

Abstract
Muraminomicin is a lipopeptidyl nucleoside antibiotic produced by Streptosporangium amethystogenes SANK 60709. Similar to several members of this antibiotic family such as A-90289 and muraymycin, the structure of muraminomicin consists of a disaccharide comprised of two modified ribofuranose units linked by an O-β(1 → 5) glycosidic bond; however, muraminomicin holds the distinction in that both ribose units are 2-deoxy sugars. The biosynthetic gene cluster of muraminomicin has been identified, cloned and sequenced, and bioinformatic analysis revealed a minimum of 24 open reading frames putatively involved in the biosynthesis, resistance, and regulation of muraminomicin. Fives enzymes are likely involved in the assembly and attachment of the 2,5-dideoxy-5-aminoribose saccharide unit, and two are now functionally assigned and characterized: Mra20, a 5′-amino-2′,5′-dideoxyuridine phosphorylase and Mra23, a UTP:5-amino-2,5-dideoxy-α-D-ribose-1-phosphate uridylyltransferase. The cumulative results are consistent with the incorporation of the ribosyl appendage of muraminomicin via the archetypical sugar biosynthetic pathway that parallels A-90289 biosynthesis, and the specificity for this appendage is dictated primarily by the two characterized enzymes.
Introduction
Antibiotics that inhibit the biosynthesis of peptidoglycan cell wall have been clinically proven to be effective; however the majority of the enzymes in this ubiquitous pathway have yet to be successfully exploited as antibiotic targets. A number of potent inhibitors of bacterial translocase I (MraY), one of these essential yet untargeted enzymes, have been discovered the past decade using an activity-based screen.1,2 One promising family of MraY inhibitors are the lipopeptidyl nucleosides, which includes muraminomicin F from Streptosporangium amethystogenes SANK 60709,3 A-90289 A from Streptomyces sp. SANK 60405,4 caprazamycin A from Streptomyces sp. MK730-62F,5 liposidomycin C from Streptomyces sp. SN-1061M,6 muraymycin A1 from Streptomyces sp. NRRL 30471,7 and FR-900493 from Bacillus cereus No. 2045.8 These nucleosides are structurally characterized by a uridine core with two 5′-modifications: a glycine unit linked by a C-C bond to form a high-carbon sugar nucleoside and an aminoribofuranoside attached at C-5′ by an O-glycosidic bond (Fig. 1).
Fig. 1.

Structures of representative lipopeptidyl nucleoside antibiotics containing an aminoribofuranoside appendage (blue).
The biosynthetic gene clusters for A-90289, caprazamycin, liposidomycin, and muraymycin have been reported.9–12 As expected, the gene clusters were found to have several shared open reading frames (orfs) that were likely involved in the assembly of the modified uridine core. Using A-90289 as a biosynthetic model, the origin and mechanism of formation of the aminoribosyl moiety was delineated to reveal five of these shared orfs are essential for the ribosylation process (Fig. S1).13 The pathway is initiated by LipL, a non-heme Fe(II) and α-ketoglutarate (αKG)-dependent dioxygenase that catalyzes oxidative dephosphorylation of uridine-5′-monophosphate (UMP) to uridine-5′-aldehyde (UA).14 Subsequently, the L-methionine:UA aminotransferase LipO catalyzes the formation of 5′-amino-5′-deoxyuridine that serves as the substrate for the phosphorylase LipP to generate 5-amino-5-deoxy-α-D-ribose-1-phosphate. This sugar-1-phosphate is then processed by two enzymes in a manner that parallels typical glycosylation event: the 5-amino-5-deoxy-α-D-ribose-1-phosphate is activated by LipM as the nucleotide-5′-diphosphate (NDP)-sugar—in this case UDP-5-amino-5-deoxy-α-D-ribose—and the sugar is transferred by LipN to an acceptor molecule to generate the final disaccharide.13 Uridine was used as a surrogate acceptor, although the identity of the true acceptor, which is potentially 5′-C-glycyluridine, remains unknown.
While the biosynthesis of the aminoribosyl moiety for the lipopeptidyl nucleoside antibiotics is expected to occur via the general ribosylation pathway that was uncovered from in vitro studies with A-90289 biosynthetic enzymes, each nucleoside has structural variations within the disaccharide core that would suggest either additional enzymes are required and/or some enzymes have an unique substrate specificity (Fig. 1). For example, the aminoribosyl moiety of muraymycin A1 is 2″-O-methylated while that of liposidomycin is O-sulfated at this position. Perhaps the most intriguing variation is that of muraminomicin, which contains 2-deoxyfuranoses within both components of the disaccharide unit. In order to pinpoint the factors controlling the incorporation of these and other structural variations observed in muraminomicin, we have identified the muraminomicin biosynthetic gene cluster and characterized two enzymes—the functional equivalents for LipM and LipP—involved in the assembly of the 5-amino-2,5-dideoxyribofuranoside. The results as described herein suggest these two enzymes play a critical role in the identity of the aminoribosyl appendage of muraminomicin.
Results and discussion
Identification of muraminomicin gene cluster
The apparent structural similarity to liposidomycin, caprazamycin, and A-90289 suggested the muraminomicin gene cluster shares most of the biosynthetic genes. We recently reported the utility of using degenerate primers for a gene annotated as a serine hydroxymethyltransferase to specifically identify the genetic locus involved in the biosynthesis of these related nucleosides,15 and this strategy was employed here to amplify DNA fragments with the expected size from Streptosporangium amethystogenes sp. SANK 60709 genomic DNA. The amplified DNA was utilized as a probe to ultimately identify 4 cosmids (pMra01-04) that were sequenced and analyzed, yielding 53 complete orfs within 73.6 kb of contiguous DNA (Fig. 2a and Table S1). Of these orfs, 22 were shared amongst all of the aforementioned clusters and a minimum of 2 orfs were unique to the muraminomicin gene cluster: mra4 encoding a hypothetical membrane protein and mra3 encoding a putative C-methyltransferase of the radical SAM superfamily. These orfs are inserted within a subcluster of genes responsible for the biosynthesis of the permethylated rhamnose, and the gene encoding a 3′-O-methyltransferase involved in A-90289 and caprazamycin biosynthesis (Fig. 2b) is missing from the muraminomicin gene cluster. Thus, it is proposed that Mra3 catalyzes C-methylation at C-6 of rhamnose to generate the unusual heptose found in muraminomicin while Mra4 functions as a 3-O-succinyltransferase (Fig. 1).
Fig. 2.

Genetic architecture of the muraminomicin gene cluster. (a) Organization of the~74-kb of sequenced DNA. (b) Comparison of the subcluster of genes involved in the biosynthesis of the rhamnosyl moiety. (c) Comparison of the subcluster of genes involved in the biosynthesis of the aminoribosyl moiety.
In contrast to the obvious differences within the genetic architecture of the locus involved in the biosynthesis of the rhamnosyl moiety, the five gene subcluster required for aminoribose formation appeared essentially the same for the A-90289 and muraminomicin gene clusters (Fig. 2c). However, minimally two differences were uncovered upon closer bioinformatics analysis: (i) the sequence identity between LipL-P and Mra20-24 were considerably lower compared to the remaining shared ORFs and (ii) the LipL homologue, Mra24, is lacking ~60 amino acids at the C-terminus that contains a His residue that is part of the HX(D/E)XnH motif (X is any amino acid), which is essential for Fe(II) binding and hence activity. Therefore, it highly likely that mra24 does not encode a functional non-heme Fe(II), αKG-dependent dioxygenase.
To provide initial evidence that the correct genetic locus was identified prior to completion of the sequencing, we introduced cosmid pMra02 into the heterologous host Streptomyces lividans TK21 with the expectation that this cosmid contains a genetic element involved in muraminomicin resistance. In contrast to controls using the empty vector, S. lividans TK21/pMra02 was resistant to muraminomicin at 100 μg/mL on solid media (Fig. S2). Retrospective analysis of this cosmid sequence reveals two potential resistance candidates mra11 and mra16 that encode a putative ABC-transporter and a protein with similarity to tunicamycin-resistance protein TmrB, respecitvely.16 Although muraminomicin was not produced by the recombinant strain, a red phenotype was observed upon introduction of pMra02 and muraminomicin selection, suggesting the cosmid and antibiotic likely upregulate additional host genes such as those potentially involved in actinorhodin biosynthesis. Encoded within pMra02 is a protein with similarity to AraC positive regulatory protein, which may be partially responsible for this phenotypic output. The identity of the individual genes involved in resistance and regulation is currently under investigation, nevertheless, the results are consistent with the role of the genetic locus in muraminomicin production.
Functional assignment of Mra20 as a low specificity pyrimidine nucleoside phosphorylase
Pyrimidine nucleoside phosphorylases catalyze the phosphorolysis of thymidine or uridine nucleosides to generate 2-deoxy-α-D-ribose-1-phosphate and thymine or α-D-ribose-1-phosphate and uracil, respectively.17,18 Although thymidine phosphorylases from several organisms are highly specific for the 2′-deoxyribosyl moiety, some are reported to have very little discrimination at this position. LipP was slightly specific for the hydroxylated ribose; additionally, efficiency with 5′-amino-5′-deoxyuridine was comparable to that of uridine.13
The mra20 gene, which is homologous to lipP, was cloned and expressed in E. coli to yield soluble protein (Fig. S3a). Using HPLC for detection, initial activity tests with Mra20 revealed uridine was converted to uracil and α-D-ribose-1-phosphate (Fig. S3b), and, identically to LipP,13 the Mra20-catalyzed reaction also proceeded with 5′-amino-5′-deoxyuridine (Fig. S3c). Additional activity tests revealed hypothetical pathway intermediates 2′-deoxyuridine 1, 5′-amino-2′,5′-dideoxyuridine 2, thymidine 3, and 5′-amino-5′-deoxythymidine 4 were also converted to uracil 5 or thymine 6 and the respective sugar-1-phosphate by Mra20 (Fig. 3), thus warranting further kinetic investigation.
Fig. 3.
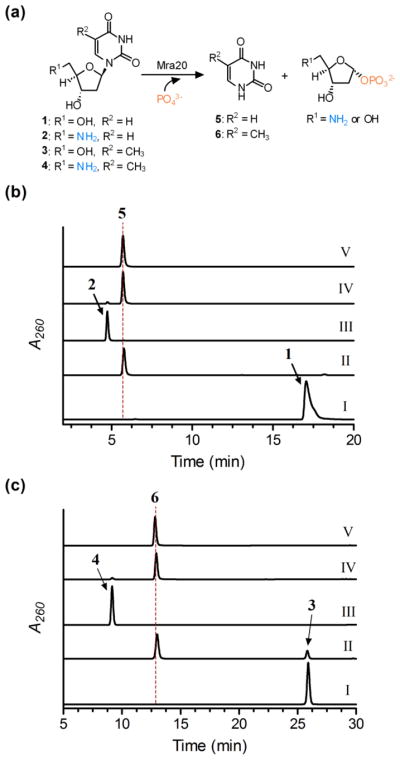
Activity of Mra20 with 2′-deoxynucleosides. (a) Phosphorolysis reaction catalysed by Mra20. (b) HPLC analysis following a 1 h reaction using 1 without Mra20 (I), 1 with Mra20 (II), 2 without Mra20 (III), 2 with Mra20 (IV), and authentic 5. (c) HPLC analysis following a 1 h reaction using 3 without Mra20 (I), 3 with Mra20 (II), 4 without Mra20 (III), 4 with Mra20 (IV), and authentic 6.
For comparative purposes, single substrate kinetic analysis was performed under the optimized conditions for LipP using UV/Vis spectroscopy for detection of the free nucleobase under alkaline conditions. All of the tested substrates displayed typical Michaelis-Menten kinetics (Figure S4), and the extracted kinetic constants revealed that Mra20 is most efficient with thymine-containing nucleosides with little preference for the C-5′ functional group (Table 1). Although the KM and kcat were both significantly increased, the efficiency with 2′-deoxyuridine derivatives were quite comparable to the respective thymine-containing nucleoside. Similar to LipP, these kinetic results suggest that Mra20 has little preference toward any hypothetical pathway intermediates, and thus cannot be utilized to delineate the timing of the biochemical events. However, in contrast to LipP, Mra20 has a modest specificity toward the 2′-deoxynucleosides relative to the hydroxylated counterpart, which is consistent with the observed 5-amino-2,5-dideoxyribosyl moiety that is found in the muraminomicins.
Table 1.
Extracted kinetic constants for Mra20
| Substrate | Km (mM) | kcat (min−1) | Relative efficiency (%) |
|---|---|---|---|
| 1 | 1.8 ± 0.3 | 28 ± 2 | 61 |
| 2 | 0.87 ± 0.24 | 16 ± 2 | 75 |
| 3 | 0.12 ± 0.04 | 2.9 ± 0.2 | 100 |
| 4 | 0.13 ± 0.05 | 3.1 ± 0.3 | 93 |
| Ua | 0.43 ± 0.07 | 0.9 ± 0.1 | 8 |
| ADUb | 0.82 ± 0.21 | 4.0 ± 0.3 | 19 |
Uridine
5′-Amino-5′-deoxyuridine
Functional assignment of Mra23 as a primary amine-requiring nucleotidylyltransferase
Bioinformatic analysis revealed Mra23 has sequence similarity to LipM and other nucleotidylyltransferases, enzymes that utilize a sugar-1-phosphate and nucleotide-5′-triphosphate to generate NDP-sugars, also known as activated sugars.19 The Mra23 homologue from the A-90289 pathway (LipM) has already been demonstrated to function as a nucleotidylyltransferase, using 5-amino-5-deoxy-α-D-ribose-1-phosphate and UTP to generate the activated sugar, and, interestingly, is only able to turn over ribose-1-phosphate with a 5-amine functionality.
Similarly to Mra20, the gene product of mra23 was soluble when produced in E.coli (Fig. S5a). The activity of Mra23 was tested with 2-deoxy-α-D-ribose-1-phosphate or 5-amino-2,5-dideoxy-α-D-ribose-1-phosphate generated in situ by Mra20 from uridine and nucleosides 2–4, and analysis of these reactions revealed a new peak was formed only in the presence of 5-amino-2,5-dideoxy-α-D-ribose-1-phosphate from 2 or 4 and UTP (Fig. 4 and Fig. 5b,c). LC-MS analysis of the purified new peak revealed an (M - H)− ion at m/z = 517.6, consistent with the molecular formula C14H23N3O14P2 of UDP-5-amino-2,5-dideoxyribose 7 (expected m/z = 518.1) (Fig. 4a and Fig. S6). We had previously characterized 7 by HR-MS and complete NMR analysis using LipP and LipM starting from 2, and thus coinjections were utilized to confirm the product of Mra20 and Mra23 (Fig. 4). In total, the data is consistent with the function of Mra23 as a UTP:5-amino-2,5-dideoxy-α-D-ribose-1-phosphate uridylyltransferase as the penultimate catalyst for formation of the disaccharide of muraminomicin.
Fig. 4.
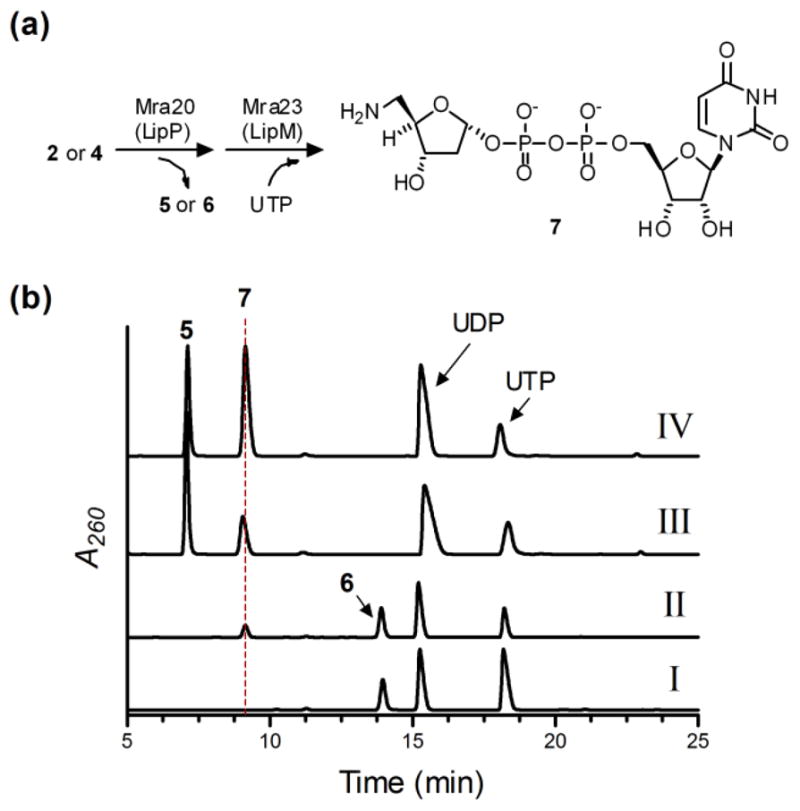
Characterization of Mra23. (a) Enzymatic preparation of the substrate using Mra20, and the reaction catalyzed by Mra23 to generate 7. (b) HPLC analysis following a 3 h reaction starting from 4 without Mra23 (I), 4 with Mra23 (II), a 12 h reaction starting from 2 with LipM from the A-90289 biosynthetic pathway replacing Mra23 (III), and a 12 h reaction starting from 2 with LipM coinjected with purified 7 generated by Mra23 (IV).
The substrate specificity of Mra23 is intriguing for a couple of reasons. Firstly, the transfer of ribosyl units onto acceptor molecules typically occurs with 5-phosphoribosyl-1-pyrophosphate, and the characterization of Mra23 and LipM has revealed a ribosyl transfer paradigm using NDP-sugars.13 This realization may ultimately provide an opportunity to diversify structures by incorporating unnatural ribosyl units in place of the native sugar via a combinatorial biosynthetic strategy. Secondly, several characterized nucleotidylyltransferases are known or have been engineered by mutagenesis to accept aminosugar-1-phosphates; however, these enzymes in general have little discrimination between the amine or hydroxyl group.20–23 Both Mra23 and LipM now establish a nucleotidylyltransferase family whose activity is absolutely dependent upon the amine functionality, and on-going structural efforts will undoubtedly establish the molecular details behind this unusual specificity.
The combined results now allow us to define a probable pathway leading to the disaccharide core starting from either TMP or 2′-deoxyUMP (Scheme 1). Through an as of yet unclear mechanism, the 5′-aldehyde is installed to serve as the substrate for the putative aminotransferase Mra21 to generate 2 or 4. Following phosphorolysis of the glycosidic bond by Mra20, the aminosugar-1-phosphate is activated as the UDP-sugar by Mra23 prior to transfer to the acceptor molecule, which is most likely 2′-deoxy-5′-C-glycyluridine.
Scheme 1.
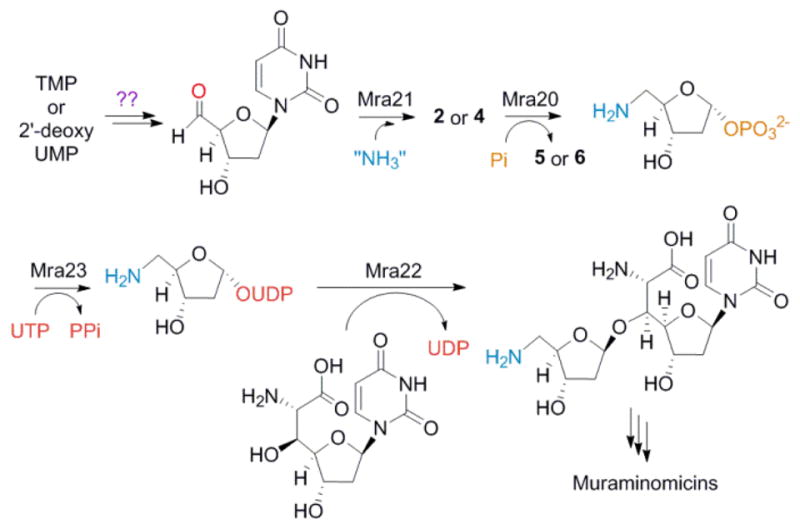
Proposed biosynthetic pathway of the disaccharide core of the muraminomicins.
CONCLUSION
The biosynthetic gene cluster for muraminomicin has been identified. Two enzymes involved in the assembly of the aminoribosyl moiety of muraminomicin have been functionally assigned: Mra20 that catalyzes phosphorolysis to initiate “salvage” of the aminoribose and Mra23 that activates this sugar for subsequent ribosyltransfer. The established specificity of Mra20 and Mra23 explains at least in part the incorporation of a 2-deoxy and 5-amino-5-deoxy sugar, respectively, into muraminomicin. The results now set the stage to explore both upstream events, such as the unclear mechanism of 5′-aldehyde formation, and the downstream event of ribosylation. Additionally, the uncovering of the genetic locus for muraminomicin biosynthetic locus provides further information for defining a complete biosynthetic mechanism for lipopeptidyl nucleoside antibiotics.
Supplementary Material
Acknowledgments
This work was supported by funding from the National Institute of Health (AI087849 for S.V.L.).
Footnotes
Electronic Supplementary Information (ESI) available: Experimental Procedures, Table S1, and Fig. S1-S6 See DOI: 10.1039/b000000x/
Notes and references
- 1.Winn M, Goss RJ, Kimura K, Bugg TD. Nat Prod Rep. 2010;27:279. doi: 10.1039/b816215h. [DOI] [PubMed] [Google Scholar]
- 2.Dini C. Curr Top Med Chem. 2005;5:1221. doi: 10.2174/156802605774463042. [DOI] [PubMed] [Google Scholar]
- 3.Muramatsu Y, Yoko F, Azusa A, Masaaki K, Toshio T, Shunichi M. 2004046368. Sankyo Co., Ltd; Patent WO. 2004
- 4.Fujita Y, Kizuka M, Funabashi M, Ogawa Y, Ishikawa T, Nonaka K, Takatsu T. J Antibiot. 2011;64:495. doi: 10.1038/ja.2011.38. [DOI] [PubMed] [Google Scholar]
- 5.Igarashi M, Takahashi Y, Shitara T, Nakamura H, Naganawa H, Miyake T, Akamatsu Y. J Antibiot. 2005;58:327. doi: 10.1038/ja.2005.41. [DOI] [PubMed] [Google Scholar]
- 6.Kimura K, Ikeda Y, Kagami S, Yoshihama M, Ubukata M, Esumi Y, Osada H, Isono K. J Antibiot. 1998;51:647. doi: 10.7164/antibiotics.51.647. [DOI] [PubMed] [Google Scholar]
- 7.McDonald LA, Barbieri LR, Carter GT, Lenoy E, Lotvin J, Petersen PJ, Siegel MM, Singh G, Williamson RT. J Am Chem Soc. 2002;124:10260. doi: 10.1021/ja017748h. [DOI] [PubMed] [Google Scholar]
- 8.Ochi K, Ezaki M, Iwani M, Komori T, Kohsaka M. EP-333177. Patent. 1989
- 9.Funabashi M, Baba S, Nonaka K, Hosobuchi M, Fujita Y, Shibata T, Van Lanen SG. ChemBioChem. 2010;11:184. doi: 10.1002/cbic.200900665. [DOI] [PubMed] [Google Scholar]
- 10.Kaysser L, Lutsch L, Siebenberg S, Wemakor E, Kammerer B, Gust B. J Biol Chem. 2009;284:14987. doi: 10.1074/jbc.M901258200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kaysser L, Siebenberg S, Kammerer B, Gust B. ChemBioChem. 2010;11:191. doi: 10.1002/cbic.200900637. [DOI] [PubMed] [Google Scholar]
- 12.Cheng L, Chen W, Zhai L, Xu D, Huang T, Lin S, Zhou X, Deng Z. Mol Biosyst. 2011;7:920. doi: 10.1039/c0mb00237b. [DOI] [PubMed] [Google Scholar]
- 13.Chi X, Pahari P, Nonaka K, Van Lanen SG. J Am Chem Soc. 2011;133:14452. doi: 10.1021/ja206304k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yang Z, Chi X, Funabashi M, Baba S, Nonaka K, Pahari P, Unrine J, Jacobsen JM, Elliott GI, Rohr J, Van Lanen SG. J Biol Chem. 2011;286:7885. doi: 10.1074/jbc.M110.203562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Funabashi M, Baba S, Cai W, Barnard-Britson S, Nonaka K, Takatsu T, Yang Z, Kizuka M, Sunkara M, Morris AJ, Van Lanen SG. manuscript submitted [Google Scholar]
- 16.Noda Y, Yoda K, Takatsuki A, Yamasaki M. J Bacteriol. 1992;174:4302. doi: 10.1128/jb.174.13.4302-4307.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pugmire MJ, Ealick SE. Biochem J. 2002;361:1. doi: 10.1042/0264-6021:3610001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sinha SC, Smith JL. Curr Opin Struct Biol. 2001;11:733. doi: 10.1016/s0959-440x(01)00274-3. [DOI] [PubMed] [Google Scholar]
- 19.Barton WA, Lesniak J, Biggins JB, Jeffrey PD, Jiang J, Rajashankar KR, Thorson JS, Nikolov DB. Nat Struct Biol. 2001;8:545. doi: 10.1038/88618. [DOI] [PubMed] [Google Scholar]
- 20.Kudo F, Kawabe K, Kuriki H, Eguchi T, Kakinuma K. J Am Chem Soc. 2005;127:1711. doi: 10.1021/ja044921b. [DOI] [PubMed] [Google Scholar]
- 21.Mizanur RM, Zea CJ, Pohl NL. J Am Chem Soc. 2004;126:15993. doi: 10.1021/ja046070d. [DOI] [PubMed] [Google Scholar]
- 22.Timmons SC, Mosher RH, Knowles SA, Jakeman DL. Org Lett. 2007;9:857. doi: 10.1021/ol0630853. [DOI] [PubMed] [Google Scholar]
- 23.Moretti R, Thorson JS. Angew Chem Int Ed. 2001;40:1502. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.