

Abstract
Since the discovery that proteins mutated in different forms of polycystic kidney disease (PKD) are tightly associated with primary cilia, strong efforts have been made to define the role of this organelle in the pathogenesis of cyst formation. Cilia are filiform microtubular structures, anchored in the basal body and extending from the apical membrane into the tubular lumen. Early work established that cilia act as flow sensors, eliciting calcium transients in response to bending, which involve the two proteins mutated in autosomal dominant PKD (ADPKD), polycystin-1 and -2. Loss of cilia alone is insufficient to cause cyst formation. Nevertheless, a large body of evidence links flow sensing by cilia to aspects relevant for cyst formation such as cell polarity, Stat6- and mammalian target of rapamycin signalling. This review summarizes the current literature on cilia and flow sensing with respect to PKD and discusses how these findings intercalate with different aspects of cyst formation.
Keywords: cilia, ADPKD, mechanosensation, polarity, mTOR, calcium
INTRODUCTION
A century passed by from the first description of primary cilia in mammalian tubular cells to the recognition that these microtubule-containing organelles protruding from the apical membrane of epithelial cells perform important functions [1]. For a long time, attention had focussed on motile cilia that occur in bundles, for instance in respiratory epithelia where they mediate airway clearance [2]. Motile cilia have been studied for several decades in unicellular organisms, particularly Chlamydomonas reinhardtii, which yielded important insights into the physiology and biochemistry of this organelle, which are for the most part applicable also to primary, non-motile, cilia [3]. The reader is referred to a number of excellent reviews on the structure and function of cilia and its relevance with disease [4–6]. The link between polycystic kidneys and cilia became apparent when the mutated gene in the oakridge polycystic kidney (orpk) mouse was found to encode a family member of the intraflagellar transport (IFT) proteins. IFT proteins are transported along microtubular rods within the cilium and are required for cilia formation and maintenance [7, 8]. Subsequently, several proteins mutated in human or murine PKD have been found to localize to cilia. They include cystin (cpk mouse), polycystin 1 and 2 (PC-1 and 2), encoded by Pkd1 and 2 and mutated in ADPKD (autosomal dominant PKD), fibrocystin mutated in ARPKD (autosomal recessive PKD) and nephrocystin and inversin mutated in nephronophthisis [9–12]. This makes a compelling argument that this organelle is at the centre of a unifying concept of cyst formation. The central role of cilia was supported further when it was demonstrated that abolishing cilia through inactivation of a ciliary motor protein selectively in the kidney resulted in cyst formation [13].
Different forms of cystic kidneys vary by age of presentation [14]. ADPKD, the most common form with an incidence of 1:1000, is a disease of adulthood. Over half of the affected patients require renal replacement therapy by the age of 60. On the other hand, a large spectrum of mostly autosomal recessive diseases occur in childhood and are frequently associated with pathologies in other organ systems such as situs inversus in nephronophthisis, retinitis pigmentosa in Joubert's disease, central nervous system malformations in Meckel syndrome, polydactyly in Bardet–Biedl syndrome (BBS) or obesity in BBS and Almström syndrome [15]. For the majority of associated pathologies, a role for cilia in their aetiology has been characterized resulting in the term ciliopathies. In the kidney, cilia extend into the tubule lumen where they are bent by flow. Indeed, groundbreaking work by Praetorius and Spring revealed that the cilium translates fluid flow into calcium signals and thus, could serve as a signal transduction platform that orchestrates tubular development, epithelial cell homeostasis and the diameter of tubules [16, 17].
Cyst formation is characterized by multiple pathogenic events including disordered cell polarity, dysregulation of various signal transduction pathways and increased proliferation [18–20]. Given the marked variation in the morphology of different cystic diseases, the genetic aspects and the varying age of presentation, it is quite likely that certain features of the pathophysiology of cyst formation diverge between the individual entities. Therefore, insights into the function of the cilium will only explain common aspects in the pathogenesis of cyst formation and have to be interpreted in the context of the pathophysiology of a specific disease or syndrome. The purpose of this review is to summarize the evidence linking the ciliary flow sensor to cyst formation, to discuss ciliary downstream signalling in response to flow and to hypothesize about the physiological functions of the ciliary flow sensor in the kidney and how its disruption may promote cystogenesis. Since vascular phenotypes are not a universal feature of PKD and shear stress in arteries is one order of magnitude higher compared with renal tubules, we will not address the role of cilia in the vasculature.
The cilium as a mechanosensor
Cilia exist on most cells in mammalian organisms [4]. In tubules, cilia are constantly exposed to urine flow, so it is intuitive that they act as flow sensors. Flow sensing is difficult to study in vivo. Alternative approaches have involved cell culture experiments in flow chambers with Madin-Darby canine kidney (MDCK) cells, immortalized cells from transgenic animals, human cyst-derived cells and ex vivo studies with explanted tubules from rodents [17, 21–24]. MDCK cells respond to bending of the cilium by a micropipette or fluid flow with influx of calcium [17, 25]. The initial signal is amplified through nucleotide release and occurs through influx of extracellular calcium via the apical surface which is followed by calcium-induced calcium release from IP3-dependent stores which is transmitted to other cells through gap junctions [26–28]. The cilium is required for calcium transients under flow because ablation of the cilium in MDCK cells by chemical means abolishes the flow response [21, 25]. This view is supported by the finding of blunted flow sensing in isolated renal collecting ducts from cilia-deficient orpk mice [29].
Direct and indirect loss of function experiments link polycystins to cilia by demonstrating a lack of or altered calcium signals in response to fluid flow. Experiments in flow chambers with isolated cells from transgenic mice that lack functional PC-1, as well as in MDCK cells inducibly depleted of PC-2, a cation channel of the TRP family (TRPP2), by small hairpin RNA, showed that Pkd1 and Pkd2 are needed for flow-induced calcium signalling [23, 30]. In a different approach, immortalized cyst lining cells from patients with a mutation in Pkd1 were compared with immortalized normal kidney cells and showed no flow-induced calcium signals [24]. PC-2 was absent in the cilia of the Pkd1-mutant cells which would explain the loss of flow-induced calcium transients. Similarly, mislocalization of PC-2 at the apical membrane was described in unciliated orpk cells [31] and expression of an N-terminal mutant of PC-2 in MDCK cells that is deficient in ciliary trafficking had a dominant-negative effect on calcium signalling under flow [32, 33]. Bearing in mind the significant heterogeneity in flow chamber set-ups, cell types, amount of shear stress and timing of calcium response in the studies described, these data suggest that PC-1 and -2 are intimately involved in flow- and cilia-dependent intracellular calcium increase.
Is the loss of the ciliary flow sensor sufficient to cause cyst formation? Several important studies suggest that this is not the case [34–36]. The genetic model of cyst formation in ADPKD is believed to lie in the loss of heterozygosity for a PKD gene in individual cells that then begin to lose polarity, proliferate and form cysts [37]. However, the inactivation of Pkd1 after completion of the postnatal stage of kidney development in mice (postnatal day 14) does not lead to accelerated cyst formation [38]. Analogous results were found after late inactivation of the ciliary motor protein Kif3a or IFT88 [35, 36], suggesting that quiescent cells in the adult kidney do not require cilia or PC-1 for maintaining tubular morphology in the intermediate term (the kidneys do form cysts in the long term). The second piece of evidence involves the osmo-sensor TRPV4, a transient receptor potential cation channel that forms a polymodal channel complex with PC-2 [34]. MDCK cells depleted of TRPV4 by inducible shRNA failed to display calcium transients in response to flow. Yet, TRPV4 morphant zebrafish did not replicate the Pkd2 phenotype of pronephric cysts [34]. Furthermore, mice lacking TRPV4 do not develop cystic kidneys [39]. Apart from the possibility of redundancy, these data argue that disruption of ciliary flow sensing alone does not suffice to transform renal epithelial cells into cyst-forming cells, and that additional factors are required. This concept is supported by the fact that the induction of ischaemia-reperfusion injury in Pkd1 or cilia-deficient mice induces rapid cyst formation [35, 40].
Polarity
A large body of evidence shows that cyst formation involves dysregulation of multiple signal transduction pathways and the disruption of cell polarity [18–20]. The Wnt pathway is a versatile signalling system regulating fate determination, proliferation and planar cell polarity that is strongly intertwined with renal development and the maintenance of kidney morphology [41]. Canonical Wnt signalling is initiated upon binding of a Wnt ligand to a Frizzled receptor causing stabilization and nuclear translocation of β-catenin as well as target gene expression [42]. A number of findings link the canonical Wnt pathway to PKD. The intracellular tail of PC-1 interacts with β-catenin involving cleavage of a C-terminal fragment and translocation to the nucleus [43, 44]. The activation as well as the inhibition of canonical Wnt signalling results in cyst formation, and increased Wnt activity has been found in gene profiling studies on cysts from patients with ADPKD [41, 45]. Secreted Frizzled-related protein 4, which specifically inhibits canonical Wnt signalling, is upregulated in human ADPKD yet recapitulates a ciliary phenotype when overexpressed in zebrafish [46], thus suggesting a finely tuned balance of canonical Wnt activity in the nephron. Several lines of evidence link cilia to canonical Wnt signalling [47]: tissue-specific inactivation of Kif3a in renal tubular cells in vivo resulted in increased nuclear β-catenin levels [13]. Work in cultured cells demonstrated that ablation of cilia by targeting Kif3a or Ift88 in mouse embryonic fibroblasts (MEFs) resulted in upregulated sensitivity to Wnt3 and increased levels of β-catenin [48]. Similarly, downregulation of genes involved in Bardet–Biedl syndrome (BBS1, BBS2 and MKKS) resulted in a hyperactive Wnt response in cultured cells [49]. Unpublished data from our group show that β-catenin-dependent reporter activity is downregulated in MDCK cells under flow. Taken together, these results imply that cilia inhibit canonical Wnt signalling. It must be noted, however, that cilia-deficient mouse embryos failed to show evidence of altered canonical Wnt signalling [50], which suggests that the regulation of β-catenin-dependent signalling by cilia involves a level of complexity that is currently not understood.
The data on how cilia affect the non-canonical, or PCP (planar cell polarity), pathway are clearer. PCP describes the ability of cells to orient along the plane relative to an axis [51]. Distinct readouts of PCP have been described including wing and eye development in Drosophila melanogaster, and in vertebrates inner ear development and convergent extension, a process describing elongation of the embryo through cell intercalation. Strong evidence implicating cilia in PCP comes from mice with missing cilia through tissue specific targeting of Ift88 and Kif3a: these animals displayed a highly disordered tissue architecture in the inner ear [52]. During normal kidney development tubular cells intercalate perpendicular to the tubular axis resulting in narrowing of tubules [53]. This process requires Wnt9b, an activator of non-canonical Wnt signals, and if disrupted leads to cyst formation. PCP is controlled by the non-canonical pathway through the activity of Disheveled (Dvl), a protein that targets the canonical regulator β-catenin for degradation and regulates the activity of Frizzled to orchestrate the polarized distribution of different sets of PCP core proteins [51]. Dvl interacts with Inversin (Inv), a ciliary protein and NPHP2 gene product mutated in nephronophthisis Type 2 [9, 54, 55]. Inv binding to Dvl promotes the degradation of β-catenin implying a role as a molecular switch between canonical and non-canonical Wnt signalling [54]. Similar functions have been attributed to BBS proteins, NPHP3, and Kif3a [48, 49, 56]. In addition, several BBS proteins (BBS1, BBS4 and BBS6) interact with the PCP pathway in mice [52, 57].
Another polarized event apart from the convergence of tubular cells during embryonic tubule elongation is oriented cell division (OCD) [58]. Studies in Drosophila and zebrafish have shown that OCD is required for PCP. Misoriented cell division results in disrupted wing shape [59] or disturbed convergent extension [60]. While tubular elongation through epithelial intercalation occurs during embryonic development [53], OCD along the axis of growing tubules is seen in postnatal kidneys [58]. OCD is disrupted in mice mutated in Pkd1, Tsc1/2 and Hnf1β and therefore, associated with cystic kidney disease [61, 62]. Cilia seem to play a role in OCD: in Kif3a-mutant mice, 46% of the mitotic angles were oriented within 20° of the collecting duct axis, in contrast to 91% in normal kidneys [35]. Furthermore, Ift20-mutant mice almost completely lacked cilia and developed renal cysts associated with a misoriented cell division axis [63]. However, loss of OCD does not suffice to trigger cyst formation. A careful study with Pkd1 and Pkd2-mutant mice did not show OCD before cystic dilatation and, conversely, in Pkhd1-mutant mice OCD was lost, but no kidney cysts developed [64]. Similarly, in Pkd1 heterozygous mice, abnormal mitotic spindle orientation has been observed at postnatal Day 2, while only occasional microscopic cysts develop [61]. A further study in Ift140-mutant mice showed disordered cilia assembly, resulting in cystic kidneys but no disoriented mitotic spindle axis was observed [65]. A possible explanation for these findings is that the axis of cell division is not the driving force in cystogenesis, but more likely an indicator of dysregulated cellular orientation and polarity.
It is evident that essential cilia proteins as well as cilia-associated PKD genes are linked to OCD and convergent extension, but is there any evidence that flow sensing by cilia affects polarity? The best argument for this notion does not come from the kidney, but from the embryonic node and from Xenopus epidermis. The node (embryonic notocord, Kupffer's vesicle in zebrafish) is a ciliated structure in the early embryo that determines left–right body asymmetry in a process where rotating 9+0 cilia create a leftward flow [66]. Disruption of this flow through interference with cilia structure or inactivation of Pkd2 leads to randomization of left–right asymmetry, in the latter case through a lack of flow-induced calcium signals in the periphery of the node [67]. Indeed, situs inversus has been described in rare patients with heterozygote PKD2 mutations [68]. Polarization of 9+2 cilia and basal body structures through flow has been observed in Xenopus skin (Figure 1) [69, 70]. The basal body is one of the two microtubule-based cylinders called centrioles with highly conserved 9-fold radial symmetry. The centriole pair displays structural and functional asymmetry [71]. In multi-ciliated cells of Xenopus larva skin and cilia lining the brain ventricles, the sensing of self-generated flow has been shown to adjust the orientation of the basal body and the direction of ciliary beating in a positive feedback manner involving PCP proteins [69, 70, 72]
FIGURE 1:

Flow transmits polarized information to the centriolar apparatus. (a) In the epidermis of Xenopus embryos, motile 9+2 cilia generate unidirectional flow (pink arrow) which is necessary for the polarization of the basal bodies (mother centriole—blue) from which the cilia originate [69]. Polarization is visualized by asymmetric localization of the basal foot, a basal body appendage (purple triangle). (b) In renal epithelial cells, passive bending of monocilia through flow (pink arrow) results in calcium transients and preferential centriole movements along the flow axis [33]. (c) In the absence of flow, or in cells expressing a polycystin-2 mutant preventing flow-induced calcium increases, no orientation bias of centriole movements is observed.
As yet, the regulation of PCP in renal epithelial cells by cilia and flow has not been demonstrated in vitro. When MDCK cells were analysed under continuous flow for up to a week, the orientation of mitotic spindles was randomized, albeit the cells were completely unciliated for the first 4 days and the number of cell divisions decreased towards zero after the majority of cell reached cilia stage [33]. In a second approach, a wound was induced in a confluent monolayer of ciliated cells under flow, but during the repair phase the mitotic spindles of the migrating cells again showed no orientation in the direction of flow, suggesting that OCD in renal epithelial cells cannot be modelled in vitro (OCD has been described in endothelial cells in the absence of cilia [73, 74]). However, polarization did occur at the centrioles [33]. The mother centriole, also called the basal body, where the primary cilium originates, anchors cytoplasmic microtubules at the sub-distal appendages to attach to the plasma membrane and is mainly stationary. In contrast, the daughter centriole, connected through fibres with the mother centriole, moves extensively during G0 and G1 [75]. Centriole movements in ciliated cells under flow increased compared with the absence of flow and showed a strong motility bias along the axis of flow (the sum of all movements was zero indicating that the centrioles moved around a central point and the position of the cells did not change, Figure 1) [33]. Interestingly, in the cells overexpressing a ciliary trafficking mutant of PC-2 that abolished flow-induced calcium currents, centriole movements were randomized. These data suggest that cilia translate the direction of flow into spatial information and to the centrioles in a calcium-dependent manner. Since polarized centriole movements have not been recorded in renal tubules in vivo, the biological significance of this finding is speculative. Nevertheless, they are proof of the principle that in addition to polarization of the centriolar apparatus of motile 9+2 cilia by self-generated flow [70], polarization can also occur in passively bent primary, 9+0 cilia in renal epithelia cells. In summary, several lines of evidence exist that cilia and PKD proteins are linked to the polarity of tubular epithelial cells and indirect evidence supports the concept that the ciliary flow sensor and downstream calcium signalling are involved in this process.
PC-1 cleavage and Stat6 signalling
PC-1 is a large orphan receptor with 11 membrane spanning domains [76]. A C-terminal portion of 200 amino acids is cleaved and translocates to the nucleus [77]. Immunoreactivity of this fragment in the nuclei is greatly enhanced in kidneys after ureteral obstruction and kidneys lacking cilia as well as in cysts where flow does not occur, suggesting that the cleavage process is regulated by flow sensing. Indeed, indirect evidence supports this model [78]. The C-terminal fragment of PC-1 interacts with the transcription factor Stat6 and its co-activator p100 to enhance Stat6-dependent transcription. Interestingly, Stat6 is found in cilia of MDCK cells when they are cultured under constant flow, but in the absence of flow, Stat6 is localized in the nucleus, but not cilia, while p100 is localized at the ciliary base under both conditions. A model derived from these observations suggests that under flow, PC-1 binds to Stat6 and p100 in the cilium [78]. Disruption of flow results in cleavage of the C-terminal PC-1 fragment and translocation of the complex to the nucleus. The C-terminal cleavage of PC-1 seems to be regulated by PC-2, however, in a calcium-independent mode [79], and direct evidence of PC-1 C-terminal cleavage under flow is lacking. Nevertheless, a recent study observed a decrease in cyst formation in PKD animals with genetic inactivation of Stat6 or treatment with a Stat6 inhibitor, supporting the biological relevance of these findings [80].
mTOR signalling
A lot of attention over the past few years has been focused on the connection between PKD and dysregulated mTOR (mechanistic target of rapamycin) signalling [81]. The interest was set off by the fact that the Tsc proteins which are mutated in tuberous sclerosis, a disease associated with polycystic kidneys, function as negative regulators of mTOR signalling [82] and by the discovery that rapamycin, an immunosuppressive drug that very effectively inhibits the mTOR pathway, prevents cyst growth in rodent models of PKD [83]. mTOR is part of two multi-protein complexes mTORC1 and 2. The rapamycin-sensitive complex mTORC1 regulates protein biosynthesis and cell size and is thus important for cell proliferation [84, 85]. mTORC1 activity is elevated in a substantial proportion of renal cysts and indeed PC-1 has been found to interact with tuberin, encoded by Tsc2, and was shown to inhibit mTORC1 activity [86, 87].
Is there a connection between mTOR activity and ciliary flow sensing? Boehlke et al. found that MDCK cells grown under continuous flow were smaller compared with cells grown in the absence of flow [30]. In a series of loss-of-function experiments and biochemical analyses, they found that the cell size under flow is regulated by mTORC1 and requires the presence of cilia (Figure 2). The mechanism for this was interesting: mTOR regulation was independent of flow-induced calcium transients or Akt, an important activator of mTORC1 activity through growth factors [85]. Instead, it was associated with activation of Lkb1, a negative mTORC1 regulator mutated in the Peutz–Jeghers syndrome [88], specifically at the basal body. An important question arising from these findings is what could be the biological significance of mTOR regulation by flow? Indeed, a physiological function can be deduced from several studies analysing the ciliary length. Several groups have shown that loss of TSC function increases the ciliary length in zebrafish, MEFs and mouse kidneys implicating mTOR in some cases [89–91]. The effect of mTOR signalling on the cilia length was analysed in detail in a recent study on zebrafish and Chlamydomonas [92]. Loss of Tsc1 function or overexpression of the mTORC1 downstream effector S6Kinase resulted in longer cilia in the Kupffer's vesicle. On the other hand, treatment of zebrafish or deflagellated recovering Chlamydomonas with the mTOR inhibitor rapamycin decreased the cilia length. The alterations in the ciliary length were physiologically relevant, resulting in altered beat frequency and loss of left–right asymmetry in zebrafish embryos. In Chlamydomonas, a bell curve relationship was discovered between the cilia length and flagellar motility, suggesting that an optimal ciliary length exists in vivo [92]. In a mathematical model, ciliary bending results in maximal membrane stress at the base of the cilium, the site where Lkb1 seems to be active and where Tsc1 is located [30, 91, 93]. Furthermore, the ciliary length is predicted to determine the amount of torque at the basal body transition zone and, therefore, affects the sensing of shear forces [13]. Taken together, these findings could suggest a physiological feedback mechanism, where a decrease in shear forces, e.g. through reduced flow, would increase mTORC1 activity leading to lengthened cilia which, in turn, would amplify the flow signal (Figure 3). Additional observations support this concept: a study from Besschetnova et al. showed that flow negatively affects the ciliary length in wild-type cells albeit not in Pkd1 mutant and constitutively PC-2 depleted cells [94]. Another study analysed biopsies of transplanted kidneys, which suffered acute tubular necrosis during transplantation and observed almost a doubling of ciliary length, which normalized after recovery of allograft function [95]. Similarly, a study analysing the length of cilia in Tsc1± or Tsc2± mice found no ciliary elongation in precystic tubules, where cilia are still subjected to flow. However, cilia were markedly longer (>200%) in the epithelia of full-blown cysts [61], where connection to the tubules is lost and no flow occurs. Taken together, flow-dependent mTOR regulation may serve to fine-tune the cilia length in an environment of alternating flow speeds and levels of shear stress.
FIGURE 2:
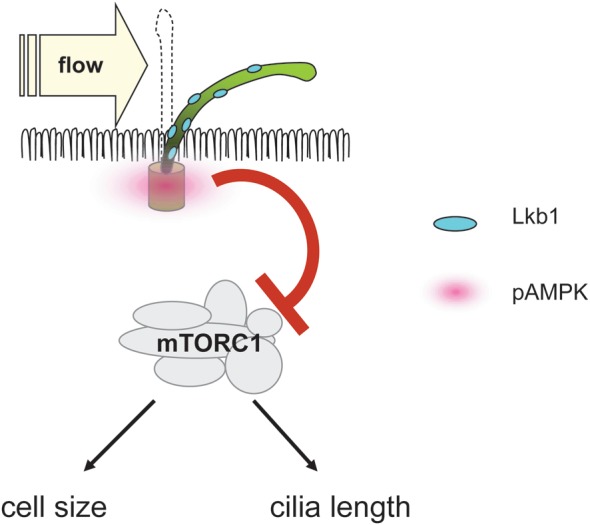
Lkb1 localizes to cilia. Bending of the cilium by flow causes increased phosphorylation of the Lkb1 target AMPK at the basal body [30]. Activated AMPK inhibits mTORC1 and decreases the cell size. In addition, mTORC1 activity regulates the cilia length [92].
FIGURE 3:
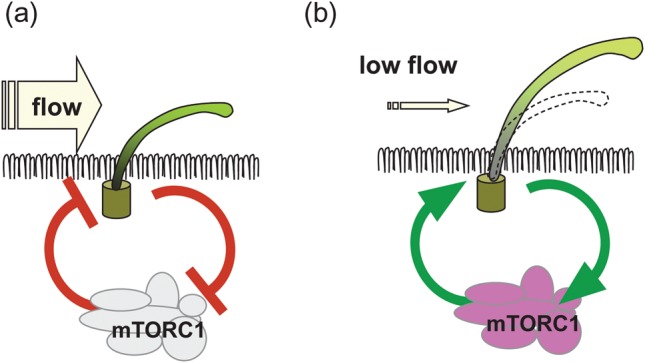
Model of a feedback loop to maintain shear stress sensing through flow-induced mTORC1 regulation. (a) During high flow, mTORC1 activity is decreased resulting in short cilia. (b) During low flow, activation of mTORC1 would result in longer cilia, which increases the amplification of the flow signal.
In hypothetical terms, flow-dependent mTOR regulation might be an initial step in cyst formation: pathologically reduced or absent flow sensing by cilia increases the cell size and therefore, would reduce the cross-section of the tubular lumen (Figure 4). The narrowing of luminal diameter would be expected to increase tubular wall stress. Conversely, maintenance of a preset level of shear stress in the setting of increased cell size would require an enlargement of the diameter of the tubular basement membrane to keep the luminal diameter steady. This could theoretically be an initial step to dilatation of the tubule. However, this model requires a second shear stress sensor other than the a priori sensing-deficient cilium. In larger cysts, where flow has ceased, the lack of flow sensing might explain mTORC1 activity in cyst lining cells.
FIGURE 4:
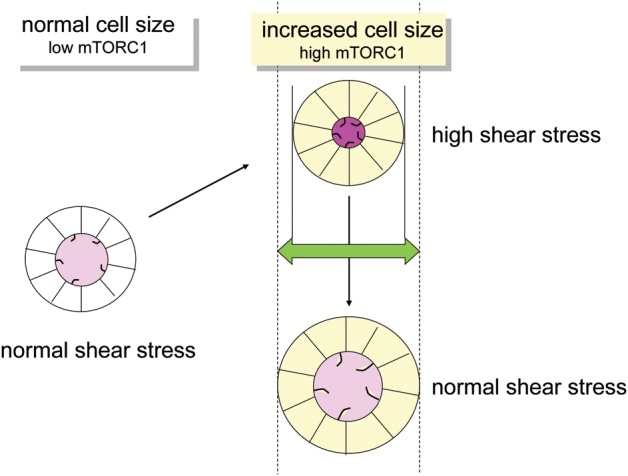
Hypothetical model showing how the cell size may affect shear stress and tubular diameter. Increased mTORC1 activity leads to bigger cell size. In the case of a fixed tubular basement membrane diameter, luminal shear stress will increase. Maintaining a normal level of shear stress requires an increase of tubular basement membrane diameter. Since the deficient ciliary flow sensor is responsible for increased mTORC1 activity and cell size dysregulation, this model requires an alternate mode of shear stress sensing.
Concluding remarks
It has been exciting times since cilia were implicated in the pathogenesis of PKD. The early and simple model that cilia translate fluid flow into calcium signals through PC-1 and -2, and that loss of either protein function converts signalling-deficient epithelial cells into a proliferating and de-differentiated phenotype that grow into cysts is clearly inadequate to match the complexities of ciliary signalling. Nevertheless, there is clear evidence from cell culture and in vivo models that flow is translated by cilia into cellular events which are associated with cyst formation, namely cell polarization, Stat6- and mTOR signalling. Renal cilia in the mammalian kidney are difficult to access. Therefore, cellular in vitro systems and model organisms such as zebrafish and Xenopus that can be easily manipulated and lend themselves to 4D imaging, will continue to be instrumental for the study of the ciliary flow sensor. Hypotheses derived from these systems need to be tested in rodent models of PKD to advance our understanding of the pathophysiology of PKD and to identify new therapeutic targets.
Funding. WK is supported by the DFG (KFO 201 and KU 1504/4-1) and the Else-Kröner Fresenius Stiftung. We thank Amandine Viau and Matias Simons for critical appraisal of the manuscript. We apologize to all the colleagues whose relevant work could not be cited for space constraints.
CONFLICT OF INTEREST STATEMENT
None declared.
REFERENCES
- 1.Zimmermann K. Beiträge zur Kenntnis einiger Drüsen und Epithelien. Arch MikroskopAnat. 1898;52:552–706. doi:10.1007/BF02975837. [Google Scholar]
- 2.Afzelius BA. A human syndrome caused by immotile cilia. Science. 1976;193:317–319. doi: 10.1126/science.1084576. doi:10.1126/science.1084576. [DOI] [PubMed] [Google Scholar]
- 3.Rosenbaum JL, Witman GB. Intraflagellar transport. Nat Rev Mol Cell Biol. 2002;3:813–825. doi: 10.1038/nrm952. doi:10.1038/nrm952. [DOI] [PubMed] [Google Scholar]
- 4.Ishikawa H, Marshall WF. Ciliogenesis: building the cell's antenna. Nat Rev Mol Cell Biol. 2011;12:222–234. doi: 10.1038/nrm3085. doi:10.1038/nrm3085. [DOI] [PubMed] [Google Scholar]
- 5.Fliegauf M, Benzing T, Omran H. When cilia go bad: cilia defects and ciliopathies. Nat Rev Mol Cell Biol. 2007;8:880–893. doi: 10.1038/nrm2278. doi:10.1038/nrm2278. [DOI] [PubMed] [Google Scholar]
- 6.Goetz SC, Anderson KV. The primary cilium: a signalling centre during vertebrate development. Nat Rev Genet. 2010;11:331–344. doi: 10.1038/nrg2774. doi:10.1038/nrg2774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pazour GJ, Dickert BL, Vucica Y, et al. Chlamydomonas IFT88 and its mouse homologue, polycystic kidney disease gene tg737, are required for assembly of cilia and flagella. J Cell Biol. 2000;151:709–718. doi: 10.1083/jcb.151.3.709. doi:10.1083/jcb.151.3.709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Murcia NS, Richards WG, Yoder BK, et al. The oak ridge polycystic kidney (orpk) disease gene is required for left-right axis determination. Development. 2000;127:2347–2355. doi: 10.1242/dev.127.11.2347. [DOI] [PubMed] [Google Scholar]
- 9.Otto EA, Schermer B, Obara T, et al. Mutations in INVS encoding inversin cause nephronophthisis type 2, linking renal cystic disease to the function of primary cilia and left-right axis determination. Nat Genet. 2003;34:413–420. doi: 10.1038/ng1217. doi:10.1038/ng1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yoder BK, Hou X, Guay-Woodford LM. The polycystic kidney disease proteins, polycystin-1, polycystin-2, polaris, and cystin, are co-localized in renal cilia. J Am Soc Nephrol. 2002;13:2508–2516. doi: 10.1097/01.asn.0000029587.47950.25. doi:10.1097/01.ASN.0000029587.47950.25. [DOI] [PubMed] [Google Scholar]
- 11.Zhang Q, Murcia NS, Chittenden LR, et al. Loss of the Tg737 protein results in skeletal patterning defects. Dev Dyn. 2003;227:78–90. doi: 10.1002/dvdy.10289. doi:10.1002/dvdy.10289. [DOI] [PubMed] [Google Scholar]
- 12.Ward CJ, Yuan D, Masyuk TV, et al. Cellular and subcellular localization of the ARPKD protein; fibrocystin is expressed on primary cilia. Hum Mol Genet. 2003;12:2703–2710. doi: 10.1093/hmg/ddg274. doi:10.1093/hmg/ddg274. [DOI] [PubMed] [Google Scholar]
- 13.Lin F, Hiesberger T, Cordes K, et al. Kidney-specific inactivation of the KIF3A subunit of kinesin-II inhibits renal ciliogenesis and produces polycystic kidney disease. Proc Natl Acad Sci USA. 2003;100:5286–5291. doi: 10.1073/pnas.0836980100. doi:10.1073/pnas.0836980100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bisceglia M, Galliani CA, Senger C, et al. Renal cystic diseases: a review. Adv Anat Pathol. 2006;13:26–56. doi: 10.1097/01.pap.0000201831.77472.d3. doi:10.1097/01.pap.0000201831.77472.d3. [DOI] [PubMed] [Google Scholar]
- 15.Tobin JL, Beales PL. The nonmotile ciliopathies. Genet Med. 2009;11:386–402. doi: 10.1097/GIM.0b013e3181a02882. doi:10.1097/GIM.0b013e3181a02882. [DOI] [PubMed] [Google Scholar]
- 16.Dorup J, Maunsbach AB. The ultrastructural development of distal nephron segments in the human fetal kidney. Anat Embryol (Berl) 1982;164:19–41. doi: 10.1007/BF00301876. doi:10.1007/BF00301876. [DOI] [PubMed] [Google Scholar]
- 17.Praetorius HA, Spring KR. Bending the MDCK cell primary cilium increases intracellular calcium. J Membr Biol. 2001;184:71–79. doi: 10.1007/s00232-001-0075-4. doi:10.1007/s00232-001-0075-4. [DOI] [PubMed] [Google Scholar]
- 18.Gallagher AR, Germino GG, Somlo S. Molecular advances in autosomal dominant polycystic kidney disease. Adv Chronic Kidney Dis. 2010;17:118–130. doi: 10.1053/j.ackd.2010.01.002. doi:10.1053/j.ackd.2010.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Torres VE, Harris PC. Autosomal dominant polycystic kidney disease: the last 3 years. Kidney Int. 2009;76:149–168. doi: 10.1038/ki.2009.128. doi:10.1038/ki.2009.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wilson PD. Apico-basal polarity in polycystic kidney disease epithelia. Biochim Biophys Acta. 2011;1812:1239–1248. doi: 10.1016/j.bbadis.2011.05.008. doi:10.1016/j.bbadis.2011.05.008. [DOI] [PubMed] [Google Scholar]
- 21.Kotsis F, Nitschke R, Boehlke C, et al. Ciliary calcium signaling is modulated by kidney injury molecule-1 (Kim1) Pflugers Arch. 2007;453:819–829. doi: 10.1007/s00424-006-0168-0. doi:10.1007/s00424-006-0168-0. [DOI] [PubMed] [Google Scholar]
- 22.Liu W, Xu S, Woda C, et al. Effect of flow and stretch on the [Ca2+]i response of principal and intercalated cells in cortical collecting duct. Am J Physiol Renal Physiol. 2003;285:F998–F1012. doi: 10.1152/ajprenal.00067.2003. [DOI] [PubMed] [Google Scholar]
- 23.Nauli SM, Alenghat FJ, Luo Y, et al. Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat Genet. 2003;33:129–137. doi: 10.1038/ng1076. doi:10.1038/ng1076. [DOI] [PubMed] [Google Scholar]
- 24.Xu C, Rossetti S, Jiang L, et al. Human ADPKD primary cyst epithelial cells with a novel, single codon deletion in the PKD1 gene exhibit defective ciliary polycystin localization and loss of flow-induced Ca2+ signaling. Am J Physiol Renal Physiol. 2007;292:F930–F945. doi: 10.1152/ajprenal.00285.2006. doi:10.1152/ajprenal.00285.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Praetorius HA, Spring KR. Removal of the MDCK cell primary cilium abolishes flow sensing. J Membr Biol. 2003;191:69–76. doi: 10.1007/s00232-002-1042-4. doi:10.1007/s00232-002-1042-4. [DOI] [PubMed] [Google Scholar]
- 26.Praetorius HA, Spring KR. A physiological view of the primary cilium. Annu Rev Physiol. 2005;67:515–529. doi: 10.1146/annurev.physiol.67.040403.101353. doi:10.1146/annurev.physiol.67.040403.101353. [DOI] [PubMed] [Google Scholar]
- 27.Praetorius HA, Leipziger J. Intrarenal purinergic signaling in the control of renal tubular transport. Annu Rev Physiol. 2010;72:377–393. doi: 10.1146/annurev-physiol-021909-135825. doi:10.1146/annurev-physiol-021909-135825. [DOI] [PubMed] [Google Scholar]
- 28.Praetorius HA, Leipziger J. Released nucleotides amplify the cilium-dependent, flow-induced [Ca2+]i response in MDCK cells. Acta Physiol (Oxf) 2009;197:241–251. doi: 10.1111/j.1748-1716.2009.02002.x. doi:10.1111/j.1748-1716.2009.02002.x. [DOI] [PubMed] [Google Scholar]
- 29.Liu W, Murcia NS, Duan Y, et al. Mechanoregulation of intracellular Ca2+ concentration is attenuated in collecting duct of monocilium-impaired orpk mice. Am J Physiol Renal Physiol. 2005;289:F978–F988. doi: 10.1152/ajprenal.00260.2004. doi:10.1152/ajprenal.00260.2004. [DOI] [PubMed] [Google Scholar]
- 30.Boehlke C, Kotsis F, Patel V, et al. Primary cilia regulate mTORC1 activity and cell size through Lkb1. Nat Cell Biol. 2010;12:1115–1122. doi: 10.1038/ncb2117. doi:10.1038/ncb2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Siroky BJ, Ferguson WB, Fuson AL, et al. Loss of primary cilia results in deregulated and unabated apical calcium entry in ARPKD collecting duct cells. Am J Physiol Renal Physiol. 2006;290:F1320–F1328. doi: 10.1152/ajprenal.00463.2005. doi:10.1152/ajprenal.00463.2005. [DOI] [PubMed] [Google Scholar]
- 32.Geng L, Okuhara D, Yu Z, et al. Polycystin-2 traffics to cilia independently of polycystin-1 by using an N-terminal RVxP motif. J Cell Sci. 2006;119:1383–1395. doi: 10.1242/jcs.02818. doi:10.1242/jcs.02818. [DOI] [PubMed] [Google Scholar]
- 33.Kotsis F, Nitschke R, Doerken M, et al. Flow modulates centriole movements in tubular epithelial cells. Pflugers Arch. 2008;456:1025–1035. doi: 10.1007/s00424-008-0475-8. doi:10.1007/s00424-008-0475-8. [DOI] [PubMed] [Google Scholar]
- 34.Kottgen M, Buchholz B, Garcia-Gonzalez MA, et al. TRPP2 and TRPV4 form a polymodal sensory channel complex. J Cell Biol. 2008;182:437–447. doi: 10.1083/jcb.200805124. doi:10.1083/jcb.200805124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Patel V, Li L, Cobo-Stark P, et al. Acute kidney injury and aberrant planar cell polarity induce cyst formation in mice lacking renal cilia. Hum Mol Genet. 2008;17:1578–1590. doi: 10.1093/hmg/ddn045. doi:10.1093/hmg/ddn045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Davenport JR, Watts AJ, Roper VC, et al. Disruption of intraflagellar transport in adult mice leads to obesity and slow-onset cystic kidney disease. Curr Biol. 2007;17:1586–1594. doi: 10.1016/j.cub.2007.08.034. doi:10.1016/j.cub.2007.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wu G, D'Agati V, Cai Y, et al. Somatic inactivation of Pkd2 results in polycystic kidney disease. Cell. 1998;93:177–188. doi: 10.1016/s0092-8674(00)81570-6. doi:10.1016/S0092-8674(00)81570-6. [DOI] [PubMed] [Google Scholar]
- 38.Piontek K, Menezes LF, Garcia-Gonzalez MA, et al. A critical developmental switch defines the kinetics of kidney cyst formation after loss of Pkd1. Nat Med. 2007;13:1490–1495. doi: 10.1038/nm1675. doi:10.1038/nm1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Taniguchi J, Tsuruoka S, Mizuno A, et al. TRPV4 as a flow sensor in flow-dependent K+ secretion from the cortical collecting duct. Am J Physiol Renal Physiol. 2007;292:F667–F673. doi: 10.1152/ajprenal.00458.2005. doi:10.1152/ajprenal.00458.2005. [DOI] [PubMed] [Google Scholar]
- 40.Takakura A, Contrino L, Zhou X, et al. Renal injury is a third hit promoting rapid development of adult polycystic kidney disease. Hum Mol Genet. 2009;18:2523–2531. doi: 10.1093/hmg/ddp147. doi:10.1093/hmg/ddp147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lancaster MA, Gleeson JG. Cystic kidney disease: the role of Wnt signaling. Trends Mol Med. 2010;16:349–360. doi: 10.1016/j.molmed.2010.05.004. doi:10.1016/j.molmed.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.van Amerongen R, Nusse R. Towards an integrated view of Wnt signaling in development. Development. 2009;136:3205–3214. doi: 10.1242/dev.033910. doi:10.1242/dev.033910. [DOI] [PubMed] [Google Scholar]
- 43.Kim E, Arnould T, Sellin LK, et al. The polycystic kidney disease 1 gene product modulates Wnt signaling. J Biol Chem. 1999;274:4947–4953. doi: 10.1074/jbc.274.8.4947. doi:10.1074/jbc.274.8.4947. [DOI] [PubMed] [Google Scholar]
- 44.Lal M, Song X, Pluznick JL, et al. Polycystin-1 C-terminal tail associates with beta-catenin and inhibits canonical Wnt signaling. Hum Mol Genet. 2008;17:3105–3117. doi: 10.1093/hmg/ddn208. doi:10.1093/hmg/ddn208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Song X, Di Giovanni V, He N, et al. Systems biology of autosomal dominant polycystic kidney disease (ADPKD): computational identification of gene expression pathways and integrated regulatory networks. Hum Mol Genet. 2009;18:2328–2343. doi: 10.1093/hmg/ddp165. doi:10.1093/hmg/ddp165. [DOI] [PubMed] [Google Scholar]
- 46.Romaker D, Puetz M, Teschner S, et al. Increased expression of secreted frizzled-related protein 4 in polycystic kidneys. J Am Soc Nephrol. 2009;20:48–56. doi: 10.1681/ASN.2008040345. doi:10.1681/ASN.2008040345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wallingford JB, Mitchell B. Strange as it may seem: the many links between Wnt signaling, planar cell polarity, and cilia. Genes Dev. 2011;25:201–213. doi: 10.1101/gad.2008011. doi:10.1101/gad.2008011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Corbit KC, Shyer AE, Dowdle WE, et al. Kif3a constrains beta-catenin-dependent Wnt signalling through dual ciliary and non-ciliary mechanisms. Nat Cell Biol. 2008;10:70–76. doi: 10.1038/ncb1670. doi:10.1038/ncb1670. [DOI] [PubMed] [Google Scholar]
- 49.Gerdes JM, Liu Y, Zaghloul NA, et al. Disruption of the basal body compromises proteasomal function and perturbs intracellular Wnt response. Nat Genet. 2007;39:1350–1360. doi: 10.1038/ng.2007.12. doi:10.1038/ng.2007.12. [DOI] [PubMed] [Google Scholar]
- 50.Ocbina PJ, Tuson M, Anderson KV. Primary cilia are not required for normal canonical Wnt signaling in the mouse embryo. PLoS One. 2009;4:e6839. doi: 10.1371/journal.pone.0006839. doi:10.1371/journal.pone.0006839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Simons M, Mlodzik M. Planar cell polarity signaling: from fly development to human disease. Annu Rev Genet. 2008;42:517–540. doi: 10.1146/annurev.genet.42.110807.091432. doi:10.1146/annurev.genet.42.110807.091432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jones C, Roper VC, Foucher I, et al. Ciliary proteins link basal body polarization to planar cell polarity regulation. Nat Genet. 2008;40:69–77. doi: 10.1038/ng.2007.54. doi:10.1038/ng.2007.54. [DOI] [PubMed] [Google Scholar]
- 53.Karner CM, Chirumamilla R, Aoki S, et al. Wnt9b signaling regulates planar cell polarity and kidney tubule morphogenesis. Nat Genet. 2009;41:793–799. doi: 10.1038/ng.400. doi:10.1038/ng.400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Simons M, Gloy J, Ganner A, et al. Inversin, the gene product mutated in nephronophthisis type II, functions as a molecular switch between Wnt signaling pathways. Nat Genet. 2005;37:537–543. doi: 10.1038/ng1552. doi:10.1038/ng1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Watanabe D, Saijoh Y, Nonaka S, et al. The left-right determinant Inversin is a component of node monocilia and other 9+0 cilia. Development. 2003;130:1725–1734. doi: 10.1242/dev.00407. doi:10.1242/dev.00407. [DOI] [PubMed] [Google Scholar]
- 56.Bergmann C, Fliegauf M, Bruchle NO, et al. Loss of nephrocystin-3 function can cause embryonic lethality, Meckel-Gruber-like syndrome, situs inversus, and renal-hepatic-pancreatic dysplasia. Am J Hum Genet. 2008;82:959–970. doi: 10.1016/j.ajhg.2008.02.017. doi:10.1016/j.ajhg.2008.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ross AJ, May-Simera H, Eichers ER, et al. Disruption of Bardet–Biedl syndrome ciliary proteins perturbs planar cell polarity in vertebrates. Nat Genet. 2005;37:1135–1140. doi: 10.1038/ng1644. doi:10.1038/ng1644. [DOI] [PubMed] [Google Scholar]
- 58.Fischer E, Legue E, Doyen A, et al. Defective planar cell polarity in polycystic kidney disease. Nat Genet. 2006;38:21–23. doi: 10.1038/ng1701. doi:10.1038/ng1701. [DOI] [PubMed] [Google Scholar]
- 59.Baena-Lopez LA, Baonza A, Garcia-Bellido A. The orientation of cell divisions determines the shape of Drosophila organs. Curr Biol. 2005;15:1640–1644. doi: 10.1016/j.cub.2005.07.062. doi:10.1016/j.cub.2005.07.062. [DOI] [PubMed] [Google Scholar]
- 60.Gong Y, Mo C, Fraser SE. Planar cell polarity signalling controls cell division orientation during zebrafish gastrulation. Nature. 2004;430:689–693. doi: 10.1038/nature02796. doi:10.1038/nature02796. [DOI] [PubMed] [Google Scholar]
- 61.Bonnet CS, Aldred M, von Ruhland C, et al. Defects in cell polarity underlie TSC and ADPKD-associated cystogenesis. Hum Mol Genet. 2009;18:2166–2176. doi: 10.1093/hmg/ddp149. doi:10.1093/hmg/ddp149. [DOI] [PubMed] [Google Scholar]
- 62.Verdeguer F, Le Corre S, Fischer E, et al. A mitotic transcriptional switch in polycystic kidney disease. Nat Med. 2010;16:106–110. doi: 10.1038/nm.2068. doi:10.1038/nm.2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jonassen JA, San Agustin J, Follit JA, et al. Deletion of IFT20 in the mouse kidney causes misorientation of the mitotic spindle and cystic kidney disease. J Cell Biol. 2008;183:377–384. doi: 10.1083/jcb.200808137. doi:10.1083/jcb.200808137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Nishio S, Tian X, Gallagher AR, et al. Loss of oriented cell division does not initiate cyst formation. J Am Soc Nephrol. 2010;21:295–302. doi: 10.1681/ASN.2009060603. doi:10.1681/ASN.2009060603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jonassen JA, SanAgustin J, Baker SP, et al. Disruption of IFT complex A causes cystic kidneys without mitotic spindle misorientation. J Am Soc Nephrol. 2012;23:641–651. doi: 10.1681/ASN.2011080829. doi:10.1681/ASN.2011080829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hirokawa N, Tanaka Y, Okada Y. Cilia, KIF3 molecular motor and nodal flow. Curr Opin Cell Biol. 2012;24:31–39. doi: 10.1016/j.ceb.2012.01.002. doi:10.1016/j.ceb.2012.01.002. [DOI] [PubMed] [Google Scholar]
- 67.McGrath J, Somlo S, Makova S, et al. Two populations of node monocilia initiate left-right asymmetry in the mouse. Cell. 2003;114:61–73. doi: 10.1016/s0092-8674(03)00511-7. doi:10.1016/S0092-8674(03)00511-7. [DOI] [PubMed] [Google Scholar]
- 68.Bataille S, Demoulin N, Devuyst O, et al. Association of PKD2 (polycystin 2) mutations with left–right laterality defects. Am J Kidney Dis. 2011;58:456–460. doi: 10.1053/j.ajkd.2011.05.015. doi:10.1053/j.ajkd.2011.05.015. [DOI] [PubMed] [Google Scholar]
- 69.Mitchell B, Jacobs R, Li J, et al. A positive feedback mechanism governs the polarity and motion of motile cilia. Nature. 2007;447:97–101. doi: 10.1038/nature05771. doi:10.1038/nature05771. [DOI] [PubMed] [Google Scholar]
- 70.Wallingford JB. Planar cell polarity signaling, cilia and polarized ciliary beating. Curr Opin Cell Biol. 2010;22:597–604. doi: 10.1016/j.ceb.2010.07.011. doi:10.1016/j.ceb.2010.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bornens M. The centrosome in cells and organisms. Science. 2012;335:422–426. doi: 10.1126/science.1209037. doi:10.1126/science.1209037. [DOI] [PubMed] [Google Scholar]
- 72.Guirao B, Meunier A, Mortaud S, et al. Coupling between hydrodynamic forces and planar cell polarity orients mammalian motile cilia. Nat Cell Biol. 2010;12:341–350. doi: 10.1038/ncb2040. doi:10.1038/ncb2040. [DOI] [PubMed] [Google Scholar]
- 73.McCue S, Dajnowiec D, Xu F, et al. Shear stress regulates forward and reverse planar cell polarity of vascular endothelium in vivo and in vitro. Circ Res. 2006;98:939–946. doi: 10.1161/01.RES.0000216595.15868.55. doi:10.1161/01.RES.0000216595.15868.55. [DOI] [PubMed] [Google Scholar]
- 74.Van der Heiden K, Egorova AD, Poelmann RE, et al. Role for primary cilia as flow detectors in the cardiovascular system. Int Rev Cell Mol Biol. 2011;290:87–119. doi: 10.1016/B978-0-12-386037-8.00004-1. doi:10.1016/B978-0-12-386037-8.00004-1. [DOI] [PubMed] [Google Scholar]
- 75.Piel M, Meyer P, Khodjakov A, et al. The respective contributions of the mother and daughter centrioles to centrosome activity and behavior in vertebrate cells. J Cell Biol. 2000;149:317–330. doi: 10.1083/jcb.149.2.317. doi:10.1083/jcb.149.2.317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hofherr A, Kottgen M. TRPP channels and polycystins. Adv Exp Med Biol. 2011;704:287–313. doi: 10.1007/978-94-007-0265-3_16. doi:10.1007/978-94-007-0265-3_16. [DOI] [PubMed] [Google Scholar]
- 77.Chauvet V, Tian X, Husson H, et al. Mechanical stimuli induce cleavage and nuclear translocation of the polycystin-1 C terminus. J Clin Invest. 2004;114:1433–1443. doi: 10.1172/JCI21753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Low SH, Vasanth S, Larson CH, et al. Polycystin-1, STAT6, and P100 function in a pathway that transduces ciliary mechanosensation and is activated in polycystic kidney disease. Dev Cell. 2006;10:57–69. doi: 10.1016/j.devcel.2005.12.005. doi:10.1016/j.devcel.2005.12.005. [DOI] [PubMed] [Google Scholar]
- 79.Bertuccio CA, Chapin HC, Cai Y, et al. Polycystin-1 C-terminal cleavage is modulated by polycystin-2 expression. J Biol Chem. 2009;284:21011–21026. doi: 10.1074/jbc.M109.017756. doi:10.1074/jbc.M109.017756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Olsan EE, Mukherjee S, Wulkersdorfer B, et al. Signal transducer and activator of transcription-6 (STAT6) inhibition suppresses renal cyst growth in polycystic kidney disease. Proc Natl Acad Sci USA. 2011;108:18067–18072. doi: 10.1073/pnas.1111966108. doi:10.1073/pnas.1111966108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Huber TB, Walz G, Kuehn EW. mTOR and rapamycin in the kidney: signaling and therapeutic implications beyond immunosuppression. Kidney Int. 2011;79:502–511. doi: 10.1038/ki.2010.457. doi:10.1038/ki.2010.457. [DOI] [PubMed] [Google Scholar]
- 82.Inoki K, Li Y, Zhu T, et al. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol. 2002;4:648–657. doi: 10.1038/ncb839. doi:10.1038/ncb839. [DOI] [PubMed] [Google Scholar]
- 83.Tao Y, Kim J, Schrier RW, et al. Rapamycin markedly slows disease progression in a rat model of polycystic kidney disease. J Am Soc Nephrol. 2005;16:46–51. doi: 10.1681/ASN.2004080660. doi:10.1681/ASN.2004080660. [DOI] [PubMed] [Google Scholar]
- 84.Foster DA, Yellen P, Xu L, et al. Regulation of G1 cell cycle progression: distinguishing the restriction point from a nutrient-sensing cell growth checkpoint(s) Genes Cancer. 2010;1:1124–1131. doi: 10.1177/1947601910392989. doi:10.1177/1947601910392989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Polak P, Hall MN. mTOR and the control of whole body metabolism. Curr Opin Cell Biol. 2009;21:209–218. doi: 10.1016/j.ceb.2009.01.024. doi:10.1016/j.ceb.2009.01.024. [DOI] [PubMed] [Google Scholar]
- 86.Distefano G, Boca M, Rowe I, et al. Polycystin-1 regulates extracellular signal-regulated kinase-dependent phosphorylation of tuberin to control cell size through mTOR and its downstream effectors S6K and 4EBP1. Mol Cell Biol. 2009;29:2359–2371. doi: 10.1128/MCB.01259-08. doi:10.1128/MCB.01259-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Shillingford JM, Murcia NS, Larson CH, et al. The mTOR pathway is regulated by polycystin-1, and its inhibition reverses renal cystogenesis in polycystic kidney disease. Proc Natl Acad Sci USA. 2006;103:5466–5471. doi: 10.1073/pnas.0509694103. doi:10.1073/pnas.0509694103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Shaw RJ. LKB1 and AMP-activated protein kinase control of mTOR signalling and growth. Acta Physiol (Oxf) 2009;196:65–80. doi: 10.1111/j.1748-1716.2009.01972.x. doi:10.1111/j.1748-1716.2009.01972.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Armour EA, Carson RP, Ess KC. Cystogenesis and Elongated Primary Cilia in Tsc1-Deficient Distal Convoluted Tubules. Am J Physiol Renal Physiol. 2012;303:F584–592. doi: 10.1152/ajprenal.00141.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.DiBella LM, Park A, Sun Z. Zebrafish Tsc1 reveals functional interactions between the cilium and the TOR pathway. Hum Mol Genet. 2009;18:595–606. doi: 10.1093/hmg/ddn384. doi:10.1093/hmg/ddn384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hartman TR, Liu D, Zilfou JT, et al. The tuberous sclerosis proteins regulate formation of the primary cilium via a rapamycin-insensitive and polycystin 1-independent pathway. Hum Mol Genet. 2009;18:151–163. doi: 10.1093/hmg/ddn325. doi:10.1093/hmg/ddn325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Yuan S, Li J, Diener DR, et al. Target-of-rapamycin complex 1 (Torc1) signaling modulates cilia size and function through protein synthesis regulation. Proc Natl Acad Sci USA. 2012;109:2021–2026. doi: 10.1073/pnas.1112834109. doi:10.1073/pnas.1112834109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Rydholm S, Zwartz G, Kowalewski JM, et al. Mechanical properties of primary cilia regulate the response to fluid flow. Am J Physiol Renal Physiol. 2010;298:F1096–1102. doi: 10.1152/ajprenal.00657.2009. [DOI] [PubMed] [Google Scholar]
- 94.Besschetnova TY, Kolpakova-Hart E, Guan Y, et al. Identification of signaling pathways regulating primary cilium length and flow-mediated adaptation. Curr Biol. 2010;20:182–187. doi: 10.1016/j.cub.2009.11.072. doi:10.1016/j.cub.2009.11.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Verghese E, Ricardo SD, Weidenfeld R, et al. Renal primary cilia lengthen after acute tubular necrosis. J Am Soc Nephrol. 2009;20:2147–2153. doi: 10.1681/ASN.2008101105. doi:10.1681/ASN.2008101105. [DOI] [PMC free article] [PubMed] [Google Scholar]