study has demonstrated an interaction of H3K4me3 with the TAF3 subunit of TFIID (Vermeulen et al., 2007).
TFIID is a general initiation factor (GTF) that, through core promoter recognition, plays a key role in PIC assembly. TFIID is comprised of the TATA-binding polypeptide (TBP) and ~14 TBP-associated factors (TAFs) that interact with transcriptional activators, core promoter elements, and other GTFs (Burley and Roeder, 1996; Juven-Gershon et al., 2008; Thomas and Chiang, 2006). TAF3, while showing tissue-specific functions through interactions with the TBP-related factor TRF3 (Deato et al., 2008; Deato and Tjian, 2007; Hart et al., 2009), is nonetheless an integral component of TFIID (Gangloff et al., 2001). H3K4me3 interactions with TAF3 (Vermeulen et al., 2007) offer a mechanism for TFIID recruitment/stabilization that is complementary to mechanisms involving TBP and TAF interactions with core promoter elements (Juven-Gershon et al., 2008) and have been suggested to be important specifically for TATA-less promoters (Vermeulen et al., 2007). However, this interesting possibility has not been tested and, importantly, there is no corresponding information on either global or gene-specific functions of H3K4me3-TAF3 interactions or on their underlying mechanism of action.
Following a global analysis of H3K4me3-TAF3 interactions, we focus on possible gene-specific effects of these interactions in the regulation of gene expression by the tumor suppressor p53. As a transcriptional activator, p53 strikes a balance between cell cycle arrest and programmed cell death through the differential activation of cognate genes (reviewed in Vousden and Prives, 2009). The kinetics of p53 target gene activation vary, ranging from a rapid induction of cell cycle control genes to a delayed induction of proapoptotic genes (Zhao et al., 2000). However, the mechanisms that regulate the distinct transcription programs remain to be fully elucidated. A potential role for H3K4me3 in p53-mediated gene activation is supported by a DNA damage-enhanced accumulation of H3K4me3 at the GADD45A promoter (An and Roeder, 2004). Also, ubiquitylated H2B, which directly stimulates hSET1-dependent H3K4 di- and trimethylation (Shilatifard, 2006) accumulates at the p21 promoter in response to DNA damage (Kim et al., 2009). However, the downstream mechanisms underlying H3K4me3 function in p53-dependent activation remain to be determined.
Here, we show that H3K4me3, through interactions with TAF3, directs global TFIID recruitment. In subsequent biochemical and cell-based analyses of p53 function, we show that H3K4me3-TAF3/TFIID interactions both enhance p53-dependent transcription by directly stimulating PIC formation and facilitate selective gene activation by p53 in response to DNA damage.
Results
H3K4me3 Facilitates Global TAF3/TFIID Recruitment
To investigate a role for H3K4me3 in the genome wide recruitment of TAF3, we employed ChIP-seq to examine the distribution of TAF3, RNAPII, and H3K4me3 in human colorectal carcinoma HCT116 cells (Bunz et al., 1998). 11,765 stringent TAF3 binding peaks (p<1E-05, FDR<5%) were identified with TAF3 bound at 46% of protein coding gene promoters. Heat maps (Figure 1A) and composite profiles (Figure S1A) demonstrate a significant colocalization of the peak binding sites for TAF3 and RNAPII with the peak sites of H3K4me3 enrichment on active promoters. The vast majority (98%) of the TAF3-occupied regions were marked with significant H3K4me3 levels and, conversely, TAF3 was bound at most (81%) H3K4me3-enriched regions (Figure 1B). There also exists a genome-wide overlap in the length and height of the H3K4me3 and TAF3 peaks (Figure S1B). Thus, our ChIP-seq results establish a strong correlation between TAF3 binding sites and sites of H3K4me3 enrichment across the genome.
Figure 1. H3K4me3 Facilitates Global TAF3/TFIID Recruitment.

(A–B) ChIP-Seq and (C–E) HIT-seq analyses. (A) Heat maps showing the distributions of RNAPII, TAF3, and H3K4me3 within 2 kb of the TSSs of 17,347 Refseq genes, rank-ordered by the size of RNAPII peaks. (B) Venn diagram showing the overlap of genes occupied by TAF3 and enriched for H3K4me3 (p<1E-05, FDR <5%). (C) HIT-seq signals for full-length WT and M880A mutant TAF3 and the corresponding isolated PHD TAF3 proteins at the indicated loci as compared to the distributions of H3K4me3, RNAPII, and expressed RNAs from published datasets (Shen et al., 2012). (D) HIT-seq binding sites with the total integrations including: TAF3 WT (22,844), TAF3 M880A mutant (39,695), TAF3 PHD WT (26,087), and TAF3 PHD M880A mutant (35,363). The number of TAF3 peaks that overlap with H3K4me3 or the TSS (+/− 2 kb) are shown. (E) Line graph showing the distribution of WT and mutant TAF3 binding sites (for both full-length and isolated TAF3 PHD proteins) and the distribution of H3K4me3 (Shen et al., 2012) relative to gene expression levels. ~18,000 mouse Refseq genes were separated into bins of 100 based on expression level. The binding sites associated with the genes in each bin were enumerated and adjusted for the genome size in each bin and the binding activity was normalized and calculated as counts per kb per million tags (CPKM). See also Figure S1.
We next used HIV Integration Targeting (HIT-seq) (Ferris et al., 2010) to determine the direct contribution of H3K4me3 to TAF3/TFIID recruitment. We compared the genome-wide distribution of WT TAF3 to the TAF3 M880A PHD point mutant, which disrupts TAF3 binding to H3K4me3 (van Ingen et al., 2008; Vermeulen et al., 2007). Consistent with our ChIP-seq data, our HIT-seq analyses showed a strong correlation of TAF3 HIT-seq peaks with RNAPII recruitment and active gene expression (Figure 1C, TAF3 WT), a significant overlap of the TAF3 binding sites with the sites of H3K4me3 enrichment (Figure 1C, D), and a strong correlation in the sizes of the H3K4me3 and TAF3 peaks (Figure 1C). In comparison to their WT counterparts, full-length M880A TAF3 and the isolated M880A TAF3 PHD finger showed moderately reduced (31%), and more substantially reduced (94%) binding, respectively, to the TSS region of the promoter (Figure 1D). This difference likely reflects the incorporation of mutant TAF3, but not the mutant TAF3 PHD finger, into TFIID and its passive recruitment through alternate recruitment mechanisms. These data suggest that H3K4me3-TAF3/TFIID interactions broadly contribute to TAF3 promoter occupancy. However, the magnitude of the decrease in TAF3 binding caused by the loss of H3K4me3 interactions varies at individual promoters (Figure 1C), which suggests that some promoters are more dependent on H3K4me3-TAF3/TFIID interactions for TFIID recruitment.
To investigate the differential requirement for H3K4me3-TAF3/TFIID interactions at individual promoters, we used the TAF3 HIT-seq data to compare the binding of WT and mutant TAF3 near the TSSs of genes that were sorted into groups of 100 based on expression level (Figure 1E). The largest HIT-seq peaks for both intact TAF3 and the isolated PHD finger, as well as the greatest differences between the binding of the WT and M880A mutant TAF3 proteins and the corresponding WT and mutant PHD fingers, were identified at highly expressed genes (Figure 1E). Gene ontology enrichment (GO) analysis revealed that the TAF3 binding sites identified by HIT-seq were significantly enriched at genes involved in cell cycle control and transcriptional regulation (data not shown). These findings suggest that the direct interaction of TAF3 with H3K4me3 facilitates global TFIID recruitment/stabilization at the promoters of actively transcribed genes. Given the propensity for PHD-dependent binding of TAF3/TFIID to cell cycle control genes, we focused on p53-dependent transcription.
H3K4me3 Enhances PIC Formation and p53/p300-Dependent Activation
We employed a cell free transcription system (Figure 2A; Figure S2B; An and Roeder, 2004) to determine how H3K4me3 and H3K4me3-TAF3/TFIID interactions affect transcription initiation. Chromatin was assembled by recombinant factors (Figure S2A) using a pG5ML template (Figure S2C) and reconstituted histone octamers containing either unmodified H3 or the H3K4me3 analogue H3Kc4me3 (Figure S2D). Micrococcal nuclease (MNase) digestion showed that both templates are chromatinized equivalently (Figure 2B). Robust transcription from the unmodified template was dependent upon p53 and p300 (Figure 2C, lanes 1–4). In contrast, the H3Kc4me3-modified template showed a significant level of p53-dependent transcription in the absence of p300 (lane 6), likely due to endogenous cofactors in the nuclear extract (NE). This activity was further enhanced (~7-fold) by ectopic p300 to yield an absolute level of transcription ~4-fold higher than observed with the unmodified template (lane 8 versus 4), and this increase was observed over a range of chromatin assembly conditions (Figure S2E). Importantly, H3Kc4me3 contributed more significantly than H3Kc4me1 and H3Kc4me2 to the stimulation of p53-dependent transcription (Figure S2F). These results demonstrate a direct effect of H3K4me3 on p53-dependent transcription.
Figure 2. H3K4me3 Enhances PIC Formation and p53-Dependent Transcription.

(A) Schematic of the in vitro transcription assay. (B) MNase digestion showing similar nucleosome spacing for chromatin assembled with unmodified H3 and H3Kc4me3-modified octamers. (C) NE-based transcription assay with deletions and additions as indicated. Relative transcription levels were quantitated by a phosphoimager. After background subtraction, all values were normalized to lane 4. The bar graph represents the average of two independent experiments and the error bars denote the standard deviation. ND, nondetectable. (D) Schematic of the in vitro ChIP assays. (E) Schematic of the pG5ML template indicating the amplicons used for qRT-PCR. (F) ChIP on chromatin assembled with unmodified H3 or H3Kc4me3 was performed with IgG and the indicated antibodies. H3K4me3 levels are relative to H3 levels. An average of two independent ChIP experiments is shown with error bars representing the standard deviation. (G and H) Kinetic analyses of PIC formation were performed +/− sarkosyl. Relative transcription levels were quantitated by a phosphoimager. After background subtraction, all values were normalized to lane 1. The bar graph represents the average of two independent experiments. Error bars denote the standard deviation. See also Figure S2.
To determine if H3K4me3 enhances transcription by stimulating PIC formation, in vitro ChIP was performed using a purified system (Figure 2D; Figure S2G, H). Chromatin was incubated with or without p53, acetylated by p300, digested with MNase, and immunoprecipitated with antibodies against p53, H3, H3K4me3, TAF3, RNAPII, and TBP. The precipitated DNA was analyzed by quantitative RT-PCR (qRT-PCR) using primer sets (Figure 2E) that scan both a promoter region (A) containing p53 binding sites and a distal vector region (B). On the unmodified template, there was selective binding of p53 and selective p53-enhanced binding of TBP and TAF3 (TFIID) and RNAPII to the promoter region (Figure 2F). On the H3Kc4me3 template, we observed (i) comparable p53 binding to the promoter region, (ii) a broad distribution of H3Kc4me3 across the template (A and B), and (iii) a p53-dependent binding of TFIID and RNAPII to the promoter region. In the presence of p53, the H3Kc4me3 template showed a significant increase in promoter-assembled TFIID and RNAPII relative to the unmodified template (Figure 2F). This H3Kc4me3-dependent increase was detected exclusively at the promoter despite the uniform distribution of H3Kc4me3 -- indicative of the importance of the TATA box and promoter-bound p53 in contributing to TFIID stabilization at the promoter. The lack of an effect of H3Kc4me3 on p53 binding also indicates that enhanced TFIID/RNAPII recruitment is not due to an H3Kc4me3-mediated stabilization of p53. Notably, TFIID showed comparable binding to H3 peptides with natural K4me3 versus the Kc4me3 analog (Figure S2I). Together, these results establish a role for H3K4me3 in directing enhanced PIC formation/stabilization.
To further investigate the impact of H3K4me3 on the rate of PIC formation and initiation, kinetic transcription analyses were performed with addition of sarkosyl at various times during PIC formation (Figure 2G). While chromatin assembled with H3Kc4me3 supported the assembly of sarkosyl-resistant PICs as early as 30 min (Figure 2H, lane 9), unmodified chromatin was significantly less efficient in assembling a functional PIC even after 2 hours (lanes 1–5) and despite the fact that this template shows the same sensitivity to sarkosyl (Figure S2J) and directs efficient transcription in the absence of sarkosyl (Figure 2H, lane 6). Because sarkosyl restricts transcription to a single round, a comparison of transcription with and without sarkosyl reveals that once formed, PICs bound to chromatin assembled with H3Kc4me3 or unmodified H3 undergo almost the same number of rounds of transcription (~7, lanes 12 versus 11, versus ~5, lanes 6 versus 5, respectively). These data are consistent with the in vitro ChIP results that show enhanced TFIID/RNAPII recruitment at the promoter of chromatin templates assembled with H3Kc4me3 versus unmodified H3 (Figure 2F). Also consistent, B2 RNAs (Espinoza et al., 2004), had little to no effect on transcription from chromatin assembled with H3Kc4me3 as early as 30 minutes following NTP addition (Figure S2K, lane 9). By comparison, transcription from unmodified chromatin was inhibited in the presence of B2 RNA at all time points (Figure S2K, lanes 1–5). Taken together these results establish a direct link between an increase in the rate of PIC formation and the stimulation of transcription by H3K4me3.
H3K4me3 Facilitates Primary TFIID Recruitment and Cooperates with the TATA Box to Enhance PIC Assembly and Initiation
To elucidate the significance of H3K4me3 with respect to TATA-TBP/TFIID interactions, we assessed p53-dependent transcription on chromatin assembled with unmodified H3 or H3Kc4me3 and containing a wild-type (TATAA) or a mutated TATA box (TATGG). Transcription from the unmodified template was reduced 2.5-fold by the TATA mutation (Figure 3A lane 4 versus 2). The enhanced transcription from H3Kc4me3-modified templates also was reduced 2.5-fold by the mutant TATA (Figure 3A, lanes 8 versus 6), although the residual signal was nonetheless comparable to the signal from chromatin assembled with unmodified H3 and a wild-type TATA (Figure 3A, lane 8 versus 2). This indicates that H3K4me3 can partially compensate for the mutant TATA in mediating transcription. Chromatin containing both H3Kc4me3 and a wild-type TATA supported a significantly higher transcription level (Figure 3A, lane 6) than did chromatin that contained only H3Kc4me3 (Figure 3A, lane 8) or a wild-type TATA (Figure 3A, lane 2), which strongly suggests that the TATA box and H3K4me3 cooperate to enhance transcription.
Figure 3. H3K4me3 Functions Independently and Cooperatively with the TATA Box to Mediate p53-Dependent Transcription.
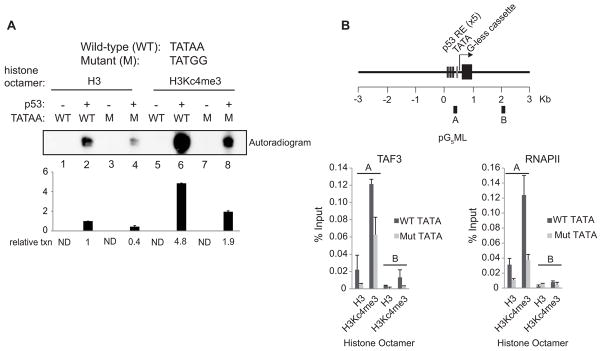
(A) NE-based transcription assays with deletions and additions as indicated. Chromatin templates contained either a wild-type or mutant TATA box and were assembled with H3 or H3Kc4me3-modified octamers. Relative transcription levels were quantitated by a phosphoimager. After background subtraction, all values were normalized to lane 2. The bar graph represents the average of two independent experiments. Error bars denote the standard deviation. ND, nondetectable. (B) In vitro ChIP on the chromatin templates described in (A). The amplicons used for real-time PCR are shown in the schematic. An average of two independent ChIP experiments that are representative of at least four is shown with error bars denoting the standard deviation.
To further investigate the role of H3K4me3 with respect to TFIID/TBP-TATA interactions, in vitro ChIP was performed. Mutation of the TATA box caused a significant decrease in TAF3/TFIID and RNAPII binding to both unmodified and H3Kc4me3 templates (Figure 3B). However, the levels of TAF3/TFIID and RNAPII bound to H3Kc4me3-modified chromatin containing a mutant TATA were equal to, or greater than, the levels bound to unmodified chromatin with a wild-type TATA (Figure 3B), showing that H3K4me3 can compensate for the mutant TATA in facilitating GTF/RNAPII recruitment. In addition, the amount of TFIID/RNAPII recruited to chromatin containing both H3Kc4me3 and a wild-type TATA was higher than that recruited to chromatin containing either H3Kc4me3 or a wild-type TATA (Figure 3B). These results parallel the transcription data (Figure 3A) and show that H3K4me3 can support a significant level of PIC assembly and transcription initiation in the presence of a mutant TATA box and that H3K4me3 can cooperate with TATA to enhance PIC assembly and transcription.
TAF3/TFIID Binding to H3K4me3 Facilitates PIC Formation and Function on Chromatin Containing a Mutant TATA
To determine if TAF3/TFIID-H3K4me3 interactions mediate the effects of H3K4me3 on p53- dependent transcription, we purified TAF3/TFIID complexes (Figure 4A and S3A, B) containing either wild-type or M880A mutant TAF3. As predicted, the WT, but not the mutant, TAF3/TFIID complex binds selectively to K4 trimethylated H3 peptides relative to unmodified H3 peptides (Figure 4B). To assess the transcription activities of the TAF3/TFIID complexes, we immunodepleted endogenous TFIID from HeLa NE (ΔIID, Figure S3C). ΔIID NE was unable to support transcription from naked DNA (Figure S3E, F; lanes 3–4) or chromatin (Figure 4C, D; lanes 3–4), whereas mock-depleted NE supported transcription from naked DNA (data not shown) and chromatin (Figure S3D) to levels comparable to control NE. To examine the rescue potential of the TAF3/TFIID complexes, equivalent amounts of each complex were examined on chromatin containing a wild-type TATA. Both the WT and mutant TAF3/TFIID complexes are equally efficient in restoring transcription to levels comparable to control NE, both on naked DNA (Figure S3E, lanes 6–9 and 11–14 versus lane 2) and on chromatin assembled with unmodified H3 or H3Kc4me3 (Figure 4C, lanes 6–9 and 11–14 versus lane 2).
Figure 4. Functional Analysis of the TAF3/TFIID Complexes.

(A) SDS/PAGE-silver stain analysis of the purified TAF3/TFIID complexes. (B) Wild-type and mutant TAF3/TFIID complexes were analyzed for binding to H3Kc4-methylated peptides and bound proteins were analyzed by immunoblots with indicated antibodies. In vitro transcription assays performed with ΔIID NE on chromatin templates assembled with H3 or H3Kc4me3-modified octamers and containing a wild-type (WT) TATA (C) or a mutant (M) TATA box (D). These assays were performed in the presence or absence of p53 and the wild-type or mutant TAF3/TFIID complexes. The amount of TFIID added in lanes 8 and 13 is comparable to the amount of endogenous TFIID in the NE in lane 2. Relative transcription levels were quantitated by a phosphoimager. After background subtraction, all values in (C) and (D) were normalized to lane 2, the upper panel of (C). Note that the assays in (C) and (D) were performed and analyzed in parallel such that the absolute levels of transcription are directly comparable. The bar graphs represent the average of two independent experiments. Error bars denote the standard deviation. ND, nondetectable. In vitro ChIP analysis on chromatin templates assembled with H3 or H3Kc4me3-modified octamers and containing a wild-type (E) or mutant (F) TATA box. The amplicons used for real-time PCR are shown in the schematic (Figure 2E, 3B). An average of two independent ChIP experiments is shown with error bars denoting the standard deviation. See also Figure S3.
Next we examined the rescue potential of the TAF3/TFIID complexes from chromatin that contains a mutant TATA. The WT and mutant TAF3/TFIID complexes showed comparable abilities to restore transcription to TFIID-depleted extracts, and were as active as the control NE, both on DNA (Figure S3F, lanes 6–9 and 11–14 versus lane 2) and on chromatin assembled with unmodified H3 (Figure 4D upper panel, lanes 6–9 and 11–14 versus lane 2). However, mutant TAF3/TFIID was significantly (~3-fold) less efficient than WT TAF3/TFIID in rescuing transcription from chromatin containing a mutant TATA and assembled with H3Kc4me3 (Figure 4D lower panel, lanes 11–14 versus 6–9 and 2).
In vitro ChIP was performed to examine the requirement for H3K4me3-TAF3/TFIID interactions in PIC formation. On chromatin with a wild-type TATA, the WT and mutant TAF3/TFIID complexes were equivalent with respect to TAF3/TFIID and RNAPII recruitment to promoters (A) both on unmodified H3 chromatin and on H3Kc4me3 chromatin, although the overall levels of recruitment were higher on the H3Kc4me3 template (Figure 4E). On chromatin templates with a mutant TATA, the WT TAF3/TFIID complex supported low levels of TAF3 and RNAPII recruitment on the unmodified H3 template and higher levels of recruitment on the H3Kc4me3 template (Figure 4F). In contrast, mutant TAF3/TFIID supported significantly reduced levels of TAF3 and RNAPII recruitment to the H3Kc4me3 template compared to the WT TAF3/TFIID complex (Figure 4F). These findings are consistent with the transcription data (Figure 4C, D) and show that H3K4me3-TAF3 interactions are important for GTF/RNAPII recruitment and in promoting transcription from templates containing a mutant TATA.
TAF3 is an Essential p53 Coactivator
We examined the significance of TAF3/TFIID and H3K4me3-TAF3/TFIID interactions in regulating the expression of endogenous p53 target genes using RNAi-mediated knockdown of TAF3 in HCT116 cells. Relative to a non-targeting control siRNA (control) (Figure 5A, left), two pairs of oligonucleotides directed against TAF3 mRNA (TAF3-1 and TAF3-2) significantly reduced both the uninduced and doxorubicin-induced level of TAF3. [The doxorubicin-induced increase in TAF3 RNA levels is specific since the RNA levels of other TAF subunits were not affected (data not shown). While doxorubicin induced TAF3 RNA levels, no change was apparent at the protein level]. Both siRNAs also significantly decreased TAF3 protein levels (Figure 5A, right) without affecting the levels of other TFIID subunits (Figure S4A) or the integrity of the TFIID complex (Figure S4B). TAF3-1 (hereafter TAF3) siRNA was selected for further studies. Following siRNA transfection, cells were treated with doxorubicin and the induction of p53 target gene expression was validated by qRT-PCR (Figure 6A) and immunoblot (Figure S4C).
Figure 5. TAF3 Functions as an Essential Coactivator for p53.

(A) HCT116 cells were transfected with non-targeting control (control) or TAF3 siRNAs (TAF3-1 and TAF3-2) and cultured with doxorubicin for 0 or 24 hr. (Left) qRT-PCR and (right) immunoblot revealed significant reduction of TAF3 compared to transfection with the control siRNA. The expression levels determined after doxorubicin treatment are relative to the levels before doxorubicin. Error bars denote the standard deviation from duplicate reactions by real time PCR. (B) Microarray analysis of genes affected by TAF3 depletion in HCT116 cells. Genes were sorted before (left-blue, No Doxo) or after (right-red, Doxo 24 h) doxorubicin treatment. Genes induced more than 2-fold after doxorubicin treatment in the control cells are represented (right-red). The numbers of affected gene probes (out of a total of 22,000) are shown with the indicated fold of down-or up-regulation due to TAF3 siRNA-mediated depletion. (C) GO analysis of the genes significantly affected by TAF3 depletion following 24 hours of doxorubicin treatment. The most highly represented categories are shown with ontology terms and the percentage of genes in each category. (D) QRT-PCR analysis of p53 target genes in control and TAF3-depleted HCT116 cells both before (Doxo 0 h) and after (24 h) doxorubicin treatment. The expression levels determined after doxorubicin are relative to the levels before doxorubicin. Error bars denote the standard deviation from duplicate reactions by real time PCR. See also Figure S4.
Figure 6. TAF3 Binding is Required for RNAPII Recruitment and Correlates with H3K4me3 Accumulation at the p21 and BTG2, but not the DR5 and FAS, Promoters.

(A) HCT116 (p53+/+ and p53−/−) cells were treated with doxorubicin (for 0, 2, 4, 8, 12, and 24 h) and qRT-PCR was performed to measure the rapid p53-dependent induction of the cell cycle genes p21 and BTG2 and the delayed induction of the proapoptotic genes DR5 and FAS. The expression levels after doxorubicin treatment are provided relative to the levels before doxorubicin. Error bars denote the standard deviation from duplicate reactions by real time PCR. (B, D) ChIP analyses with the indicated antibodies were performed using HCT116 cells that were treated with doxorubicin for 0, 4, 8, and 24 h. The amplicons used for real-time PCR are shown in the schematic of the p53 loci. ChIP for H3K4me3 was normalized to H3. An average of two independent ChIP experiments that are representative of at least three is shown with error bars denoting the standard deviation. (C) HCT116 cells were treated with non-targeting (control) or TAF3 siRNA. Following 8 h of doxorubicin, cell lysates were analyzed by ChIP with the indicated antibodies. An average of two independent ChIP experiments that are representative of at least three is shown with error bars denoting the standard deviation. See also Figure S5.
Microarray analysis comparing the transcriptome of control and TAF3 depleted cells (+/− doxorubicin) showed that TAF3 depletion significantly alters (≥ 2-fold, p<0.05) the expression of a small subset (1,234 out of 22,000) of genes, which suggests that TAF3 selectively regulates transcription. As shown in Figure 5B (red), the vast majority of the genes (n=1115, 90%) affected by TAF3 depletion are genes that are significantly induced (≥ 2-fold) by doxorubicin in the control cells. Conversely, only a small percentage (n=119, 9%) of genes were affected by TAF3 depletion in the absence of doxorubicin (Figure 5B, blue). Analysis of the doxorubicin-induced genes revealed 271 (22%) TAF3-repressed genes and 844 (68%) TAF3-activated genes (Figure 5B, red). Several key regulators of the DNA damage response were specifically identified as TAF3-activated genes, which is consistent with a role for TAF3 in mediating p53 transactivation. Overall our microarray data indicate that TAF3 is involved primarily in gene induction and, to a much lesser extent, in basal gene expression.
GO analysis of the TAF3-regulated genes did not reveal any significant functional clusters for the small group of genes in the absence of doxorubicin; however, several significant (p-value <0.01) categories were identified in the larger set of doxorubicin-induced genes. Of the 271 TAF3-repressed genes, only 36 (13%) showed significant enrichment for GO categories, which included ubiquitin ligase activity, protein biosynthesis, and steroid biosynthesis (Figure 5C, left). GO annotations related to transcription and DNA damage response pathways were not significantly identified in the TAF3-repressed gene profile; however, these annotations were among the most prominent in the TAF3-activated data set. Specifically, 817 (97%) of the 844 TAF3-activated genes were classified into ten significant categories, the most prominent of which are related to cell cycle control and transcription (including DNA metabolic processes, nucleotide binding, and transcriptional regulation) (Figure 5C, right). Although several proapoptotic genes were identified in our microarray data of TAF3-activated genes, these genes were not recognized as a functional cluster and are significantly underrepresented relative to the highly significant cell cycle category. Together, the microarray and GO data suggest that the repression and activation mechanisms that require TAF3, under doxorubicin-induced conditions, are functionally unrelated, as reflected by the lack of overlap in the functional categories between the two data sets. By comparison, the high level of significance and functional coherence of GO terms associated with the TAF3-activated genes suggests that the regulation of p53 target gene expression by TAF3 is largely mediated through DNA damage-induced activation events and not through negative gene regulation.
Consistent with our microarray results, qRT-PCR analysis of representative genes revealed that TAF3 depletion had little to no effect on uninduced gene expression but caused 2.2- to 6-fold decreases in the doxorubicin-induced mRNA levels of the cell cycle-regulated genes p21, BTG2, CDK5RAP1, and IGF1R, the auto-regulatory gene MDM2, and the proapoptotic gene PUMA (Figure 5D) -- all of which display early activation kinetics in response to p53 (Yu et al., 2001; Zhao et al., 2000). In contrast, delayed expression of the proapoptotic genes DR5, FAS, Apaf-1, sertad1, and NoxA, was not significantly affected by TAF3 depletion (Figure 5D). Consistent with this data, an immunoblot confirmed a decrease in the protein levels of p21 and BTG2, but not DR5 and FAS, as a result of TAF3 depletion (Figure S4D). The gene-selective effects of TAF3 are reproducible in osteosarcoma U2OS cells (Figure S4E), are not due to altered protein levels of p53 (Figure S4F), and are dependent on p53 since these effects were not identified in the p53-knockout HCT116 cell line (data not shown). Moreover, core promoter sequence analysis of the TAF3-dependent genes showed that TAF3 is required for the regulation of both TATA-less and TATA-containing genes (Figure 5D). Together, our global expression profiles parallel our gene-specific analysis, demonstrating that TAF3 selectively regulates rapidly induced p53 target genes.
Binding of TFIID/TAF3 and RNAPII Correlates with H3K4me3 Enrichment at the Promoters of Rapidly Induced p53 Target Genes
To assess a direct role for TAF3 in the regulation of selective p53 target genes, ChIP-qPCR was performed in HCT116 cells (+/− doxorubicin). Immunoprecipitated DNA was analyzed with primer sets (Figure 6B) specific to the promoter region (A) and a distal control region (B) of the early response cell cycle genes p21 and BTG2 and of the delayed response proapoptotic genes DR5 and FAS. ChIP-qPCR revealed pre-associated TFIID and RNAPII before doxorubicin treatment, and substantial doxorubicin-induced increases in these factors at the promoters but not at the downstream regions of all four genes (Figure 6B and Figure S5). During DNA damage-induced activation, the levels of TAF3 recruited to the promoters of p21 and BTG2 increased rapidly and peaked at 4 hr, which correlates with the TBP and RNAPII binding profiles. By comparison, TAF3, TBP, and RNAPII recruitment at the DR5 and FAS promoters was significantly more delayed, with peak binding at 8 and 24 hours (Figure 6B and Figure S5). In addition, the peak levels of TFIID and RNAPII were comparable at the p21 and BTG2 promoters but significantly (3- to 7-fold, note the different scales) higher than the peak levels at the DR5 and FAS promoters. Thus, the temporal binding profiles of TFIID and RNAPII correlate strongly with the rapid versus delayed activation kinetics of the cell cycle and proapoptotic genes, respectively (Figure 6A).
To determine if TAF3 contributes to the DNA damage-induced recruitment of RNAPII, ChIP was performed in doxorubicin-treated HCT116 cells that had been transfected with control or TAF3 siRNA. TAF3 binding in the TAF3-depleted cells is reduced significantly and comparably (approximately 65–76%), at the promoter regions of p21, BTG2, DR5, and FAS (Figure 6C). RNAPII binding to the p21 and BTG2 promoters was significantly reduced (by 80% and 50%, respectively) in TAF3-depleted versus control cells; however, the levels of RNAPII remained largely unaffected at the promoters of DR5 and FAS (Figure 6C). TBP recruitment to the promoters of all four genes was largely unaffected by TAF3 knockdown (Figure 6C). These results validate the specificity of our TAF3 antibody in ChIP and demonstrate that TAF3 directly impacts RNAPII recruitment to effect the activation of rapidly induced p53 target genes.
Having demonstrated that H3K4me3 facilitates global TFIID recruitment to promoters, particularly of cell cycle control genes, and that H3K4me3-TAF3/TFIID interactions regulate p53-dependent activation in vitro, we next investigated whether H3K4me3 enrichment overlaps with TAF3/TFIID recruitment at the promoters of select p53 target genes. ChIP revealed pre-associated H3K4me3 prior to doxorubicin treatment and a substantial enhancement in H3K4me3 in response to DNA damage (Figure 6D), which parallels the temporal changes in TAF3, TBP, and RNAPII binding at the promoter regions of p21 and BTG2 (Figure 6B and S5). In contrast, there was no parallel enrichment of H3K4me3 and TAF3/TBP (TFIID) at the promoters of DR5 and FAS. Specifically, H3K4me3 rapidly increased at the DR5 promoter in response to DNA damage (Figure 6D), while there was little to no change in H3K4me3 before or after doxorubicin treatment at the FAS promoter (Figure 6D). The doxorubicin-induced levels of H3K4me3 are significantly higher at the p21 and BTG2 versus the DR5 and FAS promoters (Figure 6D), which is consistent with our TAF3, TBP, and RNAPII ChIP results (Figure 6B and S5). Our ChIP studies thus demonstrate a role for TAF3 in the regulation of selective p53 target genes. Furthermore, the strong overlap in TAF3 binding and H3K4me3 enrichment at the TAF3-dependent genes suggests that H3K4me3-TAF3/TFIID interactions are important in regulating the expression of select p53 target genes.
The Binding of TAF3 to H3K4me3 Regulates the Expression of Select p53 Targets
To investigate a direct role for H3K4me3-TAF3/TFIID interactions in the expression of select p53 target genes, we examined (i) the effects of siRNA-mediated knockdown of WDR5, which reduces global levels of H3K4me3 (Dou et al., 2006; Steward et al., 2006) and (ii) the relative ability of siRNA-resistant wild-type TAF3 and the M880A TAF3 mutant to rescue p53 target gene expression in TAF3 siRNA-transfected cells. qRT-PCR revealed that WDR5 depletion significantly reduced H3K4me3 (Figure 7A) and the doxorubicin-induced levels of p21 and BTG2 (3-and 8-fold, respectively), while having minimal (2- and 1.4-fold, respectively) effects on the doxorubicin-induced levels of DR5 and FAS (Figure 7B). H3K4me3 also appears to repress uninduced p53 target gene expression, as revealed by the 2- to 3-fold increase in basal levels of p21, BTG2, DR5, and FAS (Figure 7B, Doxo 0h) following WDR5 knockdown. However, this effect is TAF3 independent since TAF3 depletion had little to no effect on uninduced p53 target gene expression (Figure 5D).
Figure 7. TAF3 Interacts with H3K4me3 to Facilitate the Rapid Induction of Selective p53 Target Genes.

(A) HCT116 cells were transfected with non-targeting control (control) or WDR5 siRNA and treated with doxorubicin for 0 or 24 h. Immunoblot analysis of cell lysates with the indicated antibodies. (B) QRT-PCR analysis of p53 target genes in HCT116 cells that were treated as in (A). The expression levels determined after doxorubicin are relative to the levels before doxorubicin. Error bars denote the standard deviation from duplicate reactions by real time PCR. (C, D) ChIP analysis of HCT116 cells transfected with control or WDR5 siRNA and treated with doxorubicin for 0 or 8 h. The levels of H3K4me3 are relative to H3 levels. Schematic showing the amplicons used for qRT-PCR. An average of two independent ChIP experiments that are representative of at least three is shown with error bars denoting the standard deviation. (E) (left) QRT-PCR and (right) immunoblot analysis of TAF3 in HCT116 cells that were transfected with control or TAF3 siRNA together with empty vector control or siRNA-resistant plasmids expressing wild-type or M880A mutant TAF3. The expression levels determined after doxorubicin are relative to the levels before doxorubicin and error bars denote the standard deviation from duplicate reactions by real time PCR. (F) QRT-PCR analysis of p53 target gene expression in HCT116 cells treated as described in (E). Error bars denote the standard deviation from duplicate reactions by real time PCR.
Under uninduced conditions, WDR5 knockdown resulted in an ~80–90% loss of H3K4me3 at the p21, BTG2, and DR5 promoters and a slightly lower, but still significant (~55%), decrease at the FAS promoter (Figure 7C). This decrease in H3K4me3 resulted in a partial (40 and 55%, respectively) reduction of TAF3 binding at the BTG2 and p21 promoters but had little to no effect on TAF3 binding at the FAS promoter and led to a partial (~50%) increase of TAF3 binding at the DR5 promoter (Figure 7D). These modest effects of H3K4me3 depletion on TAF3 recruitment are consistent with a role for H3K4me3 in the regulation of uninduced gene expression that is independent of TAF3 (Figure 5D). In doxorubicin-treated cells, WDR5 knockdown caused substantial losses of the doxorubicin-induced increase in TAF3 at the p21 and BTG2 promoters (70 and 80%, respectively) (Figure 7D), while having a much smaller effect on TAF3 binding at the DR5 and FAS promoters (20% and little to no change, respectively) (Figure 7D). The significant effect of H3K4me3 on TAF3 recruitment in response to DNA damage (Figure 7C, D) is in agreement with the expression data implicating both TAF3 (Figure 5D) and H3K4me3 (Figure 7B) in the doxorubicin-induced activation of both p21 and BTG2, but not DR5 and FAS. These results demonstrate that H3K4me3 is required for TFIID recruitment to cell cycle promoters in response to DNA damage.
To examine the importance of TAF3-H3K4me3 interactions, TAF3 siRNA-resistant constructs expressing wild-type or mutant M880A TAF3 were co-transfected with TAF3 siRNA into HCT116 cells. Both TAF3 expression constructs equivalently reconstituted TAF3 mRNA (Figure 7E left) and protein (Figure 7E right) levels. Notably, as shown in Figure 7F, wild-type TAF3 fully rescued the expression of the TAF3-dependent genes (p21, MDM2, BTG2, PUMA, IGF1R and CDK5RAP1), while the TAF3 PHD point mutant M880A was significantly (1.8- to 3.3-fold) less efficient. These rescue experiments confirm the specificity of our TAF3 siRNA experiments (Figure 5D) and, taken together with our in vitro analyses, unequivocally demonstrate a direct requirement for the H3K4me3-TAF3/TFIID effector pathway in the regulation of rapidly induced p53 target genes.
DISCUSSION
H3K4me3 Modulates Global TFIID Recruitment Through Direct Interactions with TAF3
We demonstrated that H3K4me3-TAF3 interactions direct global TAF3 recruitment by showing a strong genome-wide overlap of TAF3 and H3K4me3 and by a significant decrease in the recruitment of the TAF3 M880A PHD mutant relative to WT TAF3. However, while TAF3 recruitment, through H3K4me3 interactions, to the promoters of constitutively expressed genes was widespread, TAF3 depletion had little effect on constitutive gene expression. Thus, despite the specific association of TAF3 with constitutive gene promoters there was little to no change in constitutive gene expression following TAF3 knockdown. This finding is consistent with the results of a recent study of gene expression following TAF3 knockdown in ES cells (Liu et al., 2011). The results, in both cases, could reflect sufficiency of the low levels of TAF3 that remain after siRNA-mediated knockdown for steady state gene expression levels. Another possibility is that the high levels of pre-associated TAF3 prime corresponding genes and influence their expression only under certain conditions. In this regard, TAF3 could function by poising PICs for a rapid response to stress. This possibility is consistent with a report of PICs at p53 target genes prior to DNA damage (Espinosa et al., 2003) and is also supported by our finding that H3K4me3-TAF3/TFIID interactions contribute to TAF3 recruitment at the promoters of constitutive genes whose GO categories parallel the categories of TAF3-dependent genes following doxorubicin treatment. Thus, in addition to the significant genome-wide impact of H3K4me3-TAF3 interactions in mediating TFIID recruitment, our genome-wide and gene ontology analyses underscore the significance of H3K4me3-TAF3/TFIID interactions in the regulation of TFIID recruitment to p53-regulated cell cycle control genes.
H3K4me3 Directly Enhances TFIID and RNAPII Recruitment and Initiation
Given the diverse factors that interact with H3K4me3 (reviewed in Chi et al., 2010; Vermeulen and Timmers, 2010), a number of functions for H3K4me3 in transcription or transcription-coupled processes may be envisioned. Roles for H3K4me3 in transcription initiation are supported by the direct interactions between H3K4me3 methyltransferases and transcriptional activators (reviewed in Ruthenburg et al., 2007a; Vermeulen and Timmers, 2010), the contribution of H3K4me3 to the recruitment of the chromatin remodeler CHD1 to the PIC (Lin et al., 2011), and the direct interaction of H3K4me3 with TAF3 (Vermeulen et al., 2007). Our studies provide additional insights into a role for H3K4me3-TAF3 interactions in initiation. First, genome level ChIP experiments revealed the significance of H3K4me3-TAF3/TFIID interactions in global TFIID recruitment. Second, complementation assays in doxorubicin-induced cells expressing WT and mutant TAF3 highlighted the importance of H3K4me3-TAF3 interactions for the activation of a select group of p53 target genes. Third, in vitro ChIP assays demonstrated that TAF3-H3Kc4me3 interactions enhance TFIID recruitment to a model promoter and kinetic analyses showed that H3Kc4me3 increased the rate of PIC formation rather than the rounds of transcription. In addition, H3K4me3-TAF3/TFIID interactions were sufficient to nucleate PIC assembly and potentiate transcriptional activation from a TATA-mutated template that was deficient in PIC assembly and activation in the absence of H3K4me3. Overall, our results provide strong evidence for the functional significance of H3K4me3 and for H3K4me3-TAF3/TFIID interactions in stimulating initiation through an effect on TFIID recruitment and PIC formation. However, our results do not rule out the possibility that H3K4me3, through interactions with other proteins, may also act at later steps of the transcription cycle.
H3K4me3 Regulation of TFIID Recruitment to TATA-less and TATA-containing Promoters
The demonstration that only about 10% of mammalian promoters contain a classical TATA box suggested that other core promoter elements may direct transcription initiation (Carninci et al., 2006) and, indeed, initiator (INR) and downstream core promoter elements (e.g., DPE) were shown to facilitate TFIID recruitment to TATA-less promoters (reviewed in Juven-Gershon et al., 2008). We extend these findings by revealing that H3K4me3 can act in the absence of a functional TATA box to facilitate PIC formation and activation in vitro. The specificity of this H3K4me3 function, through an interaction with TAF3/TFIID, was demonstrated by showing that the wild-type, but not the M880A mutant, TAF3/TFIID complex facilitates TFIID and RNAPII recruitment and rescues transcription initiation from a TATA-mutated, H3Kc4me3-containing chromatin template.
Consistent with these in vitro results, TAF3 and H3K4me3 were shown to be required for the DNA damage-induced activation of endogenous p53 target genes BTG2, PUMA, and CDK5RAP1, all of which have TATA-less promoters. In addition, this finding supports our observation that H3K4me3 is required for TAF3/TFIID, and subsequently RNAPII recruitment to these TATA-less p53 target genes. siRNA-based TAF3 knockdown and complementation experiments further revealed that the DNA damage-induced expression of these TATA-less genes is rescued by wild-type but not M880A mutant TAF3. Collectively, our in vitro and in vivo findings define a mechanism by which H3K4me3-TAF3/TFIID interactions, perhaps acting in concert with other TFIID-core promoter interactions, are sufficient to direct TFIID recruitment and subsequent transcription of both a TATA-mutated in vitro chromatin template and endogenous p53 target genes containing TATA-less promoters.
Our cell-based assays of doxorubicin-induced genes also showed a cooperativity between H3K4me3 and the TATA box that enhances TFIID and RNAPII recruitment/stabilization and transcription. However, while siRNA rescue experiments demonstrated a direct contribution of H3K4me3-TAF3/TFIID interactions to the activation of TATA-containing p53 target genes in cells, we were unable to detect a significant contribution to p53-dependent transcription from a model TATA- and INR-containing promoter in vitro. This inconsistency between the in vivo and in vitro results regarding the requirement for H3K4me3-TAF3/TFIID interactions likely reflects the use, in vitro, of a model p53-responsive promoter (with five promoter-proximal p53 sites and a very strong core promoter) and the lack of other natural constraints (limiting factors, alternative chromatin structures, etc.).
Our current studies provide insight into the mechanisms of TFIID recruitment by showing that the stability and/or specific recruitment of TFIID is enhanced by bivalent interactions involving H3K4me3 and the TATA box. Based on previously described TFIID-core promoter interactions (reviewed in Juven-Gershon et al., 2008) and studies reported here, we support a model (Figure S6) for the functional interplay between histone modifications and core DNA sequences that emphasizes a link between the total number of TFIID interactions with the core promoter and the corresponding level of transcription initiation. This model is consistent with previous reports that have shown that multiple core promoter elements are involved in mediating cooperative GTF recruitment (Hsu et al., 2008; O’Shea-Greenfield and Smale, 1992; Smale and Kadonaga, 2003) and that multiple histone modifications facilitate the multivalent recruitment of transcription factors (Ruthenburg et al., 2007b; Vermeulen and Timmers, 2010).
H3K4me3-TAF3/TFIID Interactions Enhance the Recruitment of TAF3 for the Expression of Select p53 Target Genes
In response to stress, p53-dependent signaling pathways direct damaged cells to undergo growth arrest or apoptosis (Levine, 1997; Prives and Hall, 1999). While several mechanisms have been implicated in gene-selective functions of p53 (reviewed in Espinosa et al., 2003; Oda et al., 2000; Sykes et al., 2006; Tang et al., 2006), the full spectrum of mechanisms that control gene activation by p53 and determine the appropriate response remain to be fully elucidated. Our studies reveal a transcriptional mechanism in which H3K4me3-TAF3 interactions are required to regulate the expression of select p53 target genes. Importantly, our microarray data identified a specific subset of genes affected by TAF3 knockdown, which supports a gene-specific, rather than a global, effect of TAF3 on gene expression. The vast majority of the DNA damage-induced genes that were affected by TAF3 depletion are key regulators of the cell cycle pathway and are rapidly induced in response to DNA damage (Zhao et al., 2000). In contrast, proapoptotic genes that display delayed induction (Zhao et al., 2000), were significantly under-represented in our microarray data. Consistent with our global studies, qPCR analysis of a subset of p53 target genes confirmed this gene-selective coactivator role for TAF3. Our cell-based findings are corroborated by our in vitro mechanistic studies showing the importance of H3K4me3 in coordinating rapid PIC formation and enhanced transcriptional activation.
Our studies also reveal that, in response to DNA damage, a significant increase in TAF3/TFIID recruitment correlates with a significant increase in H3K4me3 accumulation at the promoters of several cell cycle genes. In contrast, this overlap was not identified at several proapoptotic gene promoters. Strikingly, the significant increase in H3K4me3 accumulation and TAF3 recruitment that was observed at the cell cycle promoters also correlates strongly with the rapid increase (from a low basal level) in the mRNA levels of these genes. More specifically, either H3K4me3 depletion or the loss of H3K4me3-TAF3/TFIID interactions was found to disrupt TFIID and RNAPII recruitment to the cell cycle promoters while having no effect on their recruitment to proapoptotic promoters. The recovery of cell cycle gene expression by siRNA-resistant wild-type, but not M880A mutant, TAF3 in TAF3-depleted cells underscores the importance of H3K4me3-TAF3 interactions in regulating cell cycle gene responses. Taken together, our studies reveal that TAF3, and associated H3K4me3 interactions, have a direct role in gene-specific regulation within the p53 network.
Experimental Procedures
Chromatin Assembly and Transcription Assays
Methyl-lysine analogs were installed as described (Simon et al., 2007). Chromatin was assembled and transcription assays were performed as described (An and Roeder, 2004). See Supplemental Information for details.
In Vitro ChIP
ChIP assays were performed as described (Kundu et al., 2000). For details and primer sequences see Supplemental Information and Table S2.
RNA Interference Experiments
HCT116 and U2OS cells were transfected with a non-targeting siRNA control, a human WDR5 siRNA SMART pool, or TAF3 siRNA duplexes (listed in Table S1) (all siRNAs are from Thermo Scientific). 48 hours after transfection, 0.5 μM doxorubicin (Sigma) was added. siRNA rescue experiments were performed by co-transfecting TAF3 siRNA with pcDNA5/FRT/TO-TAF3 siRNA resistant plasmids using Dharmafect Duo.
RNA Purification and Quantitative Real-Time PCR
Total RNA was prepared using the RNeasy kit (QIAGEN), reverse-transcribed with the SuperScript III First-Strand Synthesis System (Invitrogen), and amplified with SYBR Green PCR master mix (Applied Biosystems). Details and primer sequences can be found in Supplemental Information and Table S3.
Gene Expression Microarray Analysis
Standard methods for labeling and hybridization to the Illumina HumanHT-12 v3 Beadchip were performed. For details see Supplemental Information.
In Vivo ChIP, ChIP-seq, and HIT-seq
For experimental details and primer sequences see Supplemental Information, Table S4, and Table S6.
Accession Numbers
Our microarray and ChIP-seq datasets have been deposited in the database under GSE43542.
Supplementary Material
H3K4me3 facilitates global TFIID recruitment to the core promoters of active genes.
By stimulating PIC formation H3K4me3 enhances p53-dependent activation.
H3K4me3 acts both independently and cooperatively with the TATA box.
Interactions between H3K4me3 and TAF3/TFIID regulate gene-selective functions of p53.
Acknowledgments
We are grateful to Tomoyoshi Nakadai and Zhu Xie for technical assistance and Sohail Malik, Beth Moorefield, Christina Hughes, and Michael Rauchman for insightful discussions of the project. This work was supported by NRSA Training Grant #CA 09673 to S.L., the Intramural Research Program of the NIH, Center for Cancer Research, National Cancer Institute, Frederick National Lab to S.H.H., and by grants from the NIH (CA129325) and the STARR Cancer Consortium to R.G.R.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- An W, Roeder RG. Reconstitution and transcriptional analysis of chromatin in vitro. Methods Enzymol. 2004;377:460–474. doi: 10.1016/S0076-6879(03)77030-X. [DOI] [PubMed] [Google Scholar]
- Bannister AJ, Kouzarides T. Regulation of chromatin by histone modifications. Cell Res. 2011;21:381–395. doi: 10.1038/cr.2011.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein BE, Kamal M, Lindblad-Toh K, Bekiranov S, Bailey DK, Huebert DJ, McMahon S, Karlsson EK, Kulbokas EJ, 3rd, Gingeras TR, et al. Genomic maps and comparative analysis of histone modifications in human and mouse. Cell. 2005;120:169–181. doi: 10.1016/j.cell.2005.01.001. [DOI] [PubMed] [Google Scholar]
- Bunz F, Dutriaux A, Lengauer C, Waldman T, Zhou S, Brown JP, Sedivy JM, Kinzler KW, Vogelstein B. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science. 1998;282:1497–1501. doi: 10.1126/science.282.5393.1497. [DOI] [PubMed] [Google Scholar]
- Burley SK, Roeder RG. Biochemistry and structural biology of transcription factor IID (TFIID) Annu Rev Biochem. 1996;65:769–799. doi: 10.1146/annurev.bi.65.070196.004005. [DOI] [PubMed] [Google Scholar]
- Campos EI, Reinberg D. Histones: annotating chromatin. Annual review of genetics. 2009;43:559–599. doi: 10.1146/annurev.genet.032608.103928. [DOI] [PubMed] [Google Scholar]
- Carninci P, Sandelin A, Lenhard B, Katayama S, Shimokawa K, Ponjavic J, Semple CA, Taylor MS, Engstrom PG, Frith MC, et al. Genome-wide analysis of mammalian promoter architecture and evolution. Nat Genet. 2006;38:626–635. doi: 10.1038/ng1789. [DOI] [PubMed] [Google Scholar]
- Chi P, Allis CD, Wang GG. Covalent histone modifications--miswritten, misinterpreted and mis-erased in human cancers. Nat Rev Cancer. 2010;10:457–469. doi: 10.1038/nrc2876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deato MD, Marr MT, Sottero T, Inouye C, Hu P, Tjian R. MyoD targets TAF3/TRF3 to activate myogenin transcription. Mol Cell. 2008;32:96–105. doi: 10.1016/j.molcel.2008.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deato MD, Tjian R. Switching of the core transcription machinery during myogenesis. Genes Dev. 2007;21:2137–2149. doi: 10.1101/gad.1583407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dou Y, Milne TA, Ruthenburg AJ, Lee S, Lee JW, Verdine GL, Allis CD, Roeder RG. Regulation of MLL1 H3K4 methyltransferase activity by its core components. Nat Struct Mol Biol. 2006;13:713–719. doi: 10.1038/nsmb1128. [DOI] [PubMed] [Google Scholar]
- Espinosa JM, Verdun RE, Emerson BM. p53 functions through stress- and promoter-specific recruitment of transcription initiation components before and after DNA damage. Mol Cell. 2003;12:1015–1027. doi: 10.1016/s1097-2765(03)00359-9. [DOI] [PubMed] [Google Scholar]
- Espinoza CA, Allen TA, Hieb AR, Kugel JF, Goodrich JA. B2 RNA binds directly to RNA polymerase II to repress transcript synthesis. Nat Struct Mol Biol. 2004;11:822–829. doi: 10.1038/nsmb812. [DOI] [PubMed] [Google Scholar]
- Ferris AL, Wu X, Hughes CM, Stewart C, Smith SJ, Milne TA, Wang GG, Shun MC, Allis CD, Engelman A, et al. Lens epithelium-derived growth factor fusion proteins redirect HIV-1 DNA integration. Proc Natl Acad Sci U S A. 2010;107:3135–3140. doi: 10.1073/pnas.0914142107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gangloff YG, Pointud JC, Thuault S, Carre L, Romier C, Muratoglu S, Brand M, Tora L, Couderc JL, Davidson I. The TFIID components human TAF(II)140 and Drosophila BIP2 (TAF(II)155) are novel metazoan homologues of yeast TAF(II)47 containing a histone fold and a PHD finger. Mol Cell Biol. 2001;21:5109–5121. doi: 10.1128/MCB.21.15.5109-5121.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart DO, Santra MK, Raha T, Green MR. Selective interaction between Trf3 and Taf3 required for early development and hematopoiesis. Dev Dyn. 2009;238:2540–2549. doi: 10.1002/dvdy.22083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heintzman ND, Stuart RK, Hon G, Fu Y, Ching CW, Hawkins RD, Barrera LO, Van Calcar S, Qu C, Ching KA, et al. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat Genet. 2007;39:311–318. doi: 10.1038/ng1966. [DOI] [PubMed] [Google Scholar]
- Hsu JY, Juven-Gershon T, Marr MT, 2nd, Wright KJ, Tjian R, Kadonaga JT. TBP, Mot1, and NC2 establish a regulatory circuit that controls DPE-dependent versus TATA-dependent transcription. Genes Dev. 2008;22:2353–2358. doi: 10.1101/gad.1681808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juven-Gershon T, Hsu JY, Theisen JW, Kadonaga JT. The RNA polymerase II core promoter - the gateway to transcription. Curr Opin Cell Biol. 2008;20:253–259. doi: 10.1016/j.ceb.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Guermah M, McGinty RK, Lee JS, Tang Z, Milne TA, Shilatifard A, Muir TW, Roeder RG. RAD6-Mediated transcription-coupled H2B ubiquitylation directly stimulates H3K4 methylation in human cells. Cell. 2009;137:459–471. doi: 10.1016/j.cell.2009.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kundu TK, Palhan VB, Wang Z, An W, Cole PA, Roeder RG. Activator-dependent transcription from chromatin in vitro involving targeted histone acetylation by p300. Mol Cell. 2000;6:551–561. doi: 10.1016/s1097-2765(00)00054-x. [DOI] [PubMed] [Google Scholar]
- Levine AJ. p53, the cellular gatekeeper for growth and division. Cell. 1997;88:323–331. doi: 10.1016/s0092-8674(00)81871-1. [DOI] [PubMed] [Google Scholar]
- Lin JJ, Lehmann LW, Bonora G, Sridharan R, Vashisht AA, Tran N, Plath K, Wohlschlegel JA, Carey M. Mediator coordinates PIC assembly with recruitment of CHD1. Genes Dev. 2011;25:2198–2209. doi: 10.1101/gad.17554711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Scannell DR, Eisen MB, Tjian R. Control of Embryonic Stem Cell Lineage Commitment by Core Promoter Factor, TAF3. Cell. 2011;146:720–731. doi: 10.1016/j.cell.2011.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Shea-Greenfield A, Smale ST. Roles of TATA and initiator elements in determining the start site location and direction of RNA polymerase II transcription. J Biol Chem. 1992;267:1391–1402. [PubMed] [Google Scholar]
- Oda K, Arakawa H, Tanaka T, Matsuda K, Tanikawa C, Mori T, Nishimori H, Tamai K, Tokino T, Nakamura Y, et al. p53AIP1, a potential mediator of p53-dependent apoptosis, and its regulation by Ser-46-phosphorylated p53. Cell. 2000;102:849–862. doi: 10.1016/s0092-8674(00)00073-8. [DOI] [PubMed] [Google Scholar]
- Prives C, Hall PA. The p53 pathway. J Pathol. 1999;187:112–126. doi: 10.1002/(SICI)1096-9896(199901)187:1<112::AID-PATH250>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Ruthenburg AJ, Allis CD, Wysocka J. Methylation of lysine 4 on histone H3: intricacy of writing and reading a single epigenetic mark. Mol Cell. 2007a;25:15–30. doi: 10.1016/j.molcel.2006.12.014. [DOI] [PubMed] [Google Scholar]
- Ruthenburg AJ, Li H, Patel DJ, Allis CD. Multivalent engagement of chromatin modifications by linked binding modules. Nat Rev Mol Cell Biol. 2007b;8:983–994. doi: 10.1038/nrm2298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y, Yue F, McCleary DF, Ye Z, Edsall L, Kuan S, Wagner U, Dixon J, Lee L, Lobanenkov VV, et al. A map of the cis-regulatory sequences in the mouse genome. Nature. 2012 doi: 10.1038/nature11243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shilatifard A. Chromatin modifications by methylation and ubiquitination: implications in the regulation of gene expression. Annu Rev Biochem. 2006;75:243–269. doi: 10.1146/annurev.biochem.75.103004.142422. [DOI] [PubMed] [Google Scholar]
- Simon MD, Chu F, Racki LR, de la Cruz CC, Burlingame AL, Panning B, Narlikar GJ, Shokat KM. The site-specific installation of methyl-lysine analogs into recombinant histones. Cell. 2007;128:1003–1012. doi: 10.1016/j.cell.2006.12.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smale ST, Kadonaga JT. The RNA polymerase II core promoter. Annu Rev Biochem. 2003;72:449–479. doi: 10.1146/annurev.biochem.72.121801.161520. [DOI] [PubMed] [Google Scholar]
- Steward MM, Lee JS, O’Donovan A, Wyatt M, Bernstein BE, Shilatifard A. Molecular regulation of H3K4 trimethylation by ASH2L, a shared subunit of MLL complexes. Nat Struct Mol Biol. 2006;13:852–854. doi: 10.1038/nsmb1131. [DOI] [PubMed] [Google Scholar]
- Sykes SM, Mellert HS, Holbert MA, Li K, Marmorstein R, Lane WS, McMahon SB. Acetylation of the p53 DNA-binding domain regulates apoptosis induction. Mol Cell. 2006;24:841–851. doi: 10.1016/j.molcel.2006.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Y, Luo J, Zhang W, Gu W. Tip60-dependent acetylation of p53 modulates the decision between cell-cycle arrest and apoptosis. Mol Cell. 2006;24:827–839. doi: 10.1016/j.molcel.2006.11.021. [DOI] [PubMed] [Google Scholar]
- Taverna SD, Li H, Ruthenburg AJ, Allis CD, Patel DJ. How chromatin-binding modules interpret histone modifications: lessons from professional pocket pickers. Nat Struct Mol Biol. 2007;14:1025–1040. doi: 10.1038/nsmb1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas MC, Chiang CM. The general transcription machinery and general cofactors. Critical reviews in biochemistry and molecular biology. 2006;41:105–178. doi: 10.1080/10409230600648736. [DOI] [PubMed] [Google Scholar]
- van Ingen H, van Schaik FM, Wienk H, Ballering J, Rehmann H, Dechesne AC, Kruijzer JA, Liskamp RM, Timmers HT, Boelens R. Structural insight into the recognition of the H3K4me3 mark by the TFIID subunit TAF3. Structure. 2008;16:1245–1256. doi: 10.1016/j.str.2008.04.015. [DOI] [PubMed] [Google Scholar]
- Vermeulen M, Mulder KW, Denissov S, Pijnappel WW, van Schaik FM, Varier RA, Baltissen MP, Stunnenberg HG, Mann M, Timmers HT. Selective anchoring of TFIID to nucleosomes by trimethylation of histone H3 lysine 4. Cell. 2007;131:58–69. doi: 10.1016/j.cell.2007.08.016. [DOI] [PubMed] [Google Scholar]
- Vermeulen M, Timmers HT. Grasping trimethylation of histone H3 at lysine 4. Epigenomics. 2010;2:395–406. doi: 10.2217/epi.10.11. [DOI] [PubMed] [Google Scholar]
- Vousden KH, Prives C. Blinded by the Light: The Growing Complexity of p53. Cell. 2009;137:413–431. doi: 10.1016/j.cell.2009.04.037. [DOI] [PubMed] [Google Scholar]
- Yu J, Zhang L, Hwang PM, Kinzler KW, Vogelstein B. PUMA induces the rapid apoptosis of colorectal cancer cells. Mol Cell. 2001;7:673–682. doi: 10.1016/s1097-2765(01)00213-1. [DOI] [PubMed] [Google Scholar]
- Zhao R, Gish K, Murphy M, Yin Y, Notterman D, Hoffman WH, Tom E, Mack DH, Levine AJ. Analysis of p53-regulated gene expression patterns using oligonucleotide arrays. Genes Dev. 2000;14:981–993. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.