

Abstract
Many DNA transactions are crucial for maintaining genomic integrity and faithful transfer of genetic information but remain poorly understood. An example is the interplay between nucleotide excision repair (NER) and transcription, also known as transcription-coupled DNA repair (TCR). Discovered decades ago, the mechanisms for TCR have remained elusive, not in small part due to the scarcity of structural studies of key players. Here we summarize recent structural information on NER/TCR factors, focusing on bacterial systems, and integrate it with existing genetic, biochemical, and biophysical data to delineate the mechanisms at play. We also review emerging, alternative modalities for recruitment of NER proteins to DNA lesions.
NER: a versatile pathway
To deal with DNA insults, organisms have evolved a hierarchy of DNA repair pathways, which, to ensure genomic stability, are sometimes overlapping and redundant. Among all DNA repair mechanisms, NER is the most versatile; it can repair a large repertoire of chemically and structurally distinct DNA lesions, and it does so by a ‘cut and patch’ mechanism that exists in all domains of life (reviewed in [1]). In this process, the DNA phosphodiester backbone is hydrolyzed 3′ and 5′ of the lesion by machinery termed ‘excinuclease’ owing to its dual endonuclease activity that excises DNA. Then, the short lesion-containing oligonucleotide is removed, and the resulting gap is filled [2,3]. Thus, the pathway consists of five steps: damage recognition, incision, excision, repair synthesis, and ligation. Bacterial NER is carried out by a relatively simple Uvr system, whereas in higher organisms the NER machinery is significantly more complex [2]. In many respects, damage recognition is the most complex step, not only because of the broad spectrum of lesions recognized, which include UV-induced damage (such as cyclobutane pyrimidine dimers; CPDs), oligopeptide crosslinks, possibly some oxidative DNA lesions and base excision intermediates [2,4], but also because of its interplay with other DNA-based processes such as transcription. Here, we focus on the bacterial NER system – consisting of Uvr(A)BC – and its integration with RNA synthesis via transcription–repair coupling factors (TRCFs). Bacterial NER factors and TRCFs are simpler and better understood compared to their eukaryotic counterparts, and have recently been the subject of long-awaited structural studies that have brought insight into how they might function.
Link to transcription
Soon after it was established that recovery of RNA synthesis upon UV irradiation preceded DNA damage removal, it was first proposed that transcribed regions of the genome were repaired preferentially [5]. The preferential repair of the template strand [read by RNA polymerase (RNAP)], or TCR, was first uncovered in eukaryotes [6], and only later in bacteria [7]. TCR was reported to result in a 10-fold higher rate of repair [5], but the relative increases in repair rates are likely to be organism-specific and highly variable. Eukaryotic TCR is considerably more complex and less understood, and has been the subject of recent reviews [2,8,9]. In Archaea, TCR has not been detected [10,11], and no homologs of known TRCFs, such as bacterial Mfd, Saccharomyces cerevisiae Rad26, or mammalian Cockayne syndrome proteins A and B (CSA/CSB) have been uncovered. Bacterial TRCFs have been subjected to structural studies, and much of the existing mechanistic understanding of TCR has been derived from the Mfd-dependent system described below.
Mfd: structure and function
Mfd belongs to the DExH/D family of SF2 ATPases (see Glossary) and is akin to chromatin-remodeling factors in its ability to remodel protein–DNA interfaces and inability to promote DNA strand separation. The two basic functions of Mfd are: (i) to forward translocate and ultimately dislodge the stalled RNAPs via energy-consuming translocation on dsDNA upstream of RNAP; and (ii) to recruit the Uvr(A)BC machinery via binding to UvrA. Once the UvrAB complex has been recruited, repair likely happens as in global NER (Figure 1). Mfd has a multimodular architecture, with eight domains connected by flexible linkers (Figure 2a) [12–15]. At the N terminus, domains D1–D2 resemble the homonymous domains of UvrB; D3 is a weakly conserved domain of unknown function, which has been shown by small angle X-ray scattering (SAXS) to be mobile during catalysis [16]. Between D3 and D4 (also known as the RID, RNAP-interacting domain) is a flexible linker that spans over 40 Å. The RID is a Tudor-like domain connected to the DNA- and ATP-binding translocase module (D5 and D6) via a ‘relay helix’, believed to move and communicate with other structural elements such as the ‘hook helices’ and the TRG motif. The latter elements sense the bound nucleotide, are mobile, and wrap around the relay helix (Figure 2a) [12,17]. D5 and D6 constitute a typical RecA-type ATP-hydrolyzing engine, whereas D7 appears to play important roles in the auto-inhibition of UvrA binding, ATP hydrolysis, and dsDNA translocation [16]. Overall, because of its modular architecture, Mfd appears primed for large conformational changes. Nucleotide-dependent structural changes within Mfd have indeed been observed at low resolution using SAXS [16], but high-resolution structures of alternative functional states are still lacking.
Figure 1.

Schematic representation of bacterial transcription-coupled DNA repair (TCR). Distinct modalities exist for dealing with DNA damage encountered by the transcription machinery, which can (i) release, (ii) reposition, or (iii) destroy the stalled RNA polymerase (RNAP) in order to expose the lesion to nucleotide excision repair (NER) proteins. Bacterial, Mfd-dependent TCR is an example of (i) [44], whereas eukaryotic TCR might occur by either (ii) or (iii) [8]. Mfd-dependent repair occurs through the following sequence of events: (a) elongating RNAP (green) stalls at DNA lesions in the template strand (yellow star) and might backtrack; this recruits Mfd [colored by module in cyan (D1–D3), magenta (RNAP-interacting domain; RID), brown (D5–D6), and red (D7)], which promotes forward translocation of RNAP through ATP hydrolysis by the translocase module. Annealing of the upstream edge of the transcription bubble eventually results in its collapse and ternary elongation complex (TEC) dissociation. Next, the UvrAB complex (the UvrB dimer is lime and UvrA is pink) is recruited by virtue of the unmasking of the UvrA-binding surface in D2 (cyan) by motion of D7. (b) The TCR pathway continues with formation of an UvrB–DNA preincision complex, recruitment of UvrC (red) by UvrB and dual incision of the DNA. The damage-containing oligonucleotide is then excised and removed via UvrD (orange), after which the gap is filled via the action of DNA polI (blue) and ligase (coffee). Global NER differs in the initial localization of the damage by the UvrAB complex, which does not depend on RNAP (as depicted in a), while subsequent to formation of the UvrB–DNA preincision complex (depicted in b), both global NER and TCR are thought to occur similarly. For simplicity, the UvrB preincision complex is depicted with two UvrB copies; however, given the competitive nature of UvrB/Mfd binding to UvrA [16,44], that remains a matter of debate.
Figure 2.

Mfd structure and selected structural interfaces involved in transcription-coupled DNA repair (TCR). (a) Domain organization and crystal structure of Escherichia coli Mfd (PDB ID 2EYQ) seen from the top (left) and sideways (right). The molecule adopts a modular architecture with functional modules connected by flexible loops and hinges [12], shown as black tubes. Within the RNA polymerase (RNAP)-interacting domain (RID), substitution of L499 (spheres) compromises TCR in vivo and in vitro due to a defect of this variant in binding to RNAP [12]. Also shown in spheres are A167 and G1051, which are substituted for cysteine in order to create an intramolecular disulfide tethering D2 to D7 and resulting in masking of the UvrA binding surface [seen in (e)] located at the interface between D2 and D7 [16]. An asterisk indicates the ATP binding site and mobile structural elements presumed to change conformation during the mechanochemical cycle are labeled. Adapted from [12]. (b) Crystal structure of Thermus thermophilus ternary elongation complex assembled on a synthetic nucleic acid scaffold (PDB ID 2O5I) color coded by subunit as follows: β (cyan), β′ (pink), α (magenta and yellow), ω (olive), nascent RNA (blue), template dsDNA (orange). The crab-claw-shaped enzyme is positioned such that the downstream DNA is shown going into the page. Shown in teal is the β1 fragment that associates with the RID domain of Mfd [19]. The IKE sequence motif within the β1 fragment (initially identified in E. coli as I117 K118 E119) [29] and corresponding to Thermus aquaticus and T. thermophilus I108 K109 E110 [28]) is important for the Mfd–RNAP interaction and is shown as red spheres. (c) Close-up view of the hybrid T. thermophilus Mfd–RID and T. aquaticus RNAP-β1 fragment (PDB ID 3MLQ) [28]. The interaction is bipartite: the core interaction comprises evolutionarily conserved residues located on the edge β-strands and forming an intermolecular β-sheet; a second, phylum-specific peripheral interaction is mediated by R341 (corresponding to E. coli L499), and contacts the conserved IKE motif (red) and phylum-specific Q99. (d) Close-up view of the Mfd D2–D7 interaction inhibitory for UvrA binding. Conserved D7 residues are shown in black; D2 residues that support the interaction are shown in lime. Substitutions of orange residues are functionally important for TCR in vivo and in vitro [54]. Reproduced from [16]. (e) Close-up of the transcription–repair coupling factor (TRCF)–UvrA interface. Residues in UvrA that bind UvrB are shown in blue [59]. Other residues are colored as in (d). Reproduced from [16].
Escherichia coli Mfd has a single ATP-binding site located between D5 and D6 [12]. In E. coli, nucleotide binding does not modulate oligomerization as reported in other species [18], because Mfd remains monomeric irrespective of nucleotide status [16]. Recently, SAXS studies have demonstrated that ADP/ATP binding reorganizes multiple interdomain contacts [16], but does not lead to a large-scale unraveling of the structure as proposed previously [12]. It is thought that binding to a stalled ternary elongation complex (TEC) may trigger structural rearrangements that stimulate several Mfd functions, including ATP hydrolysis, DNA translocation, and possibly, lesion binding [16]. The former likely promotes destabilization of the TEC, whereas the latter might be important for the precise targeting of UvrAB. These multiple distinct structural rearrangements during the TRCF functional cycle are likely key to understanding the tight temporal and contextual regulation of TRCF activities.
TEC recognition and TCR initiation
What triggers TCR? It has generally been assumed that an RNAP that is stalled at a DNA lesion recruits TRCFs, which in turn recruit the NER machinery. Indeed, it has been established that TCR requires active transcription by RNAP [8], but it remains unknown what features of the stalled TEC are specifically recognized. These features might not be universally conserved, especially considering that prokaryotic and eukaryotic TRCFs share little sequence similarity outside of their ATP-binding domains and differ in several respects. Although both Mfd [19] and CSB are able to push a stalled RNAP forward, CSB does not induce RNAPII release [20].
It has been proposed that RNAP backtracking, which accompanies RNAP stalling at obstacles ranging from DNA lesions to DNA-bound proteins [21,22], might play a defining role in TEC recognition by TRCFs [12,23]. Mfd is active at class II pause sites where RNAP is prone to backtracking, but not at hairpin-dependent class I pause sites, where no backtracking occurs [12]. However, TECs that are transiently paused at some lesions, such as abasic sites [24], or are stalled by nucleotide deprivation [19], are not backtracked, and yet they are also recognized and displaced by Mfd [24]. Crystallographic analyses of yeast CPD-bound RNAPII (no structural models of similar bacterial TECs are available) did not reveal major conformational differences from an active RNAPII [23], raising questions about how (and if) TRCFs discriminate active from stalled TECs. The caveat of this structural study is the lack of the non-template DNA strand and upstream DNA in the crystallized TEC, which likely constitute important recognition elements for Mfd [19].
If damage-stalled RNAPs resemble active RNAPs, how do TRCFs find the right targets? A simple kinetic model is that Mfd could only displace RNAP stalled for a sufficiently long time, no matter what the reason. This model is supported by reports that Mfd stochastically releases RNAPs stalled by nucleoside triphosphate (NTP) limitation. In this model, an apparent resistance of some TECs to Mfd could be explained by their excessive stability; if the destabilization effect of Mfd were modest it would not produce a measurable effect (Box 1). Alternatively, this resistance could be due to conformational changes in the TEC, for example, upon hairpin formation [25], that prevent Mfd binding or action. Although comparative analysis of many different TECs would be required to address this question, the observation that potentiating the DNA translocation activity of Mfd allows it to release (normally resistant) transcription initiation complexes [26], supports a simple model of Mfd action.
Box 1. Intrinsic and factor-dependent termination.
In the TEC, the energetically costly maintenance of the ~17-nt bubble is balanced by two favorable contributions: the formation of the RNA:DNA hybrid, and RNAP interactions with the nucleic acid (NA) chains. The free energy of the TEC can be described as: ΔGTEC = ΔGbubble + ΔGRNA:DNA hybrid + ΔGRNAP:NA [64,65]; hence, the complex could be destabilized by extending the bubble, shortening the hybrid, or disrupting RNAP/NA contacts. The RNA:DNA hybrid is the major stability determinant, but RNAP interactions with the nascent RNA and with the duplex DNA in the downstream channel also make significant contributions to TEC stability (see [66] and references therein). In the absence of a roadblock, the decision between elongation and termination pathways is thought to be kinetically controlled, and a dramatic destabilization of the TEC is required to bring its normal dwell time (>105 s) into the range characteristic for nucleotide addition (<10−1 s). Yager and von Hippel have argued that at most template positions the barrier to termination exceeds that to elongation by ~18 kcal/mol [64]. Recent studies have suggested that this barrier might be lower because termination competes with RNA extension in paused, rather than in rapidly elongating, complexes [66].
E. coli RNAP is released by the intrinsic terminator or the Rho helicase. Three models of termination have been proposed (reviewed in [66]). In the hybrid shearing model, the upstream portion of the hybrid is disrupted by hairpin formation or Rho. In the allosteric model, changes in the active site cleft induced by RNAP contacts to the hairpin or Rho weaken interactions with the hybrid. In the hypertranslocation model, RNAP is pushed forward without nucleotide addition, extending the bubble and leaving the hybrid behind. Termination by a single-subunit T7 RNAP was argued to follow either the shearing or the hypertranslocation pathway [67,68]. For E. coli RNAP, bulk biochemical data that support all three mechanisms are available [37–39,69] and single-molecule studies have revealed that although termination at intrinsic signals with weak rU:dA hybrids proceeds via shearing, release at terminators that have imperfect U-runs (and thus more stable hybrids) may be accompanied by hypertranslocation [70]. Therefore, the mechanisms of RNA release can be different even in the simplest case of hairpin-induced termination. It is likely that alternative pathways may be utilized by accessory factors. Although disruption of the hybrid may be the most effective trigger for dissociation, proteins that act on stalled TECs face a less arduous task and might target less crucial interactions while still enabling RNAP removal within the physiologically relevant time window. For example, a 3.5 kcal/mol decrease in the termination barrier would be expected to facilitate TEC dissociation 300-fold; an effect observed in vitro in the presence of Mfd [16].
Despite significant efforts, the field is still lacking a structural model of Mfd (or any other TRCF) bound to a stalled TEC. The limited knowledge that we do have comes from low-resolution domain mapping [27], two-hybrid screens [19], saturation mutagenesis [12], and a crystal structure of a minimal thermophilic Mfd–RNAP complex consisting of an N-terminal fragment of the β subunit (β1) and RID of Mfd [28]. Together with subsequent in vivo and in vitro analyses of E. coli Mfd and RNAP mutants that are defective in Mfd binding, these studies have highlighted the crucial role of the so-called IKE sequence motif in the β1-region and of L499 in Mfd (Figure 2b,c) [12,29]. The interaction is bipartite, comprising a central interaction, conserved across phyla, and a phylum-specific peripheral interaction (Figure 2c) [28]. Curiously, a small register shift of residues 103–111, the β1 region that harbors the key IKE motif in Thermus aquaticus, was observed in the RID-β1 crystal structure [28]. Although this shift has not been observed in any of the many RNAP structures available, it has been suggested that β1 might exist in a dynamic equilibrium of multiple states and that, rather than inducing it, Mfd traps and stabilizes the shifted state [28]. In this scenario, the operative mechanism for recognition might be conformational selection rather than induced fit.
Although it is not clear if this peripheral structural change can serve as an allosteric release signal, Mfd/TEC interaction constitutes more than a simple binding event. The TEC appears to trigger conformational changes in Mfd that stimulate ATP hydrolysis in a manner reminiscent of chromatin remodeling factors, which are often stimulated preferentially by nucleosome substrate over naked DNA [30]. In fact, Mfd is unusual among SF2 DNA translocases (stimulated by DNA), in that dsDNA has a negligible effect on ATP hydrolysis [14,31]. ATPase stimulation (and consequent translocation on dsDNA) can only be achieved with the physiological substrate, the TEC [16,31], and does not appear to require a separation in space between D2 and D7 [16], as previously proposed [14]. An oxidized TRCF variant in which D2 and D7 are linked via a disulfide bridge has almost wild-type RNAP release activity and exhibits ATPase stimulation by TECs, although it is greatly impaired in dsDNA binding and (non-stimulated) ATP turnover [16]. This suggests that the previously characterized hyperactive variants lacking D1–D3 or D7, which are capable of translocating on naked DNA, might not necessarily mimic the TEC-bound state as suggested [14,31], and that translocase activation might occur differently in truncated Mfd (measured on naked DNA via triplex-destabilizing assays) compared to full-length TEC-bound Mfd [14,31]. These studies, forming the basis of the ‘clamp model’ (discussed later) [14], were carried out in the absence of TEC/UvrA and might not reflect physiological conditions. Although the structure of the minimal TRCF–RNAP β1 complex clearly confirms existing biochemistry and genetics, it nevertheless remains rather limited in bringing insights into how the TEC may impact the structure and function of full-length TRCF.
TEC destabilization via the dsDNA motor activity of TRCF
TEC release is no easy feat; the TEC is one of the most stable known protein–nucleic acid complexes, with a half-life on the order of hours to days in vitro [16,32,33], and TEC dissociation requires massive destabilization of a network of contacts among RNAP and the nucleic acid chains (Box 1). It should be noted that release of the stalled TEC is essential for TCR and also for several DNA-repair-independent functions of Mfd, greatly extending the role of the factor. These functions are all based on the removal of RNAP stalled by protein roadblocks, which can include transcriptional regulators [34,35], but also the replication machinery in those cases in which the replication fork collides head-on with transcribing RNAPs [36].
Several, not necessarily mutually exclusive models have been put forth to explain the mechanism of TEC dissociation by a termination hairpin or an accessory protein, such as Rho (Box 1), which unlike Mfd, exerts its effects by translocating on the nascent RNA (Figure 3a, top). However, for Mfd, only data supporting the forward translocation model have been reported so far [19,37]. According to this model, when a TEC is stalled by NTP deprivation in vitro (or by a DNA lesion in the cell), the rotational motion of Mfd tracking on the DNA is believed to be converted to torque on the upstream DNA (Figure 3a, bottom). It is thought that this torque causes bubble collapse through reannealing of the upstream edge of the bubble and coordinated unwinding of the RNA–DNA hybrid; the key stability determinant of the TEC [38]. This mechanical model for Mfd- (and Rho-)mediated termination is supported by observations that neither protein can efficiently dissociate heteroduplex TECs (as monitored by RNA release) in which bubble collapse is prevented by a mismatch at the upstream edge of the transcription bubble [37]. However, a hyperactive Mfd variant destabilizes promoter complexes lacking the RNA:DNA hybrid, therefore, hybrid unwinding appears not to be essential for release [26].
Figure 3.

Forward translocation in enzymatic transcription termination and structure of the UvrAB machinery. (a) Schematic representation of the forward translocation model of transcription termination mediated by Rho (top, blue) and Mfd (bottom, magenta). Arrows indicate the relative motions of RNA polymerase (RNAP; green arrow) and nucleic acid (red for RNA, black for dsDNA). Rectangle indicates the location of the RNAP active site. Mfd [with translocase (T) domains and RNAP-interacting domain (RID)] is shown as acting on a backtracked RNAP, in which the 3′-end of the nascent RNA is extruded. Adapted from [71]. (b) The continued pushing by Mfd rescues the backtracked RNAP into productive chain elongation (top) or, in the presence of DNA damage, induces transcription bubble collapse and partial unraveling of the RNA–DNA hybrid (bottom) [19,37]. (c) Superposition of UvrB-homology module of Escherichia coli transcription–repair coupling factor (TRCF) (with D1a in navy, D2 in cyan and D1b in blue) and the homologous region of Bacillus caldotenax UvrB (gray, PDB ID 2FDC). UvrB-bound DNA is colored in lime. Mfd lacks the conserved β-hairpin (fuchsia), which in UvrB is critical for binding the DNA lesion [56,72]. (d) Architecture of the UvrA2B2 (PDB ID 3UWX). Surfaces involved in DNA binding are highlighted in blue, and the approximate location of the DNA lesion is indicated by an asterisk. (e) UvrA monomer (PDB ID 2R6F) colored by domain: ATP-binding domain I (red), UvrB-binding domain (yellow), insertion domain (green), signature domain I (pink), signature domain II (teal) and ATP-binding domain II (navy). The two ATPase sites are labeled. (f) Conformational changes in the UvrA dimer (PDB ID 3UWX and 2R6F) underlying DNA binding and lesion recognition [51]. The proposed path of the DNA helix is indicated by a lime circle, and selected residues involved in DNA binding and undergoing large motions are indicated as blue spheres.
Although mechanical forces clearly play a defining role in termination, conformational changes within the stalled RNAP induced by a termination factor may also contribute to RNAP release. It is possible that Rho and Mfd might utilize direct interactions with RNAP to destabilize the TEC [12,39]. In the case of Rho, the interaction surface is unknown, but the location of key Mfd RID/β1 contacts (Figure 2b) has led to a model in which remodeling of the TEC by Mfd can induce an opening motion of the RNAP pincers and subsequent release of the nucleic acid chains [29]. This model is consistent with the key role of the RNAP clamp in TEC processivity and stability. The closed clamp is thought to correspond to rapidly moving RNAP, whereas opening of the clamp would favor TEC isomerization into a paused state, and perhaps termination [40,41]. The β N-terminal domain might be a common target for regulators that control clamp movements and thus RNAP processivity [40,41], whereas the RID fold appears to define a more general platform for interactions with RNAP, such as by the large family of CarD regulators of rRNA transcription [42].
Importantly, specific contacts with RNAP appear dispensable for Mfd-mediated release: TCR has also been observed for genes transcribed by T7 RNAP [43]. It is likely that the IKE/RID contacts, together with other, yet unknown interactions, trigger a conformational change in Mfd that unmasks its cryptic motor activity, which would be deleterious to the cell if left unregulated. Notably, overexpressed Mfd and hyperactive Mfd variants do not appear to be toxic to E. coli [26]. Furthermore, disruption of the hook–relay interface (Figure 2a) by a W550A substitution allows Mfd to displace RNAP lacking the IKE motif [26]. Thus, the IKE/RID contacts might merely contribute processivity when the translocase domains disengage the dsDNA, whereas other hypothetical interactions with RNAP might be more tightly coupled to dsDNA translocation. An experimentally based model of an Mfd-bound TEC will be required to understand Mfd–TEC interactions and whether Mfd-mediated TEC destabilization shares (or not) features of the several models of transcription termination available.
Recruitment of the Uvr(A)BC NER machinery
As early as the 1990s, it was observed that Mfd and UvrB binding to UvrA are mutually exclusive [44]. In light of the homology in the D2 region and in vitro studies with Mfd truncations [27], it was proposed that Mfd binding to UvrA resembles UvrB binding to UvrA. We now know that the structural similarity between Mfd and UvrB extends over a much longer region of Mfd comprising D1–D2 (Figure 3c) [12]. A recent X-ray crystal structure of a core UvrA–Mfd complex [16] established that, indeed, Mfd and UvrB share the same mode of UvrA recognition. Notably, binding of UvrA residues 131–250 occurs on the D2 face that packs against the C-terminal D7 domain via residues that are conserved both within and across the Mfd and UvrB families (Figure 2d,e). This autoinhibition of UvrA binding via D2–D7 contacts was initially observed crystallographically [12,34], and recently by SAXS under near-physiological conditions [16], establishing that it is of biological relevance.
These studies concluded that, at least in its nucleotide-free state, Mfd is autoinhibited with respect to UvrA binding. Both in vivo and in vitro studies lend support to this conclusion [12,27,45]. However, binding to UvrA was directly detected in pull-down assays in the absence and presence of nucleotides [16,27]. Therefore, TRCF must exist in a dynamic equilibrium of conformations, at least one of which supports UvrA binding (termed the open conformation due to the swinging motion of D7 required for accommodating UvrA). This conformation is populated even in the absence of nucleotides, but ATP and UvrA binding shift the conformational equilibrium towards the open structure [16]. Could ATP binding be the trigger for UvrA recruitment? This seems unlikely for several reasons. (i) If true, UvrA would be bound by Mfd even when not engaged with RNAP and committed to TCR, significantly lowering the available UvrA pool in the cell. TRCF is expressed at relatively high levels in E. coli, whereas UvrA levels are normally low, and only increase 10-fold upon induction of the SOS response [1]; a C-terminal truncation lacking D7 confers UV sensitivity to cells [27], suggesting that in vivo and in the full-length protein, UvrA binding is normally masked. (ii) Mfd participates in processes that rely on RNAP removal (requiring ATP binding and hydrolysis) [35,36,46–48], in which recruitment of UvrAB and subsequent DNA excision is dispensable, if not detrimental. (iii) SAXS reconstructions of ATP-bound Mfd reveal a repositioning of D7 that might increase the solvent exposure of the UvrA binding surface, but would likely be insufficient to abolish the clash between Mfd and full-length UvrA. Several crystal structures of UvrA/B and UvrAB (Figure 3) are now available [49–53] for construction of molecular models.
The stalled RNAP might not trigger the changes required for UvrA binding either, because a TRCF variant carrying a D2–D7 interdomain disulfide (which would prevent UvrA binding to D2) has an RNAP-release activity comparable to wild type [16]. This is in contrast to earlier studies with Mfd truncations lacking D7 or D1–D3, which were found to be hyperactive and derepressed (with respect to DNA translocation), suggesting that Mfd action is based on a single conformational switch in which the D2–D7 interface is broken to enable synchronous RNAP binding, translocase activation, and UvrA recruitment [14,31]. At the structural level, this ‘clamp model’ suggests that Mfd might be restrained in an inactive conformation by the UvrB-homology module that clamps onto the C-terminal region of Mfd [14]. Furthermore, later studies have proposed that the regulation of UvrA binding is inconsequential [54]. In other words, the point at which UvrA is recruited is not important. This model was based on the use of deregulated TRCF variants carrying mutations in D7 that break the evolutionarily conserved D2/D7 interactions, but do not impair TCR in vitro [54]. However, a completely deregulated system, which would bind UvrA indiscriminately, is expected to undermine the DNA-repair-independent functions of TRCF in the cell.
How and when is inhibition of UvrA binding relieved in TRCF? To reconcile the ability of the D2–D7 crosslinked variant to release transcript from the TEC, it was recently suggested that the trigger for UvrA binding might occur late in the pathway, during or even subsequent to RNAP release, and possibly involving the DNA lesion [16]. TEC destabilization is a complex process that is not well understood and release of the RNA could precede removal of RNAP from DNA. During forward translocation of the TEC, and even once its destabilization commences, Mfd might maintain contacts with the enzyme and DNA, allowing it to ‘slide’ on the DNA, engage the lesion or the proximal DNA, then reposition D7 (and possibly the UvrB-homology module or D2) for UvrA recruitment to occur (Figure 1). Binding of Mfd to damaged DNA has not been studied, and the commonly adopted view of DNA lesion detection in TCR is ‘by proxy’, via TRCF interaction with RNAP rather than the lesion. Although DNA damage is buried under the RNAP footprint at an early stage, the lesion could become accessible to Mfd subsequent to TEC remodeling and possibly release. Strand-specific and global NER differ in the requirements for DNA damage discrimination in that the damage-recognition function of UvrA is less important in TCR (Box 2) [54]. This suggests that, in addition to RNAP, Mfd might also sense DNA lesions (likely via distortions in the DNA geometry), possibly collaboratively with the other TCR components. Other DNA–protein contacts outside of D5/D6 might exist in Mfd, which could potentially mediate DNA lesion detection. For example, the D2–D7 crosslinked mutant has a drastically reduced affinity for DNA, yet it can release TECs [16]. Such interactions with DNA might be transient, could possibly contribute to damage recognition rather than high-affinity binding, and could be difficult to detect. This might explain why Selby and Sancar only detected dsDNA binding to D5 and D6 [27]. Furthermore, this preferential binding, if any, might only be uncovered in a system containing RNAP on damaged transcription bubble mimics.
Box 2. Structure and dynamics of the UvrAB machinery.
Structural studies of UvrA/UvrB in isolation [49,50,52,53,57,73] and in complex with each other [51,59] have revealed their architecture (see Figure 3c–f in main text) and enabled generation of a mechanistic model of how the UvrAB complex scans for damage, rearranges, and finally disassembles to leave a tight preincision complex that recruits UvrC and initiates the excision process (Figure I).
Crystallized UvrAB has a 2:2 stoichiometry and assumes a ‘closed groove’ conformation with a central narrow and deep groove that accommodates about 45 bp of B-form DNA, out of which, 32 bp are contained within the UvrA dimer (see Figure 3d in main text). Notably, UvrB is located at the periphery of the complex, and UvrA2 assumes a closed conformation that cannot accommodate DNA lesions, unlike the ‘open tray conformation’ observed in the isolated UvrA2 dimer (see Figure 3f in main text). This UvrA conformation can bind to damaged DNA. Notably, the two conformers are held in place by different interfaces, and UvrA–UvrB contacts include, in addition to a previously known contact patch, Signature Domain II (SDII), part of the proximal ATPase site.
It is notable that UvrA2B2 contains six ATP binding sites, which are all occupied in the crystal structure. Yet, the unambiguous identification of nucleotides in each site is not possible due to limited resolution. Comparison with existing higher resolution structures of UvrA bound to nucleotides [51] suggests that the machinery may undergo cycles of narrowing and widening of the DNA-binding groove caused by NTP dynamics while scanning the genome for lesions. In TCR, due to the ‘by proxy’ lesion detection via RNAP, the proximal ATPase site, the insertion domain and the DNA damage recognition functions of UvrA (see Figure 3e in main text) appear to be less important [54]. Once a lesion is located, SDII might rotate upon ATP hydrolysis at the proximal site and could cause eviction of UvrA from the UvrA2B2–lesion complex because SDII contacts UvrB. Simple geometrical considerations reveal that the two UvrB molecules left behind after UvrA departure are located far away, about 80 Å from the expected position of the lesion. This implies that UvrB needs to translocate towards the lesion by virtue of its 5′ → 3′ helicase activity, and that then once at the lesion, the two UvrB molecules may converge via dimerization of their C-terminal domains to then recruit UvrC (Figure I) [51]. The sequence of events leading to preincision complex formation in TCR remains largely unknown. However, the ATPase activity of UvrB as well as its ability to discriminate damage and locally separate DNA strands via its β-hairpin are required for both global NER and TCR [54].
In this context, it is interesting to consider the other domains of the UvrB homology module (D1a and D1b). These have been somewhat neglected and their functional involvement remains obscure. No ATP or DNA binding has been detected [13], although this module contains the region corresponding to the DNA and ATP binding sites of UvrB [55]. In E. coli Mfd, the ATP-binding site is degenerate and nonfunctional, and residues crucial for damage recognition and DNA melting are missing. In UvrB, these residues form a β-hairpin, which inserts itself in between the DNA strands to open up the helix locally [56,57]. In Mfd, this β-hairpin is replaced by a loop (Figure 3c), which might reflect different requirements for the recognition of local DNA distortions around the lesion. The lesion itself might be transiently presented in a bubble (previously opened up by RNAP) rather than in double-stranded form as in global NER. Nevertheless, the helix-destabilizing function of UvrB (compromised by the Y101A F108A substitutions, Figure 3c) is required for efficient TCR and NER [54].
In cells, UvrA exists mostly as a UvrAB complex with complicated dynamics (Box 2), and several studies, including a recent crystallographic study, have suggested that the complex has a 2:2 UvrA:UvrB [51,58] rather than a 2:1 stoichiometry as originally proposed [3,59]. This could have important mechanistic consequences. Atomic force microscopy has suggested that the presence of two UvrB copies ensures alternative DNA wrapping and probing for damage in both DNA strands [58]. TCR circumvents the probing for damage in the non-template strand, and thus Mfd might compete out one UvrB, while leaving the second one in place for probing of the template strand and formation of the UvrB–DNA preincision complex.
Of all the mechanisms for UvrA recruitment, the one based on recruitment after RNAP release [16] allows for the most precise and coordinated recruitment of the NER machinery. Validation of this model will require detection of the expected coupling intermediates (e.g., Mfd–DNA–UvrAB versus TEC–Mfd–UvrAB), which has been attempted unsuccessfully in the past [20]. Nevertheless, Mfd does not appear to be unique in its ability to target UvrAB to DNA damage; novel factors have recently been identified that recruit NER machinery to lesions via direct binding to UvrA (Box 3) [60,61]. Therefore, it appears that damage recognition in NER resorts to accessory factors that target lesions of various structures, different genomic contexts, and can even regulate the choice between global NER and TCR [62].
Box 3. Alternative modalities of recruiting NER enzymes.
After identification of Mfd as a transcription–repair coupling factor [74], a question that persisted was why the mfd mutant is only slightly sensitive to UV radiation. The general belief is that in bacteria TCR might not be as important because global NER is more efficient, and possibly also because eukaryotic genes require more time to be transcribed, increasing the probability of stalling. By uncovering a novel TCR mechanism, recent studies [61] have prompted reconsideration. The new key player is NusA, a protein that binds RNAP, and is well known for its roles in transcription termination, pausing, and antitermination.
Both in vivo and in vitro, NusA-mediated TCR appears to operate on chemically-induced DNA damage such as nitrofurazone-induced adducts [61]. This distinction has interesting mechanistic implications; these adducts do not enter the RNAP active site, but remain partially exposed because transcription is stalled four nucleotides upstream from the lesion. RNAP backtracking could also occur, although this has not been established for the N2-furfuryl-dG lesion in question. This mechanism is Mfd independent, but also relies on recruiting the NER machinery via UvrA binding. Indeed, if the adducts are exposed and not buried under RNAP, it can be envisioned that UvrAB might be able to recognize and load even in the presence of TECs bound upstream. Therefore, Mfd-dependent processes (e.g., release of RNAP) might not be required, and this conjecture is supported by epistasis analysis [61]. A question that remains unanswered is how NusA-dependent TCR is triggered. Is NusA preferentially recruited to the stalled RNAP, or does the pathway use a prebound NusA? Given the known dynamic association of NusA with elongating RNAPs [75], the latter scenario appears more likely.
Yet another mechanism for targeting Uvr(A)BC to DNA lesions (not necessarily located in the transcribed strand) has recently been discovered [60]. This involves YbaZ, an alkyltransferase-like (ATL) protein. Unlike AT proteins, ATL proteins do not serve as suicidal traps for the adduct because they lack the alkyl receptor Cys residue. By contrast, they enhance the repair of O6-alkylguanine adducts by direct binding of the lesion and UvrA [60,76]. In fact, ATL proteins such as YbaZ utilize DNA bending and a rare nonenzymatic nucleotide flipping mechanism to channel specific, weakly distorting base damage into the NER pathway [77]. Despite recent structural studies [77,78], the molecular details of recruitment of the NER machinery by YbaZ await elucidation.
Figure I.
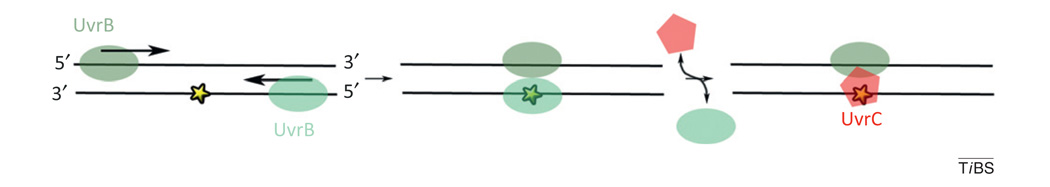
Hypothetical model for UvrB loading during nucleotide excision repair. After UvrA departure, UvrB remains deposited on the DNA away from the lesion (yellow star). UvrB then translocates towards and dimerizes at the damaged site. UvrC then replaces one of the UvrB copies to initiate excision. Adapted from [51].
Concluding remarks
A substantial body of knowledge has accumulated since the isolation of the mfd mutant [63]. Extensive genetic, biochemical, and structural data are now available to test more refined TCR models. The missing piece remains an experiment-based model for Mfd bound to a stalled TEC. This would clarify aspects of TEC recognition, dsDNA translocation and RNAP release. The pathway for Mfd-mediated termination will likely depend on the nucleic acid context, the rates of RNA chain extension, and factor translocation, and might combine features of several termination mechanisms proposed so far. Furthermore, complementary studies, such as at the level of single molecules, might provide essential kinetic information about the rates of the different steps in TCR, which remain unknown, as well as insight into the ordered assembly (and disassembly) of the different intermediates. NER machinery recruitment is emerging as a more complex process than initially thought, with multiple factors targeting the NER assembly to specific classes of DNA lesions. NER targeting and damage detection mechanisms will hence remain a fertile area of investigation.
Glossary
- Backtracking
the backward sliding of RNAP on the template (such as upon encountering a DNA lesion, such as a CPD, or a regulatory pause site) that results in extrusion of the 3′-OH of the nascent RNA from the active site and transcriptional arrest. Backtracked RNAPs are rescued by transcript cleavage factors (GreA/GreB, TFIIS in eukaryotes) that stimulate the intrinsic endonucleolytic activity of RNAP to trim the extruded RNA and restore its reactive alignment in the catalytic center.
- DExH/D ATPases
a subset of SF2 ATPases that are characterized by the DExH/D sequence motif, many of which are involved in chromatin remodeling and RNA metabolism.
- DNA alkyltransferase (AT)
a class of proteins (including the well-characterized Ada protein) that repair alkylating damage within DNA, for example the highly mutagenic O6-methylguanine adduct. In this case, ATs transfer the methyl group to a conserved Cys residue within the protein, thus serving as suicidal stoichiometric agents rather than enzymes.
- Intrinsic termination
does not require any accessory proteins and is mediated by the formation of an RNA hairpin followed by a U-rich region in the nascent transcript, which slows down and destabilizes the TEC.
- Pausing
a transcriptional process in which RNAP halts temporarily at a regulatory site. At class I sites, the nascent stable RNA hairpin stabilizes an inactivated enzyme intermediate. At class II sites, a weak RNA–DNA hybrid favors RNAP backtracking.
- Rho
a bacterial transcription termination factor that is essential for RNA surveillance. It is an ATP-dependent RNA translocase that binds to untranslated mRNAs and releases them from RNAP. Similarly to Mfd, Rho might trigger forward translocation of RNAP.
- SF2 ATPase
ATP-hydrolyzing enzyme belonging to superfamily 2 of ATPases, such as chromatin remodeling factors. Many SF2 ATPases are stimulated by nucleic acids or nucleoprotein complexes, such as nucleosomes, and are devoid of helicase activity but are able to translocate on dsDNA.
- SOS response
a global response to DNA damage, first identified by Miroslav Radman in 1975, in which error-prone DNA repair and mutagenesis are induced. SOS response is mediated by the RecA protein (Rad51 in eukaryotes), which detects accumulating amounts of ssDNA and inactivates the LexA repressor, thus upregulating LexA-controlled genes, including NER genes, such as uvrA, uvrB, and uvrD.
References
- 1.Reardon JT, Sancar A. Nucleotide excision repair. Prog. Nucleic Acid Res. Mol. Biol. 2005;79:183–235. doi: 10.1016/S0079-6603(04)79004-2. [DOI] [PubMed] [Google Scholar]
- 2.Nouspikel T. DNA repair in mammalian cells: nucleotide excision repair: variations on versatility. Cell. Mol. Life Sci. 2009;66:994–1009. doi: 10.1007/s00018-009-8737-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Selby CP, Sancar A. Structure and function of the (A)BC excinuclease of Escherichia coli. Mutat. Res. 1990;236:203–211. doi: 10.1016/0921-8777(90)90005-p. [DOI] [PubMed] [Google Scholar]
- 4.Truglio JJ, et al. Prokaryotic nucleotide excision repair: the UvrABC system. Chem. Rev. 2006;106:233–252. doi: 10.1021/cr040471u. [DOI] [PubMed] [Google Scholar]
- 5.Bohr VA, et al. DNA repair in an active gene: removal of pyrimidine dimers from the DHFR gene of CHO cells is much more efficient than in the genome overall. Cell. 1985;40:359–369. doi: 10.1016/0092-8674(85)90150-3. [DOI] [PubMed] [Google Scholar]
- 6.Mellon I, et al. Selective removal of transcription-blocking DNA damage from the transcribed strand of the mammalian DHFR gene. Cell. 1987;51:241–249. doi: 10.1016/0092-8674(87)90151-6. [DOI] [PubMed] [Google Scholar]
- 7.Mellon I, Hanawalt PC. Induction of the Escherichia coli lactose operon selectively increases repair of its transcribed DNA strand. Nature. 1989;342:95–98. doi: 10.1038/342095a0. [DOI] [PubMed] [Google Scholar]
- 8.Hanawalt PC, Spivak G. Transcription-coupled DNA repair: two decades of progress and surprises. Nat. Rev. Mol. Cell Biol. 2008;9:958–970. doi: 10.1038/nrm2549. [DOI] [PubMed] [Google Scholar]
- 9.Tornaletti S. DNA repair in mammalian cells: transcription-coupled DNA repair: directing your effort where it’s most needed. Cell. Mol. Life Sci. 2009;66:1010–1020. doi: 10.1007/s00018-009-8738-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dorazi R, et al. Equal rates of repair of DNA photoproducts in transcribed and non-transcribed strands in Sulfolobus solfataricus. Mol. Microbiol. 2007;63:521–529. doi: 10.1111/j.1365-2958.2006.05516.x. [DOI] [PubMed] [Google Scholar]
- 11.Romano V, et al. Lack of strand-specific repair of UV-induced DNA lesions in three genes of the archaeon Sulfolobus solfataricus. J. Mol. Biol. 2007;365:921–929. doi: 10.1016/j.jmb.2006.10.045. [DOI] [PubMed] [Google Scholar]
- 12.Deaconescu AM, et al. Structural basis for bacterial transcription-coupled DNA repair. Cell. 2006;124:507–520. doi: 10.1016/j.cell.2005.11.045. [DOI] [PubMed] [Google Scholar]
- 13.Assenmacher N, et al. Structural basis for transcription-coupled repair: the N terminus of Mfd resembles UvrB with degenerate ATPase motifs. J. Mol. Biol. 2006;355:675–683. doi: 10.1016/j.jmb.2005.10.033. [DOI] [PubMed] [Google Scholar]
- 14.Murphy MN, et al. An N-terminal clamp restrains the motor domains of the bacterial transcription-repair coupling factor Mfd. Nucleic Acids Res. 2009;37:6042–6053. doi: 10.1093/nar/gkp680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Deaconescu AM, Darst SA. Crystallization and preliminary structure determination of Escherichia coli Mfd, the transcription-repair coupling factor. Acta Crystallogr. Sect. F: Struct. Biol. Cryst. Commun. 2005;61:1062–1064. doi: 10.1107/S1744309105035876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Deaconescu AM, et al. Nucleotide excision repair (NER) machinery recruitment by the transcription-repair coupling factor involves unmasking of a conserved intramolecular interface. Proc. Natl. Acad. Sci. U.S.A. 2012;109:3353–3358. doi: 10.1073/pnas.1115105109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chambers AL, et al. A DNA translocation motif in the bacterial transcription–repair coupling factor, Mfd. Nucleic Acids Res. 2003;31:6409–6418. doi: 10.1093/nar/gkg868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Prabha S, et al. Distinct properties of hexameric but functionally conserved Mycobacterium tuberculosis transcription-repair coupling factor. PLoS ONE. 2011;6:e19131. doi: 10.1371/journal.pone.0019131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Park J-S, et al. E. coli transcription repair coupling factor (Mfd protein) rescues arrested complexes by promoting forward translocation. Cell. 2002;109:757–767. doi: 10.1016/s0092-8674(02)00769-9. [DOI] [PubMed] [Google Scholar]
- 20.Selby CP, Sancar A. Characterization of transcription-repair coupling factors in E coli and humans. Methods Enzymol. 2003;371:300–324. doi: 10.1016/S0076-6879(03)71023-4. [DOI] [PubMed] [Google Scholar]
- 21.Dutta D, et al. Linking RNA polymerase backtracking to genome instability in E. coli. Cell. 2011;146:533–543. doi: 10.1016/j.cell.2011.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Epshtein V, Nudler E. Cooperation between RNA polymerase molecules in transcription elongation. Science. 2003;300:801–805. doi: 10.1126/science.1083219. [DOI] [PubMed] [Google Scholar]
- 23.Brueckner F, et al. CPD damage recognition by transcribing RNA polymerase II. Science. 2007;315:859–862. doi: 10.1126/science.1135400. [DOI] [PubMed] [Google Scholar]
- 24.Smith AJ, Savery NJ. Effects of the bacterial transcription-repair coupling factor during transcription of DNA containing non-bulky lesions. DNA Repair (Amst.) 2008;7:1670–1679. doi: 10.1016/j.dnarep.2008.06.020. [DOI] [PubMed] [Google Scholar]
- 25.Tagami S, et al. Crystal structure of bacterial RNA polymerase bound with a transcription inhibitor protein. Nature. 2010;468:978–982. doi: 10.1038/nature09573. [DOI] [PubMed] [Google Scholar]
- 26.Smith AJ, et al. Multipartite control of the DNA translocase, Mfd. Nucleic Acids Res. 2012 doi: 10.1093/nar/gks775. http://dx.doi.org/10.1093/nar/gks775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Selby CP, Sancar A. Structure and function of transcription-repair coupling factor. I. Structural domains and binding properties. J. Biol. Chem. 1995;270:4882–4889. doi: 10.1074/jbc.270.9.4882. [DOI] [PubMed] [Google Scholar]
- 28.Westblade LF, et al. Structural basis for the bacterial transcription-repair coupling factor/RNA polymerase interaction. Nucleic Acids Res. 2010;38:8357–8369. doi: 10.1093/nar/gkq692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Smith AJ, Savery NJ. RNA polymerase mutants defective in the initiation of transcription-coupled DNA repair. Nucleic Acids Res. 2005;33:755–764. doi: 10.1093/nar/gki225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hauk G, et al. The chromodomains of the Chd1 chromatin remodeler regulate DNA access to the ATPase motor. Mol. Cell. 2010;39:711–723. doi: 10.1016/j.molcel.2010.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Smith AJ, et al. Controlling the motor activity of a transcription-repair coupling factor: autoinhibition and the role of RNA polymerase. Nucleic Acids Res. 2007;35:1802–1811. doi: 10.1093/nar/gkm019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Selby CP, et al. RNA polymerase II stalled at a thymine dimer: footprint and effect on excision repair. Nucleic Acids Res. 1997;25:787–793. doi: 10.1093/nar/25.4.787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rhodes G, Chamberlin MJ. Ribonucleic acid chain elongation by Escherichia coli ribonucleic acid polymerase. I. Isolation of ternary complexes and the kinetics of elongation. J. Biol. Chem. 1974;249:6675–6683. [PubMed] [Google Scholar]
- 34.Deaconescu AM, et al. The bacterial transcription repair coupling factor. Curr. Opin. Struct. Biol. 2007;17:96–102. doi: 10.1016/j.sbi.2007.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Belitsky BR, Sonenshein AL. Roadblock repression of transcription by Bacillus subtilis CodY. J. Mol. Biol. 2011;411:729–743. doi: 10.1016/j.jmb.2011.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pomerantz RT, O’Donnell M. Direct restart of a replication fork stalled by a head-on RNA polymerase. Science. 2010;327:590–592. doi: 10.1126/science.1179595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Park JS, Roberts JW. Role of DNA bubble rewinding in enzymatic transcription termination. Proc. Natl. Acad. Sci. U.S.A. 2006;103:4870–4875. doi: 10.1073/pnas.0600145103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sidorenkov I, et al. Crucial role of the RNA:DNA hybrid in the processivity of transcription. Mol. Cell. 1998;2:55–64. doi: 10.1016/s1097-2765(00)80113-6. [DOI] [PubMed] [Google Scholar]
- 39.Epshtein V, et al. An allosteric mechanism of Rho-dependent transcription termination. Nature. 2010;463:245–249. doi: 10.1038/nature08669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Svetlov V, Nudler E. Clamping the clamp of RNA polymerase. EMBO J. 2011;30:1190–1191. doi: 10.1038/emboj.2011.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sevostyanova A, et al. The beta subunit gate loop is required for RNA polymerase modification by RfaH and NusG. Mol. Cell. 2011;43:253–262. doi: 10.1016/j.molcel.2011.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stallings CL, et al. CarD is an essential regulator of rRNA transcription required for Mycobacterium tuberculosis persistence. Cell. 2009;138:146–159. doi: 10.1016/j.cell.2009.04.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ganesan AK, Hanawalt PC. Transcription-coupled nucleotide excision repair of a gene transcribed by bacteriophage T7 RNA polymerase in Escherichia coli. DNA Repair (Amst.) 2010;9:958–963. doi: 10.1016/j.dnarep.2010.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Selby CP, Sancar A. Molecular mechanism of transcription-repair coupling. Science. 1993;260:53–58. doi: 10.1126/science.8465200. [DOI] [PubMed] [Google Scholar]
- 45.Selby CP, Sancar A. Structure and function of transcription-repair coupling factor. II. Catalytic properties. J. Biol. Chem. 1995;270:4890–4895. doi: 10.1074/jbc.270.9.4890. [DOI] [PubMed] [Google Scholar]
- 46.Zalieckas JM, et al. Transcription-repair coupling factor is involved in carbon catabolite repression of the Bacillus subtilis hut and gnt operons. Mol. Microbiol. 1998;27:1031–1038. doi: 10.1046/j.1365-2958.1998.00751.x. [DOI] [PubMed] [Google Scholar]
- 47.Ayora S, et al. The Mfd protein of Bacillus subtilis 168 is involved in both transcription-coupled DNA repair and DNA recombination. J. Mol. Biol. 1996;256:301–318. doi: 10.1006/jmbi.1996.0087. [DOI] [PubMed] [Google Scholar]
- 48.Washburn RS, et al. Role of E coli transcription-repair coupling factor Mfd in Nun-mediated transcription termination. J. Mol. Biol. 2003;329:655–662. doi: 10.1016/s0022-2836(03)00465-0. [DOI] [PubMed] [Google Scholar]
- 49.Pakotiprapha D, et al. Crystal structure of Bacillus stearothermophilus UvrA provides insight into ATP-modulated dimerization, UvrB interaction, and DNA binding. Mol. Cell. 2008;29:122–133. doi: 10.1016/j.molcel.2007.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Timmins J, et al. Structural and mutational analyses of Deinococcus radiodurans UvrA2 provide insight into DNA binding and damage recognition by UvrAs. Structure. 2009;17:547–558. doi: 10.1016/j.str.2009.02.008. [DOI] [PubMed] [Google Scholar]
- 51.Pakotiprapha D, et al. Structure and mechanism of the UvrA-UvrB DNA damage sensor. Nat. Struct. Mol. Biol. 2012;19:291–298. doi: 10.1038/nsmb.2240. [DOI] [PubMed] [Google Scholar]
- 52.Rossi F, et al. The biological and structural characterization of Mycobacterium tuberculosis UvrA provides novel insights into its mechanism of action. Nucleic Acids Res. 2011;39:7316–7328. doi: 10.1093/nar/gkr271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jaciuk M, et al. Structure of UvrA nucleotide excision repair protein in complex with modified DNA. Nat. Struct. Mol. Biol. 2011;18:191–197. doi: 10.1038/nsmb.1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Manelyte L, et al. Regulation and rate enhancement during transcription-coupled DNA repair. Mol. Cell. 2010;40:714–724. doi: 10.1016/j.molcel.2010.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Theis K, et al. The nucleotide excision repair protein UvrB, a helicase-like enzyme with a catch. Mutat. Res. 2000;460:277–300. doi: 10.1016/s0921-8777(00)00032-x. [DOI] [PubMed] [Google Scholar]
- 56.Skorvaga M, et al. Identification of residues within UvrB that are important for efficient DNA binding and damage processing. J. Biol. Chem. 2004;279:51574–51580. doi: 10.1074/jbc.M409266200. [DOI] [PubMed] [Google Scholar]
- 57.Truglio J, et al. Structural basis for DNA recognition and processing by UvrB. Nat. Struct. Mol. Biol. 2006;13:360–364. doi: 10.1038/nsmb1072. [DOI] [PubMed] [Google Scholar]
- 58.Verhoeven EE, et al. The presence of two UvrB subunits in the UvrAB complex ensures damage detection in both DNA strands. EMBO J. 2002;21:4196–4205. doi: 10.1093/emboj/cdf396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pakotiprapha D, et al. A structural model for the damage-sensing complex in bacterial nucleotide excision repair. J. Biol. Chem. 2009;284:12837–12844. doi: 10.1074/jbc.M900571200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mazon G, et al. The alkyltransferase-like ybaZ gene product enhances nucleotide excision repair of O(6)-alkylguanine adducts in E coli. DNA Repair (Amst.) 2009;8:697–703. doi: 10.1016/j.dnarep.2009.01.022. [DOI] [PubMed] [Google Scholar]
- 61.Cohen SE, et al. Roles for the transcription elongation factor NusA in both DNA repair and damage tolerance pathways in Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 2010;107:15517–15522. doi: 10.1073/pnas.1005203107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Latypov VF, et al. Atl1 regulates choice between global genome and transcription-coupled repair of O(6)-alkylguanines. Mol. Cell. 2012;47:50–60. doi: 10.1016/j.molcel.2012.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Witkin EM. Radiation-induced mutations and their repair. Science. 1966;152:1345–1353. doi: 10.1126/science.152.3727.1345. [DOI] [PubMed] [Google Scholar]
- 64.von Hippel PH, Yager TD. Transcript elongation and termination are competitive kinetic processes. Proc. Natl. Acad. Sci. U.S.A. 1991;88:2307–2311. doi: 10.1073/pnas.88.6.2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yager TD, von Hippel PH. A thermodynamic analysis of RNA transcript elongation and termination in Escherichia coli. Biochemistry. 1991;30:1097–1118. doi: 10.1021/bi00218a032. [DOI] [PubMed] [Google Scholar]
- 66.Peters JM, et al. Bacterial transcription terminators: the RNA 3′-end chronicles. J. Mol. Biol. 2011;412:793–813. doi: 10.1016/j.jmb.2011.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Datta K, von Hippel PH. Direct spectroscopic study of reconstituted transcription complexes reveals that intrinsic termination is driven primarily by thermodynamic destabilization of the nucleic acid framework. J. Biol. Chem. 2008;283:3537–3549. doi: 10.1074/jbc.M707998200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhou Y, et al. Dissociation of halted T7 RNA polymerase elongation complexes proceeds via a forward-translocation mechanism. Proc. Natl. Acad. Sci. U.S.A. 2007;104:10352–10357. doi: 10.1073/pnas.0606306104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Epshtein V, et al. An allosteric path to transcription termination. Mol. Cell. 2007;28:991–1001. doi: 10.1016/j.molcel.2007.10.011. [DOI] [PubMed] [Google Scholar]
- 70.Larson MH, et al. Applied force reveals mechanistic and energetic details of transcription termination. Cell. 2008;132:971–982. doi: 10.1016/j.cell.2008.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Roberts J, Park JS. Mfd, the bacterial transcription repair coupling factor: translocation, repair and termination. Curr. Opin. Microbiol. 2004;7:120–125. doi: 10.1016/j.mib.2004.02.014. [DOI] [PubMed] [Google Scholar]
- 72.Skorvaga M, et al. The beta -hairpin motif of UvrB is essential for DNA binding, damage processing, and UvrC-mediated incisions. J. Biol. Chem. 2002;277:1553–1559. doi: 10.1074/jbc.M108847200. [DOI] [PubMed] [Google Scholar]
- 73.Truglio JJ, et al. Interactions between UvrA and UvrB: the role of UvrB’s domain 2 in nucleotide excision repair. EMBO J. 2004;23:2498–2509. doi: 10.1038/sj.emboj.7600263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Selby CP, et al. Escherichia coli mfd mutant deficient in “mutation frequency decline” lacks strand-specific repair: in vitro complementation with purified coupling factor. Proc. Natl. Acad. Sci. U.S.A. 1991;88:11574–11578. doi: 10.1073/pnas.88.24.11574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yang X, et al. The structure of bacterial RNA polymerase in complex with the essential transcription elongation factor NusA. EMBO Rep. 2009;10:997–1002. doi: 10.1038/embor.2009.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chen CS, et al. A proteome chip approach reveals new DNA damage recognition activities in Escherichia coli. Nat. Methods. 2008;5:69–74. doi: 10.1038/NMETH1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Tubbs JL, et al. Flipping of alkylated DNA damage bridges base and nucleotide excision repair. Nature. 2009;459:808–813. doi: 10.1038/nature08076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Aramini JM, et al. Structural basis of O6-alkylguanine recognition by a bacterial alkyltransferase-like DNA repair protein. J. Biol. Chem. 2010;285:13736–13741. doi: 10.1074/jbc.M109.093591. [DOI] [PMC free article] [PubMed] [Google Scholar]