

Abstract
Functional defects in ATPase class I type 8B membrane 1 (ATP8B1 or familial intrahepatic cholestasis 1, FIC1) lead to cholestasis by mechanism(s) that are not fully understood. One proposed pathophysiology involves aberrant signaling to the bile acid sensor, the farnesoid X receptor (FXR), via protein kinase C ζ (PKCζ). The following cell line-based studies investigated whether phospholipase D2 may transduce a signal from FIC1 to FXR. PLD2 gain of function led to activation of the bile salt export pump (BSEP) promoter, a well-characterized FXR response. BSEP activation by PLD2 could be blocked by abrogating either PKCζ or FXR signaling. PLD2 loss of function led to a reduction in BSEP promoter activity. In addition, a variety of proteins that are activated by FXR, including BSEP, were reduced in HepG2 cells treated with PLD2 siRNA. Similar effects were observed in freshly isolated human hepatocytes. Activation of BSEP by FIC1 gain of function was blocked when PLD2 but not PLD1 was silenced. Overexpression of wild-type but not Byler mutant FIC1 led to an increase in membrane associated PLD activity. An intermediate level of activation of PLD activity was induced when a benign recurrent intrahepatic cholestasis FIC1 mutant construct was expressed. These studies show that FIC1 signals to FXR via a signaling pathway including PLD2 and PKCζ.
Keywords: bile, cholestasis, intestine, ileum, lipid, phoshatidic acid, liver, gallbladder, promoter
Byler disease is a form of intrahepatic cholestasis with extrahepatic manifestations (1, 2). Mutations in ATPase class I type 8B member 1 (ATP8B1) underlie many of the reported cases of Byler disease (3, 4). Byler disease has also been referred to as progressive familial intrahepatic cholestasis type 1, and as such, ATP8B1 is known as FIC1 and the disease as FIC1 disease. The clinical severity of liver disease can be correlated with predictions of the effects of specific mutations and from analyses of the proposed functions of FIC1 (5–8). Severe disease is known as Byler disease, whereas milder disease may be intermittent with limited if any progression to cirrhosis and thus known as benign recurrent intrahepatic cholestasis (BRIC). This categorization may be an oversimplification with a clinical continuum of disease being reported (9). FIC1 is expressed in a wide variety of tissues, which in part explains the extrahepatic manifestations of FIC1 disease (3). A mouse model of FIC1 disease is complex in its hepatic presentation and does not completely recapitulate the liver disease observed in humans, although pulmonary issues and hearing defects have been described in these mice (10–12).
The exact molecular pathophysiology of the liver disease associated with mutations in ATP8B1 remains an area of on-going investigations and controversy. FIC1 is a member of the family of P4-ATPases and has been shown to facilitate transport of aminophospholipids, presumably by flipping of aminophospholipids from the outer to inner hemi-leaflet of the lipid bilayer (13–15). One explanation for the cholestasis in FIC1 disease is that loss of normal phospholipid asymmetry at the canalicular membrane leads to diminished membrane resistance to hydrophobic bile salts, reduced canalicular cholesterol and ectoenzymes, and lessened functional capacity of the canalicular bile salt export pump (BSEP, ABCB11) (16–19). Reduced FIC1 expression may lead to apical membrane alterations independent of aminophospholipid transport (20). An alternative explanation for the pathophysiology of FIC1-related diseases to changes in bile acid-related signal transduction pathways. Abrogation of FIC1 activity has been reported by several but not all groups to lead to diminished signaling via the bile acid sensor, the farnesoid X receptor (FXR) (17, 21–24). As FXR activates BSEP and inactivates the ileal bile acid transporter ASBT, FIC1 disease would be associated with a reduction in canalicular bile acid excretion and augmentation of intestinal reabsorption of bile acids. This related pathophysiology is supported in part by cell line-based studies and in limited fashion using human tissues (7, 21, 22, 25). The signal transduction pathway influenced by FIC1 involves protein kinase C ζ (PKCζ), whereby PKCζ phosphorylates and activates FXR (7, 25). The following studies were stimulated by the observation that phospholipase D2 has been shown to activate PKCζ (26).
EXPERIMENTAL PROCEDURES
Cells and cell culture
A mutant Chinese hamster ovary cell line that has defective uptake of phosphatidylserine (UPS cells, generous gift from Richard Pagano, Mayo Medical Center, Rochester, MN) was used for FIC1 gain of function in these studies because it has been shown to lack endogenous FIC1 expression (7, 13, 27). UPS cells can be readily transfected with and express FIC1, enabling assessment of the role of PLD2 in FIC1-mediated signaling, as has been done for PKCζ (7). UPS cells were grown and maintained in Ham's F-12 medium supplemented with 5% fetal calf serum. HepG2 (HB-8065) cells were obtained from the American Type Culture Collection (Rockville, MD) and maintained in Eagle's minimum essential medium (Invitrogen) containing 2 mM L-glutamine, 1.5 g/liter sodium bicarbonate, 0.1 mM nonessential amino acids, 1.0 mM sodium pyruvate, and 10% fetal calf serum (Invitrogen). Normal human hepatocytes (obtained through the Liver Tissue Cell Distribution System, Pittsburgh, PA; NIH Contract #N01-DK-7-0004 / HHSN267200700004C) were cultured in Clonetics HMM medium (Lonza, Walkersville, MD), supplemented with 5% fetal calf serum. UPS cells were cultured at 33°C, while HepG2 cells and primary human hepatocytes were cultured at 37°C, both in 5% CO2. In studies of the effect of the FXR ligand chenodeoxycholic acid (CDCA), media was changed to 0.5% charcoal-treated fetal calf serum to avoid the effects of FXR ligands, e.g., bile acids, in fetal calf serum (7).
Plasmid constructs, silencing vectors, enzyme inhibitors, and antibodies
Two hundred thirty-one base pairs of the human BSEP promoter (−145 to +86) linked to a luciferase expression vector (28) (hBSEP) was used as a readout of the FXR activity as previously described (7, 25). FXREµ is a construct where the cis-element for FXR is mutated, thereby abolishing FXR-mediated responses (28).
Gain-of-function studies utilized a previously described expression construct for FIC1 (13) and a generous gift from Dr. Sung Ho Ryu for PLD2 (26, 29). FIC1 mutant expression constructs that mimic Byler and BRIC disease were also utilized as previously described (7, 30).
Loss of function was achieved using silencing constructs or enzyme inhibitors. Specific siRNA constructs were used to silence FIC1 (21), PLD2 (sc-44001; Santa Cruz Biotechnology, Santa Cruz, CA), PLD1 (sc-44000; Santa Cruz Biotechnology), PKCζ (7), and FXR (31). A scrambled anti-sense construct, siScr, was utilized as a control (21). PKCζ pseudosubstrate inhibitor (PSI) (myristoylated, 100 µM) (32, 33), purchased from EMD Biosciences Inc. (San Diego, CA), was used to inhibit PKCζ. 5-Fluoro-2-Indoyl-1H-indole-2-carboxamide (FIPI), purchased from Cayman Chemicals (Ann Arbor, MI), was used to inhibit phospholipase D (34).
Antibodies used for Western analyses were obtained from Santa Cruz Biotechnology Inc., and were directed against the following proteins: PLD2 (sc-25513), PLD1 (sc-25512), FIC1 (sc-134967), PKCζ (sc-216), phospho-PKCζ (Thr 410) (sc-12894-R), Na/K-ATPase α (sc-48345), FXR (sc-1204), short heterodimer partner (SHP) (sc-15283), organic solute transporter (Ost)β (sc-163192), scavenger receptor (SR)-B1 (67098), BSEP (sc-74500), and β-actin (sc-81178).
Transient transfection assays
The procedures for transient transfection and luciferase analysis of cells were performed as described previously (35). Confluent cells (5 × 106) were transfected with 3 μg of human BSEP-luciferase reporter and 0.3 μg of a control plasmid (pRL-TK; Promega). Luciferase activities were determined by the dual luciferase reporter assay system (Promega) using a Turner 20/20 Luminometer (Turner BioSystems). All transfections were performed in triplicate and repeated in three separate sets of experiments.
Protein preparation, immunoprecipitation, and Western blotting
Whole-cell lysates were prepared from cells suspended in 25 mM Tris-HCI (pH 7.8), 0.5 mM EDTA, and protease inhibitors (Roche, Nutley, NJ). Cells were lysed by four cycles of freezing and thawing, followed by centrifugation at 16,000 g at 4°C for 15 min (36). Cellular membranes were obtained by centrifugation of whole-cell lysate at 3,000 g for 10 min at 4°C, followed by a second spin of the supernatant at 30,000 g for 20 min (37). Western blot analyses were performed as previously described (38). Sample loading was examined with anti-actin or anti Na/K-ATPase antibody.
Phospholipase D assay
Phospholipase D activity was measured using an Amplex Red Phospholipase D Assay Kit (A12219) according to the manufacturer's instructions (Life Technologies, Grand Island, NY).
Statistical analysis
Data was expressed as the means ± SD from at least nine experimental measurements. Differences between experimental groups were evaluated for statistical significance using either ANOVA or Student t-test, where P < 0.05 was considered to be statistically significant (InStat software, GraphPad Inc., San Diego CA).
RESULTS
PLD2 gain of function
The effect of gain of function of PLD2 was assessed in UPS cells. Human BSEP promoter activity was increased in UPS cells transfected with either FIC1 or PLD2; there was a synergistic activation when both FIC1 and PLD2 were overexpressed (Fig. 1A). Previous investigations have shown that FIC1 activates a signal transduction pathway involving PKCζ and FXR (7, 21, 25). The role of PKCζ in mediating the signal induced by PLD2 overexpression was assessed by either silencing PKCζ (siPKCζ) or inhibiting its activity using a myristolated pseudosubstrate inhibitor (PSI) (Fig. 1B). Human BSEP promoter activity was reduced in response to both methods of inhibiting PKCζ activity. In the setting of PKCζ inhibition, PLD2 no longer activated the BSEP promoter. Similar studies were performed examining the effect of blocking the function of FXR (Fig. 1C). Silencing FXR resulted in a marked reduction of BSEP promoter activity. An equivalent effect was observed when a dominant negative FXR was overexpressed. When FXR function was blocked, overexpression of PLD2 did not lead to a statistically significant increase in BSEP promoter activity. The role of FXR was further assessed using a modified human BSEP promoter construct, in which the FXR binding site in the promoter was mutated by site-directed mutagenesis (28) (Fig. 1D). As seen before, the wild-type BSEP promoter was activated by the FXR ligand chenodeoxycholic acid (CDCA) and by overexpression of PLD2. In these investigations, basal activation of the BSEP promoter was minimized by removing fetal calf serum and associated bile acids from the culture media (7). Synergistic activation of the BSEP promoter was observed when cells were transfected with PLD2 and treated with CDCA. A similar effect has been previously observed for PKCζ (25). The basal activity of the FXRE mutant BSEP promoter construct, FXREµ, was reduced and was not activated by CDCA and/or PLD2.
Fig. 1.

Effect of phospholipase D2 gain of function on human BSEP promoter activity. UPS cells were transfected with expression constructs for PLD2 and/or FIC1. The human BSEP promoter was used as a readout of FXR activity. Activity in untreated UPS cells was set at 100% and all results were normalized to renilla luciferase from a pRL-TK construct as a control for transfection efficiency. The scale of the y axis is logarithmic. Error bars represent the SD for nine measurements. A: Effect of PLD2 and FIC1. Human BSEP promoter activity was significantly increased by overexpression of PLD2 and/or FIC1 with synergistic activation by both PLD2 and FIC1. All comparisons were significant at P < 0.001. B: Effect of PKCζ inhibition on PLD2-mediated signaling. UPS cells were transfected with no PLD2 construct (control) or a wild-type PLD2 expression construct. PKCζ was inhibited by either siRNA silencing (siPKCζ) or by treatment with 100 µM myristolated pseudosubstrate inhibitor (PSI). PLD2 activated the BSEP promoter in the untreated UPS cells but not in the cells where PKCζ was either silenced or inhibited. C: Effect of FXR inhibition on PLD2-mediated signaling. UPS cells were transfected with no PLD2 construct (control) or a wild-type PLD2 expression construct. FXR was inhibited by either siRNA silencing (siFXR) or overexpression of a dominant negative FXR (dn FXR). Basal activity of the BSEP promoter was markedly reduced and not significantly increased when FXR was inhibited. D: Effect of an FXR response element mutant BSEP promoter on PLD2 mediated signaling. UPS cells were transfected with a wild-type human BSEP promoter reporter construct or one in which the FXR binding cis-element was mutated (FXREµ). Media for these studies included 0.5% charcoal fetal calf serum to minimize the effect of bile acids in fetal calf serum. Cells were untreated (control), treated with 50 µM CDCA and/or transfected with wild-type PLD2. Treatment with CDCA and/or PLD2 significantly increased the wild-type human BSEP promoter activity, but not the FXR response element mutant. Synergistic activation of wild-type human BSEP promoter was seen when CDCA and PLD2 were combined. All comparisons of the wild-type BSEP promoter were significant at a P < 0.001. There were no significant differences for any of the mutant promoter responses.
PLD2 loss of function
Loss of PLD2 function was assessed by either silencing PLD2 or chemically inhibiting its activity in UPS cells. PLD1 was silenced as a control for the effect of generalized loss of phospholipase D activity. The specificity of the PLD silencing constructs used in these investigations was assessed by Western blotting of total cellular homogenates of UPS cells (Fig. 2). The PLD1 and PLD2 found endogenously in UPS cells can be effectively silenced with commercially available constructs, whereas a scrambled anti-sense construct had no effect. The dose-dependence of the effect of the PLD inhibitor FIPI on BSEP promoter activity was assessed to identify the appropriate concentration for analysis of FIC1 signaling (supplementary Fig. I). Dose-dependent reduction in BSEP promoter activity was observed with FIPI concentrations between 1 and 20 nM. Greater than 50% reduction of activity was observed after 1 nM FIPI treatment, and this concentration was used for subsequent investigation of FIC1-mediated signaling. Silencing or inhibiting PLD2 in UPS cells was associated with a reduction in BSEP promoter activity (Fig. 3A). No effect was observed when PLD1 was silenced or when cells were treated with a scrambled silencing construct (Fig. 3A). FIC1 overexpression in UPS cells lead to marked activation of the BSEP promoter in UPS cells that were untreated (9.2 ± 0.2-fold activation) and those transfected with siPLD1 (9.2 ± 0.2) or a scrambled antisense vector (9.2 ± 0.1) (Fig. 3A, B). In contrast, silencing PLD2 (5.0 ± 0.3) or treatment with 1 nM FIPI (5.6 ± 0.3) was associated with a reduction in the activation of BSEP by FIC1 (Fig. 3A, B).
Fig. 2.
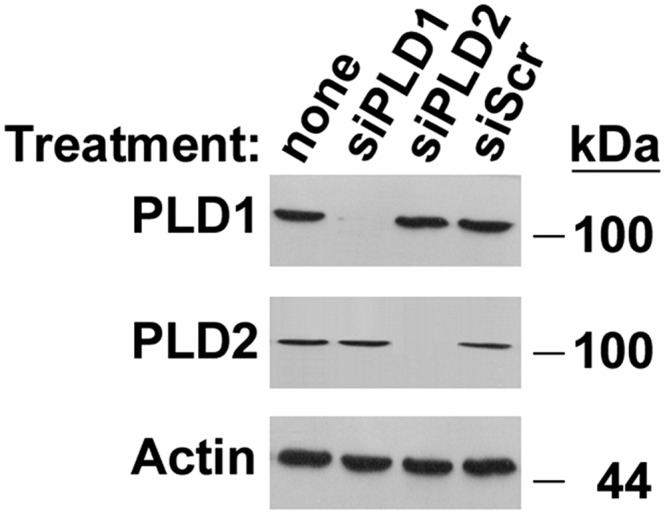
Silencing of PLD1 and PLD2. UPS cells were treated with siPLD1, siPLD2, or a scrambled anti-sense control (siScr). Western blotting was performed with cellular homogenates.
Fig. 3.

Effect of PLD inhibition on FIC1-mediated activation of the human BSEP promoter. UPS cells were untransfected (control) or transfected (FIC1) with a wild-type FIC1 construct. Cells were treated with siPLD1, siPLD2, a scrambled anti-sense control (siScr), or 1 nM FIPI. A: Human BSEP promoter activity. The human BSEP promoter was used as a readout of FXR activity. Activity in untreated UPS cells was set at 100% and all results were normalized to renilla luciferase from a pRL-TK construct as a control for transfection efficiency. The scale of the y axis is logarithmic. Error bars represent the SD for nine measurements. Basal BSEP promoter activity was significantly lower in the UPS cells treated with siPLD2 or FIPI but not siPLD1 or siScr. In each of the treatment groups, FIC1 overexpression led to a statistically significant increase in BSEP promoter activity. B: FIC1 mediated activation of human BSEP promoter activity. The degree of activation (i.e., FIC1 treated/untreated promoter activity) is significantly lower in the siPLD2- and FIPI-treated cells compared with the rest of the experimental groups that are equivalent.
The effect of loss of PLD2 function was also assessed in HepG2 cells and primarily derived human hepatocytes, permitting assessment of the effects of alteration in PLD2 expression on native human BSEP protein expression. As seen in UPS cells, silencing PLD2 and/or FIC1 in HepG2 cells led to a significant reduction in BSEP promoter activity (Fig. 4). In HepG2 cells, silencing PLD2 was associated with a significant reduction in PKCζ and a reduction in the expression of a variety of proteins whose expression is activated by FXR, including FXR (21), SHP (39, 40), OSTβ (31), SR-B1 (41), and BSEP protein (28)(Fig. 5A, B). Similar findings were observed when PLD2 was silenced in primarily derived human hepatocytes (Fig. 5C).
Fig. 4.

Effect of FIC1 and PLD2 loss of function on human BSEP promoter activity in HepG2 cells. HepG2 cells were transfected with the human BSEP promoter as a readout of FXR activity. Activity in untreated HepG2 cells was set at 100% and all results were normalized to renilla luciferase from a pRL-TK construct as a control for transfection efficiency. Cells were then untreated (control) or treated with siPLD2, siFIC1, siPLD2 + siFIC1, or a scrambled siRNA control. The scale of the y axis is linear. Error bars represent the SD for nine measurements. Silencing either PLD2 or FIC1 resulted in a statistically significant reduction in BSEP promoter activity.
Fig. 5.

Effect of PLD2 silencing on hepatocyte protein expression. A: Western blot analysis of PLD2 silencing in HepG2 cells. Three separate sets of experiments are shown. Cells were untreated (none) or treated with siPLD2 or a scrambled control (siScr). Total cellular homogenates were probed for PLD2, PKCζ, FXR, SHP, BSEP, Ostβ, SR-B1, and actin (loading control). Densitometric quantification of band intensity is shown below each band. B: Depiction of statistical analysis of Western blotting of HepG2 cells. Protein levels are expressed as a percentage of the untreated cells. Bars represent the mean for each protein; error bars are SD. With the exception of actin, all of the proteins were reduced by a significant amount compared with either untreated cells or siScr-treated cells. C: Western blot analysis of PLD2 silencing in freshly isolated human hepatocytes.
FIC1 gain of function
The effect of FIC1 expression on PLD2 activity was assessed in UPS cells, which do not express endogenous FIC1. Western blotting was performed on crude membranes from UPS cells transfected with wild-type FIC1 or mutant constructs associated with severe disease (Byler disease) or intermittent disease (BRIC). As we have previously observed (42), FIC1 protein expression was observed in crude membranes prepared from UPS cells treated with either the wild-type or BRIC FIC1 expression constructs (Fig. 6A). The level of membrane expression in the BRIC-transfected cells was less than that of the wild-type FIC1-transfected cells. PLD2 membrane protein expression was slightly increased in the cells transfected with wild-type FIC1, whereas expression was similar in the cells expressing the other FIC1 proteins. This finding is distinct from previously observed similar levels of overall increases in FIC1 protein expression observed in total cellular homogenates treated with wild-type, BRIC, or Byler FIC1 constructs (7). The level of PKCζ and phosphorylated PKCζ (Threonine 410) correlated with the level of expression of FIC1 in the crude membranes. PLD activity was assessed in the crude membranes from the transfected UPS cells. Overexpression of wild-type FIC1 in UPS cells led to a 9-fold increase in PLD activity in crude membrane preparations, whereas BRIC FIC1 expression yielded only a 2-fold increase in PLD activity. Overexpression of the Byler FIC1 protein had no effect of membrane PLD activity (control UPS cells 10.4 ± 0.7, wild-type FIC1-transfected 95.8 ± 14.0, BRIC FIC1-transfected 20.2 ± 1.9, Byler FIC1-transfected 11.3 ± 1.1 mU PLD activity/ml/20 µg membrane protein, n = 9 for each assay, P < 0.001 for control versus wild-type, wild-type versus Byler, and wild-type versus BRIC, Fig. 6B).
Fig. 6.

Effect of FIC1 overexpression in UPS cells. UPS cells were untransfected (−) or transfected with wild-type FIC1 (wt), a Byler disease FIC1 construct (By), or a BRIC disease FIC1 construct (BR). A: Western blot analysis. Crude membranes were probed by Western blot. B: PLD enzyme activity. Depiction of statistical analysis. Mean PLD activity for each treatment is depicted by the bar, with error bars representing SD. PLD activity was highest in the UPS cells transfected with wild-type FIC1. BRIC-transfected cells had an intermediate level of activity, whereas untreated and Byler-transfected cells were equal.
DISCUSSION
These studies suggest the signal transduction pathway activated by FIC1 involves the following cascade: PLD2, PKCζ, and FXR. This represents a novel and distinct role for PLD2. PLD catalyzes the hydrolysis of glycerophospholipids, yielding phosphatidic acid and a head group. A wide spectrum of potential biological functions has been linked to PLD activity, including regulation of vesicle trafficking, development, cytoskeletal structure, morphogenesis, growth, and proliferation (43–46). Phosphatidic acid is a well-described signaling molecule, so it is not surprising that PLD activity could modulate a diverse set of signaling pathways (47). PLD also affects other signaling molecules via direct interactions that may not be dependent upon the catalytic function of PLD (26). PLD2 was first identified by homology screening using a PLD1 cDNA probe (48). Although PLD1 and PLD2 have similar enzymatic functions, PLD2 has distinct characteristics that are relevant for this signaling pathway. PLD1 is predominantly peri-nuclear, whereas PLD2 is primarily found at the plasma membrane. PLD2 is expressed in a wide variety of tissues, including the ileum, pancreas, lung, and liver, which are all potentially relevant for FIC1-mediated signaling effects (29, 49, 50).
PLD2 is regulated by a diverse set of molecules and proteins, including but not limited to fatty acids, phosphoinositides, growth factors, GTP binding proteins, and PKC (43, 46, 51). FIC1 is an integral plasma membrane protein that influences the lipid bilayer, presumably by altering the asymmetric distribution of aminophospholipids. It is not exactly certain how FIC1 activity would influence PLD2, although given the plethora of effects of changes in lipid bilayer composition and the multitude of ways in which PLD2 can be regulated, a regulatory interaction is quite plausible. PLD2 is endogenously expressed in UPS cells; thus, FIC1 may modulate its activity and/or membrane localization. In these studies, PLD2 function was assessed by measuring overall PLD activity in crude membrane fractions. This approach was used as a means of assessing specific effects on PLD2 function, as we are not aware of an available enzyme assay for PLD2 independent of PLD1. Overexpression of FIC1 led to a marked enhancement in PLD2 activity, which was greater than the slight increase in its membrane expression. This finding suggests that FIC1 does not necessarily regulate PLD2 expression but likely modulates its functional activity. FIC1 may modulate the activity of BSEP by modulating cholesterol in the lipid bilayer (18). Overexpression of a nonfunctional FIC1 (Byler) protein had no effect on PLD2 function, whereas a partially functional FIC1 (BRIC) protein had an intermediate effect. The same pattern of responses has been observed for the downstream effects mediated by FXR (7). The absence of FIC1 activity in UPS cells can be compensated for by overexpression of PLD2, suggesting that the level of PLD2 expression may be rate limiting in UPS cells. Conversely, when PLD2 function is abolished by either silencing its expression or inhibiting its activity, the effect of FIC1 on the human BSEP promoter is abrogated.
The signaling pathway influenced by PLD2 overlaps with the previously described pathway involving FIC1, PKCζ, and FXR (7, 21, 25). The effects of PLD2 on FXR-mediated responses can be readily blocked by a variety of approaches that abrogate the effects of either PKCζ or FXR. Similarly, the effects of FIC1 on FXR-mediated responses are blocked when PLD is silenced. It is remarkable that this effect is isoform specific, as silencing PLD1 has no effect, in contrast to the pronounced effect of silencing PLD2. Given the overlap in enzymatic function between PLD1 and PLD2, one would speculate that this is the result of a physical interaction between membrane-associated PLD2 and PKCζ. The effects of FIPI indicate that it is not simple interaction but also relies on PLD function. Phosphatidic acid treatment leads to phosphorylation and activation of PKCζ (52). The biological relevance of these results has been confirmed in HepG2 cells and freshly isolated human hepatocytes in which the canalicular bile salt transporter BSEP is expressed. Silencing PLD2 leads to a reduction in BSEP protein expression. Other FXR-responsive genes, including SHP, Ostß, and SR-B1, are also regulated by the level of expression of PLD2.
In summary, these studies have demonstrated a new signaling pathway that specifically involves PLD2. FIC1 alters PLD2 function, leading to PKCζ-mediated activation of FXR with subsequent effects on downstream bile acid homeostatic molecules such as BSEP. These observations are novel and will require further investigation to determine the exact molecular mechanisms involved in the interaction between FIC1 and PLD2 and between PLD2 and PKCζ.
Supplementary Material
Footnotes
Abbreviations:
- ATP8B1
- ATPase class I type 8B membrane 1
- BRIC
- benign recurrent intrahepatic cholestasis
- BSEP
- bile salt export pump
- CDCA
- chenodeoxycholic acid
- FIC1
- familial intrahepatic cholestasis 1
- FIPI
- 5-fluoro-2-indoyl-1H-indole-2-carboxamide
- FXR
- farnesoid X receptor
- OSTβ
- organic solute transporter β
- PKC
- protein kinase C
- PLD
- phospholipase D
- PSI
- pseudosubstrate inhibitor
- SHP
- short heterodimer partner
- SR-B1
- scavenger receptor-B1
- UPS
- uptake of phosphatidylserine
This work was supported in part by National Institutes of Health Grant DK-080808 to B.L.S. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the National Institutes of Health.
The online version of this article (available at http://www.jlr.org) contains supplementary data in the form of one figure.
REFERENCES
- 1.Clayton R. J., Iber F. L., Ruebner B. H., McKusick V. A. 1965. Byler disease. Fatal familial intrahepatic cholestasis in an Amish kindred. J. Pediatr. 67: 1025–1028 [PubMed] [Google Scholar]
- 2.Alissa F. T., Jaffe R., Shneider B. L. 2008. Update on progressive familial intrahepatic cholestasis. J. Pediatr. Gastroenterol. Nutr. 46: 241–252 [DOI] [PubMed] [Google Scholar]
- 3.Bull L. N., van Eijk M. J. T., Pawlikowska L., DeYoung J. A., Juiun J., Liao M., Klomp L. W. J., Lomri N., Berger R., Scharschmidt B. F., et al. 1998. Identification of a P-type ATPase mutated in two forms of hereditary cholestasis. Nat. Genet. 18: 219–224 [DOI] [PubMed] [Google Scholar]
- 4.Pawlikowska L., Strautnieks S., Jankowska I., Czubkowski P., Emerick K., Antoniou A., Wanty C., Fischler B., Jacquemin E., Wali S., et al. 2010. Differences in presentation and progression between severe FIC1 and BSEP deficiencies. J. Hepatol. 53: 170–178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Klomp L. W., Vargas J. C., van Mil S. W., Pawlikowska L., Strautnieks S. S., van Eijk M. J., Juijn J. A., Pabon-Pena C., Smith L. B., DeYoung J. A., et al. 2004. Characterization of mutations in ATP8B1 associated with hereditary cholestasis. Hepatology. 40: 27–38 [DOI] [PubMed] [Google Scholar]
- 6.Folmer D. E., van der Mark V. A., Ho-Mok K. S., Oude Elferink R. P., Paulusma C. C. 2009. Differential effects of progressive familial intrahepatic cholestasis type 1 and benign recurrent intrahepatic cholestasis type 1 mutations on canalicular localization of ATP8B1. Hepatology. 50: 1597–1605 [DOI] [PubMed] [Google Scholar]
- 7.Frankenberg T., Miloh T., Chen F. Y., Ananthanarayanan M., Sun A. Q., Balasubramaniyan N., Arias I., Setchell K. D., Suchy F. J., Shneider B. L. 2008. The membrane protein ATPase class I type 8B member 1 signals through protein kinase C zeta to activate the farnesoid X receptor. Hepatology. 48: 1896–1905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stone A., Chau C., Eaton C., Foran E., Kapur M., Prevatt E., Belkin N., Kerr D., Kohlin T. K., Williamson P.2012. Biochemical characterization of P4-ATPase mutations identified in patients with progressive familial intrahepatic cholestasis. J. Biol. Chem. 287: 41139–41151. [DOI] [PMC free article] [PubMed]
- 9.van Ooteghem N. A., Klomp L. W., van Berge-Henegouwen G. P., Houwen R. H. 2002. Benign recurrent intrahepatic cholestasis progressing to progressive familial intrahepatic cholestasis: low GGT cholestasis is a clinical continuum. J. Hepatol. 36: 439–443 [DOI] [PubMed] [Google Scholar]
- 10.Pawlikowska L., Groen A., Eppens E. F., Kunne C., Ottenhoff R., Looije N., Knisely A. S., Killeen N. P., Bull L. N., Elferink R. P., et al. 2004. A mouse genetic model for familial cholestasis caused by ATP8B1 mutations reveals perturbed bile salt homeostasis but no impairment in bile secretion. Hum. Mol. Genet. 13: 881–892 [DOI] [PubMed] [Google Scholar]
- 11.Stapelbroek J. M., Peters T. A., van Beurden D. H., Curfs J. H., Joosten A., Beynon A. J., van Leeuwen B. M., van der Velden L. M., Bull L., Oude Elferink R. P., et al. 2009. ATP8B1 is essential for maintaining normal hearing. Proc. Natl. Acad. Sci. USA. 106: 9709–9714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ray N. B., Durairaj L., Chen B. B., McVerry B. J., Ryan A. J., Donahoe M., Waltenbaugh A. K., O'Donnell C. P., Henderson F. C., Etscheidt C. A., et al. 2010. Dynamic regulation of cardiolipin by the lipid pump Atp8b1 determines the severity of lung injury in experimental pneumonia. Nat. Med. 16: 1120–1127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ujhazy P., Ortiz D., Misra S., Li S., Moseley J., Jones H., Arias I. 2001. Familial intrahepatic cholestasis 1: studies of localization and function. Hepatology. 34: 768–775 [DOI] [PubMed] [Google Scholar]
- 14.Paulusma C. C., Folmer D. E., Ho-Mok K. S., de Waart D. R., Hilarius P. M., Verhoeven A. J., Oude Elferink R. P. 2008. ATP8B1 requires an accessory protein for endoplasmic reticulum exit and plasma membrane lipid flippase activity. Hepatology. 47: 268–278 [DOI] [PubMed] [Google Scholar]
- 15.Folmer D. E., Elferink R. P., Paulusma C. C. 2009. P4 ATPases - lipid flippases and their role in disease. Biochim. Biophys. Acta. 1791: 628–635 [DOI] [PubMed] [Google Scholar]
- 16.Paulusma C. C., Groen A., Kunne C., Ho-Mok K. S., Spijkerboer A. L., Rudi de Waart D., Hoek F. J., Vreeling H., Hoeben K. A., van Marle J., et al. 2006. Atp8b1 deficiency in mice reduces resistance of the canalicular membrane to hydrophobic bile salts and impairs bile salt transport. Hepatology. 44: 195–204 [DOI] [PubMed] [Google Scholar]
- 17.Cai S. Y., Gautam S., Nguyen T., Soroka C. J., Rahner C., Boyer J. L. 2009. ATP8B1 deficiency disrupts the bile canalicular membrane bilayer structure in hepatocytes, but FXR expression and activity are maintained. Gastroenterology. 136: 1060–1069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Paulusma C. C., de Waart D. R., Kunne C., Mok K. S., Elferink R. P. 2009. Activity of the bile salt export pump (ABCB11) is critically dependent on canalicular membrane cholesterol content. J. Biol. Chem. 284: 9947–9954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Groen A., Romero M. R., Kunne C., Hoosdally S. J., Dixon P. H., Wooding C., Williamson C., Seppen J., Van den Oever K., Mok K. S., et al. 2011. Complementary functions of the flippase ATP8B1 and the floppase ABCB4 in maintaining canalicular membrane integrity. Gastroenterology 141: 1927–1937.e1–4. [DOI] [PubMed]
- 20.Verhulst P. M., van der Velden L. M., Oorschot V., van Faassen E. E., Klumperman J., Houwen R. H., Pomorski T. G., Holthuis J. C., Klomp L. W. 2010. A flippase-independent function of ATP8B1, the protein affected in familial intrahepatic cholestasis type 1, is required for apical protein expression and microvillus formation in polarized epithelial cells. Hepatology. 51: 2049–2060 [DOI] [PubMed] [Google Scholar]
- 21.Chen F., Ananthanarayanan M., Emre S., Neimark E., Bull L. N., Knisely A. S., Strautnieks S. S., Thompson R. J., Magid M. S., Gordon R., et al. 2004. Progressive familial intrahepatic cholestasis, type 1, is associated with decreased farnesoid X receptor activity. Gastroenterology. 126: 756–764 [DOI] [PubMed] [Google Scholar]
- 22.Alvarez L., Jara P., Sanchez-Sabate E., Hierro L., Larrauri J., Diaz M. C., Camarena C., De La Vega A., Frauca E., Lopez-Collazo E., et al. 2004. Reduced hepatic expression of farnesoid X receptor in hereditary cholestasis associated to mutation in ATP8B1. Hum. Mol. Genet. 13: 2451–2460 [DOI] [PubMed] [Google Scholar]
- 23.Martinez-Fernandez P., Hierro L., Jara P., Alvarez L. 2009. Knockdown of ATP8B1 expression leads to specific downregulation of the bile acid sensor FXR in HepG2 cells: effect of the FXR agonist GW4064. Am. J. Physiol. Gastrointest. Liver Physiol. 296: G1119–G1129 [DOI] [PubMed] [Google Scholar]
- 24.Koh S., Takada T., Kukuu I., Suzuki H. 2009. FIC1-mediated stimulation of FXR activity is decreased with PFIC1 mutations in HepG2 cells. J. Gastroenterol. 44: 592–600 [DOI] [PubMed] [Google Scholar]
- 25.Chen F., Ellis E., Strom S. C., Shneider B. L. 2010. ATPase Class I Type 8B Member 1 and protein kinase C zeta induce the expression of the canalicular bile salt export pump in human hepatocytes. Pediatr. Res. 67: 183–187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim J. H., Ohba M., Suh P. G., Ryu S. H. 2005. Novel functions of the phospholipase D2-Phox homology domain in protein kinase Czeta activation. Mol. Cell. Biol. 25: 3194–3208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hanada K., Pagano R. E. 1995. A Chinese hamster ovary cell mutant defective in the non-endocytic uptake of fluorescent analogs of phosphatidylserine: isolation using a cytosol acidification protocol. J. Cell Biol. 128: 793–804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ananthanarayanan M., Balasubramanian N., Makishima M., Mangelsdorf D., Suchy F. 2001. Human bile salt export pump promoter is transactivated by farsenoid X receptor/bile acid receptor. J. Biol. Chem. 276: 28857–28865 [DOI] [PubMed] [Google Scholar]
- 29.Lopez I., Arnold R. S., Lambeth J. D. 1998. Cloning and initial characterization of a human phospholipase D2 (hPLD2). ADP-ribosylation factor regulates hPLD2. J. Biol. Chem. 273: 12846–12852 [DOI] [PubMed] [Google Scholar]
- 30.Harris M. J., Kagawa T., Dawson P. A., Arias I. M. 2004. Taurocholate transport by hepatic and intestinal bile acid transporters is independent of FIC1 overexpression in Madin-Darby canine kidney cells. J. Gastroenterol. Hepatol. 19: 819–825 [DOI] [PubMed] [Google Scholar]
- 31.Frankenberg T., Rao A., Chen F., Haywood J., Shneider B. L., Dawson P. A. 2006. Regulation of the mouse organic solute transporter alpha-beta, Ostalpha-Ostbeta, by bile acids. Am. J. Physiol. Gastrointest. Liver Physiol. 290: G912–G922 [DOI] [PubMed] [Google Scholar]
- 32.McConkey M., Gillin H., Webster C. R., Anwer M. S. 2004. Cross-talk between protein kinases Czeta and B in cyclic AMP-mediated sodium taurocholate co-transporting polypeptide translocation in hepatocytes. J. Biol. Chem. 279: 20882–20888 [DOI] [PubMed] [Google Scholar]
- 33.Muscella A., Storelli C., Marsigliante S. 2005. Atypical PKC-zeta and PKC-iota mediate opposing effects on MCF-7 Na+/K+ATPase activity. J. Cell. Physiol. 205: 278–285 [DOI] [PubMed] [Google Scholar]
- 34.Su W., Yeku O., Olepu S., Genna A., Park J. S., Ren H., Du G., Gelb M. H., Morris A. J., Frohman M. A. 2009. 5-Fluoro-2-indolyl des-chlorohalopemide (FIPI), a phospholipase D pharmacological inhibitor that alters cell spreading and inhibits chemotaxis. Mol. Pharmacol. 75: 437–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen F., Ma L., Al-Ansari N., Shneider B. 2001. The role of AP-1 in the transcriptional regulation of the rat apical sodium-dependent bile acid transporter. J. Biol. Chem. 276: 38703–38714 [DOI] [PubMed] [Google Scholar]
- 36.Malter J. S. 1989. Identification of an AUUUA-specific messenger RNA binding protein. Science. 246: 664–666 [DOI] [PubMed] [Google Scholar]
- 37.Coppola C. P., Gosche J. R., Arrese M., Ancowitz B., Madsen J., Vanderhoof J., Shneider B. L. 1998. Molecular analysis of the adaptive response of intestinal bile acid transport after ileal resection. Gastroenterology. 115: 1172–1178 [DOI] [PubMed] [Google Scholar]
- 38.Shneider B. L., Dawson P. A., Christie D. M., Hardikar W., Wong M. H., Suchy F. J. 1995. Cloning and molecular characterization of the ontogeny of a rat ileal sodium-dependent bile acid transporter. J. Clin. Invest. 95: 745–754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Goodwin B., Jones S., Price R., Watson M., McKee D., Moore L., Galardi C., Wilson J., Lewis M., Roth M., et al. 2000. A regulatory cascade of the nuclear receptors FXR, SHP-1, and LRH-1 represses bile acid biosynthesis. Mol. Cell. 6: 517–526 [DOI] [PubMed] [Google Scholar]
- 40.Lu T. T., Makishima M., Repa J. J., Schoonjans K., Kerr T. A., Auwerx J., Mangelsdorf D. J. 2000. Molecular basis for feedback regulation of bile acid synthesis by nuclear receptors. Mol. Cell. 6: 507–515 [DOI] [PubMed] [Google Scholar]
- 41.Lambert G., Amar M. J., Guo G., Brewer H. B., Jr, Gonzalez F. J., Sinal C. J. 2003. The farnesoid X-receptor is an essential regulator of cholesterol homeostasis. J. Biol. Chem. 278: 2563–2570 [DOI] [PubMed] [Google Scholar]
- 42.Siebold L., Chen F., Miloh T., Shneider B. 2011. 4-Phenylbutyrate and subenroylanilide hydroxomic acid enhance FXR mediated signaling by wild type and mutant FIC1 proteins - potential pharmacologic approaches to the treatment of Byler Disease and Benign Recurrent Intrahepatic Cholestasis. Hepatology. 54: 88A [Google Scholar]
- 43.Jang J. H., Lee C. S., Hwang D., Ryu S. H. 2012. Understanding of the roles of phospholipase D and phosphatidic acid through their binding partners. Prog. Lipid Res. 51: 71–81 [DOI] [PubMed] [Google Scholar]
- 44.Selvy P. E., Lavieri R. R., Lindsley C. W., Brown H. A. 2011. Phospholipase D: enzymology, functionality, and chemical modulation. Chem. Rev. 111: 6064–6119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Huang P., Frohman M. A. 2007. The potential for phospholipase D as a new therapeutic target. Expert Opin. Ther. Targets. 11: 707–716 [DOI] [PubMed] [Google Scholar]
- 46.McDermott M., Wakelam M. J., Morris A. J. 2004. Phospholipase D. Biochem. Cell Biol. 82: 225–253 [DOI] [PubMed] [Google Scholar]
- 47.Hancock J. F. 2007. PA promoted to manager. Nat. Cell Biol. 9: 615–617 [DOI] [PubMed] [Google Scholar]
- 48.Colley W. C., Sung T. C., Roll R., Jenco J., Hammond S. M., Altshuller Y., Bar-Sagi D., Morris A. J., Frohman M. A. 1997. Phospholipase D2, a distinct phospholipase D isoform with novel regulatory properties that provokes cytoskeletal reorganization. Curr. Biol. 7: 191–201 [DOI] [PubMed] [Google Scholar]
- 49.Meier K. E., Gibbs T. C., Knoepp S. M., Ella K. M. 1999. Expression of phospholipase D isoforms in mammalian cells. Biochim. Biophys. Acta. 1439: 199–213 [DOI] [PubMed] [Google Scholar]
- 50.Kim H., Lee J., Kim S., Shin M. K., Min do S., Shin T. 2007. Differential expression of phospholipases D1 and D2 in mouse tissues. Cell Biol. Int. 31: 148–155 [DOI] [PubMed] [Google Scholar]
- 51.Lan W., Tong L. 2012. Focus on molecules: phospholipase D. Exp. Eye Res. 103: 121–122 [DOI] [PubMed] [Google Scholar]
- 52.Limatola C., Schaap D., Moolenaar W. H., van Blitterswijk W. J. 1994. Phosphatidic acid activation of protein kinase C-zeta overexpressed in COS cells: comparison with other protein kinase C isotypes and other acidic lipids. Biochem. J. 304: 1001–1008 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.