

Abstract
We introduce a method for identifying elements of a protein structure that can be shuffled to make chimeric proteins from two or more homologous parents. Formulating recombination as a graph-partitioning problem allows us to identify noncontiguous segments of the sequence that should be inherited together in the progeny proteins. We demonstrate this noncontiguous recombination approach by constructing a chimera of β-glucosidases from two different kingdoms of life. Although the protein's alpha–beta barrel fold has no obvious subdomains for recombination, noncontiguous SCHEMA recombination generated a functional chimera that takes approximately half its structure from each parent. The X-ray crystal structure shows that the structural blocks that make up the chimera maintain the backbone conformations found in their respective parental structures. Although the chimera has lower β-glucosidase activity than the parent enzymes, the activity was easily recovered by directed evolution. This simple method, which does not rely on detailed atomic models, can be used to design chimeras that take structural, and functional, elements from distantly-related proteins.
Keywords: chimeragenesis, protein recombination, eukaryotic-prokaryotic chimera, GH1, structural conservation, graph partitioning
Introduction
Swapping sequence elements among related proteins1 can produce chimeric proteins with novel behaviors2,3 and improved properties such as enhanced stability.4 Although homologous mutations are much more conservative than random mutations, chimeras of distantly-related proteins have a low probability of retaining fold and function.5 Selecting crossover locations that minimize disruption of the folded structure increases the likelihood that a chimeric protein will be functional.
To design libraries of chimeric proteins, we have used structural information to select crossover locations that minimize the average number of non-native residue–residue contacts in the resulting chimeras.6 The sequence elements are then shuffled and reassembled in the correct order to generate the chimeric progeny. We have used this SCHEMA recombination method to make large numbers of functional enzyme chimeras, with which we have explored the benefits and costs of recombination.3,7–9 We have also shown that stabilities and other properties of these recombined enzymes—the “recombination landscape”—can be predicted with high accuracy using models built by sampling small numbers of chimeras.4,10
To date, we have only considered recombination of sequence blocks that are contiguous along the polypeptide chain. Sequence blocks that are contiguous in the primary structure, however, are not necessarily optimal elements for recombination.11 Here, we introduce a new tool for protein recombination that identifies structural blocks that can be swapped among homologous proteins with minimal disruption. Because elements that are distant in the primary structure are often brought together in the folded protein, structural blocks may not be contiguous in the polypeptide chain. This noncontiguous recombination approach enables design of chimeras and libraries of chimeras with less disruption than can be achieved by swapping blocks of sequence. Less disruption means that we can generate libraries with higher fractions of functional enzymes and enables recombination of more distant homologs.
We demonstrate this new tool by constructing a functional β-glucosidase that derives approximately half of its sequence from each of two distantly-related parents. The crystal structure of this prokaryote–eukaryote chimera illustrates the structurally conservative nature of this recombination: the hybrid structure retains the overall function as well as the detailed structural features of the parental enzymes.
Results
Noncontiguous protein recombination
The goal is to identify blocks that can be shuffled among related parent proteins to create chimeras with minimal disruption. The overall process is illustrated in Figure 1 for the simple case of two parents, but can be extended easily to any number of parents. Starting from one or more structures and a parental sequence alignment [Fig. 1(a)], noncontiguous recombination involves splitting the proteins into a set of blocks [Fig. 1(b)], which are swapped to create chimeras [Fig. 1(c)]. Similar to previous work with recombination of contiguous sequence elements, our disruption metric is the number of non-native residue–residue contacts that are broken in the recombined sequence; we call this the SCHEMA disruption.6 To minimize disruption, the residue–residue contacts that are not shared among the parents and therefore could be broken on recombination are converted into a graph, with residues as nodes and non-native contacts as edges [Fig. 1(d)]. Assigning residues to blocks is then equivalent to partitioning the graph to minimize the number of edges that are cut [Fig. 1(e)]. This is an NP-complete problem,12 but there are heuristic algorithms that can find near optimal solutions very quickly.13 We use hMETIS,14,15 a suite of graph partitioning tools. The hMETIS suite assigns each node to a partition, which corresponds to assigning each residue to a block. The noncontiguous chimeras are then assembled from the shuffled blocks, where a block can comprise multiple sequence fragments that should be inherited together [Fig. 1(f)].
Figure 1.
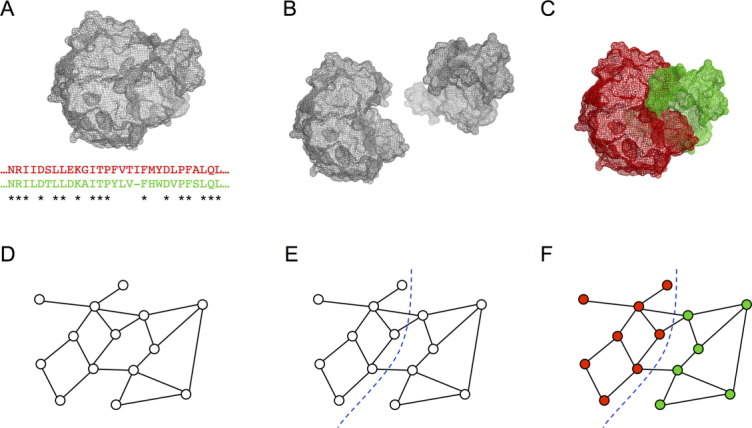
Noncontiguous recombination. (a) One or more structures and a parental sequence alignment are used to identify contacts that are not conserved and can be disrupted on recombination (SCHEMA contacts). (b) Sequence elements that should be inherited together (blocks) are identified based using the SCHEMA contact map. Optimal blocks are often noncontiguous along the polypeptide chain but are contiguous on the three-dimensional structure. (c) The chimeras are reassembled using blocks from different parents. (d) The SCHEMA contact map can be reformulated as a graph, where nodes represent residues and edges represent SCHEMA contacts. (e) To design noncontiguous recombination chimera libraries, the graph is partitioned, with each residue assigned to a block. Partitions are chosen to minimize the edges between blocks. (f) Graph schematic of a chimeric protein.
Chimeric β-glucosidase design
We chose to test this noncontiguous SCHEMA recombination approach by making a chimera of two distantly-related GH1 β-glucosidases, one from a prokaryote, the thermophilic Thermotoga maritima BglA16,17 (TmBglA), and the other from a eukaryote, the mesophilic Trichoderma reesei Bgl218,19 (TrBgl2). These enzymes share 41% sequence identity, with a conserved active site. The TIM-barrel enzyme fold has no obviously interchangeable subdomains.
We generated various two-block chimera designs that are predicted to have low disruption and picked the one shown in Figure 2 for construction and characterization. Chimera NcrBgl would have approximately half its sequence from TmBglA and half from TrBgl2; it would have 144 mutations, corresponding to ∼31% of its sequence, from the closest parent (TmBglA). Figure 2(a) shows NcrBgl on the sequence alignment of TmBglA and TrBgl2. The noncontiguous nature of the two blocks on the polypeptide chain is readily apparent—the red TrBgl2 block has seven separate sequence fragments, and the green TmBglA block has eight fragments. These blocks are contiguous, however, on the three-dimensional structure, as shown in Figure 2(b).
Figure 2.
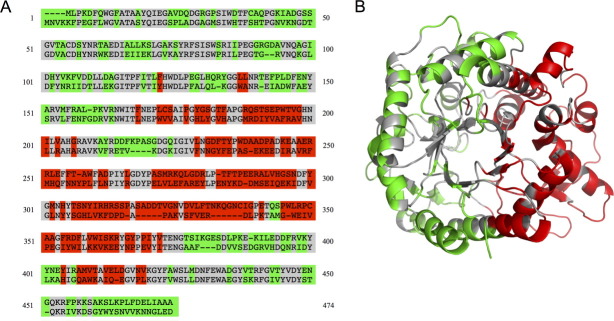
β-Glucosidase noncontiguous chimera design chosen for construction. (a) Numbered sequence alignment of the eukaryotic (top) and prokaryotic (bottom) β-glucosidases. Conserved residues are in gray, the block of eukaryotic mutations are in red, and the block of prokaryotic mutations are in green. (b) The two-block design illustrated on the structure of the prokaryotic enzyme, TmBglA (2WBG.pdb).
We predicted that this choice of crossovers should be minimally disruptive. The number of residue–residue contacts in NcrBgl that are not found in any of the parent contact maps is only 27.5, an average of 25 broken contacts based on TmBglA's structure 2WBG.pdb and 30 based on TrBgl2′s structure 3AHY.pdb. By comparison, swapping half the protein's structure randomly breaks on average 155 contacts [Fig. 3(a)], and the best design of 10,000 “random” designs breaks more than 70 contacts (see Materials and Methods section). Designs with many broken contacts are unlikely to lead to properly folded, functional enzymes.7 Figure 3(b) shows the optimized noncontiguous chimera design on a plot of the residue–residue contacts that could be broken (SCHEMA contacts). Most SCHEMA contacts are sequestered within a block in this design, and thus few contacts are disrupted on recombination.
Figure 3.
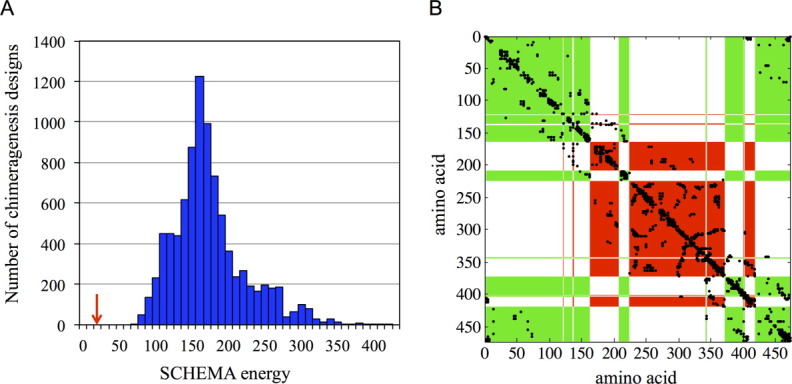
The optimal noncontiguous design breaks far fewer contacts than random two-block partitions of the structure. (a) A histogram of the SCHEMA energies of 10,000 random two-block chimeragenesis designs. The SCHEMA energy of the optimized noncontiguous design is highlighted with a red arrow. (b) The SCHEMA contact map for the optimized noncontiguous two-block design. Most of the SCHEMA contacts are within the two blocks and thus are not disrupted on recombination. The numbering is based on the parent alignment, and SCHEMA contacts are shown in black. Red and green areas show the two blocks (For greater clarity, the conserved residues have been assigned to one of the two blocks based on structural proximity.)
Structural conservation
The gene encoding the eukaryotic-prokaryotic NcrBgl chimera was synthesized and expressed under the control of an arabinose-inducible promoter in Top10 Escherichia coli cells. TrBgl2 and TmBglA break down cellobiose and other short oligosaccharides into glucose. Both parent enzymes are active over a range of pH, from 4 to 7, and TrBgl2 is active between 30 and 55°C,19 whereas TmBgl2 is highly thermostable with significant activity between 60 and 100°C.16 NcrBgl is catalytically active over the temperature range 30–60°C and is approximately a factor of 103 less active than TrBgl2 at 37°C. The activity is easily recovered, however, to TrBgl2 levels, by directed evolution (see below). We also synthesized the gene for the “mirror” chimera (with the parental identities of each block swapped), but it was not expressed as a functional protein in E. coli.
For structure determination, the NcrBgl chimera was expressed in E. coli BL21 DE3 with an N-terminal his6 tag and purified from cell lysate on a Ni-NTA column followed by an anion exchange column. Crystals were grown using the vapor-diffusion method, and NcrBgl's structure was solved from X-ray diffraction data using MOLREP20 and REFMAC521 (see Materials and Methods section).
The crystal structure of NcrBgl (4GXP.pdb), determined at 3.0 Å, shows that both blocks retain the structures of their respective parents. Chimera NcrBgl has the TIM-barrel fold and catalytic residues E170 and E374 [numbering based on the alignment shown in Fig. 2(a)] of the parent enzymes. Figure 4(a) illustrates the blocks on the parent structures and the structure of the chimera. The structural independence of recombined blocks is pronounced: there are significant differences between the aligned structures of the parents [Fig. 4(b)], particularly on the surface where there are multiple insertions and deletions in loop regions [Fig. 4(c)]. These structurally disparate regions are apparently unaffected by the chimeragenesis and maintain their backbone conformations when reassembled in the chimera.
Figure 4.
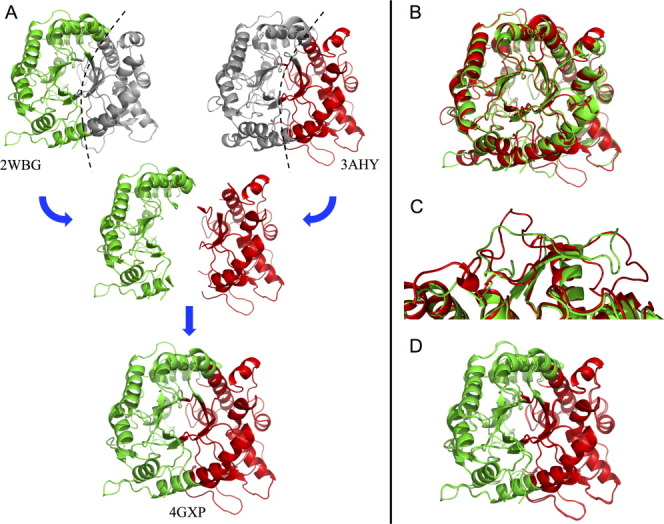
Structural elements are conserved on recombination. (a) The structure of chimera NcrBgl (4GXP.pdb), bottom, is nearly identical to the assembled structure of its component blocks from TrBgl2 (3AHY.pdb) and TmBglA (2WBG.pdb), top. The eukaryotic TrBgl2 residues and the prokaryotic TmBglA residues are highlighted in red and green, respectively. (For greater clarity, the conserved residues have been assigned to one of the two blocks based on structural proximity.) (b) A structural alignment of TmBglA 2WBG.pdb and TrBgl2 3AHY.pdb (RMSD = 3.34 Å) shows significant variation between these two homologs. (c) An example of significant variations in loop regions. (d) Model of NcrBgl constructed simply by stitching together the parental blocks closely aligns with NcrBgl's actual structure (RMSD = 1.15 Å).
We tested whether we could model the structure of the chimera by combining the parental structures of the chimera's blocks, using an alignment of the parental structures to position each block. Thus, for NcrBgl, we combined the structures of the TrBgl2 block and the TmBglA block to predict the structure of NcrBgl. This model does a good job at capturing variations in the backbone and loops [Fig. 4(d)]. Our ability to predict finer structural features is limited by the current low resolution of the chimera structure.
Recovering activity with directed evolution
We performed five rounds of random mutagenesis and screening for higher activity on the fluorescent β-glucosidase substrate, 4-nitrophenyl β-d-glucopyranosidase (pNPG) (see Materials and Methods section). Figure 5 shows the activity of the best mutant from each round, relative to NcrBgl. Activity increased almost 1000-fold in just five rounds. The resulting mutant has 149 mutations from the closest known natural sequence (TmBglA) and activity comparable to TrBgl2.
Figure 5.
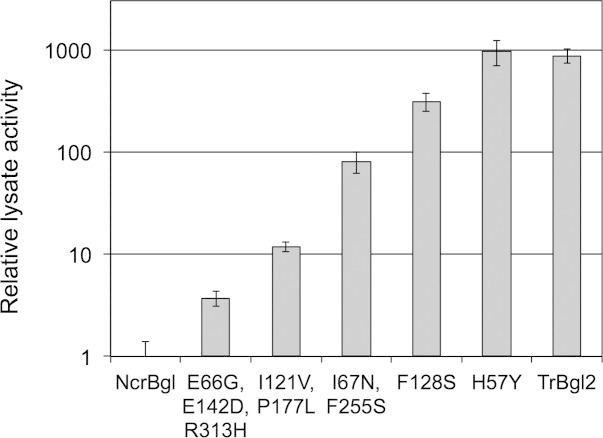
Directed evolution recovers the activity of NcrBgl to wild-type levels. Activity is measured in lysate with a 1-h assay on pNPG at 37°C and normalized relative to NcrBgl. The new mutations found at each round are listed (numbering based on the parental alignment). Five rounds of directed evolution increased the activity of NcrBgl almost 1000-fold.
Discussion
Structure-guided recombination is a powerful tool for generating novel enzymes with diverse sequences. We have presented a new method that splits proteins into elements of sequence that should be inherited together to minimize structural disruption. The resulting blocks can be noncontiguous along the polypeptide chain. We have developed tools to efficiently design chimeras and chimera libraries. These noncontiguous block designs disrupt far fewer SCHEMA contacts than equivalent designs that require contiguous sequence blocks. Indeed, contiguous block designs are a (suboptimal) subset of the noncontiguous block design space.
This approach does not rely on detailed atomistic models of the parent and progeny proteins. Indeed, the only structural information used is a set of residue–residue contacts, which, with the parent sequences, is sufficient to design functional chimeras of distantly-related proteins that do not have obvious subdomains. Simply minimizing the number of broken parental contacts seems to be sufficient to generate functional chimeras with a good success rate, as has been shown for contiguous SCHEMA recombination.7
To test the method, we designed and constructed a chimeric β-glucosidase that takes large blocks from a prokaryotic parent and a eukaryotic parent. While we designed a two-block, two-parent chimera for this example, the graph partitioning method can easily produce noncontiguous designs for libraries of chimeric proteins having multiple parents and multiple blocks.
On solving the crystal structure of the chimeric enzyme, we discovered that each block retains the structure of its corresponding parent (within the limits of the 3.0 Å resolution), suggesting that it may be possible to predict the structures of chimeric enzymes from the parent enzymes by simply combining the known parent structures. Alternatively, structures of the chimeric proteins could provide detailed and accurate information on the structures of the parent proteins. This can be very useful for eukaryotic protein structure determination, for example, where chimeragenesis enables production in a microbial recombinant host.22,23 The fact that the recombined blocks retain their parental structure could also be very useful for creating protein chimeras that acquire the functions (e.g., allosteric regulation, interactions with other proteins, or substrate specificity) of their parent blocks.
That the chimera is somewhat compromised in β-glucosidase activity compared with its parents is not surprising, considering the simplicity of the design approach and also that 144 mutations were introduced. However, the chimera was easily fine-tuned for native-like activity levels in just five rounds of random mutagenesis and screening. This example offers promise for exploring distant parts of sequence space, perhaps never explored by nature, for novel enzymes.
Materials and Methods
Noncontiguous recombination
A structure-based sequence alignment of the parental enzymes TmBglA16,17 and TrBgl218,19 was created using PROMALS3D.24 For a given structure, two residues are in contact if any atoms from each residue were within 4.5 Å of each other, excluding hydrogen atoms. A SCHEMA contact map contains those contacts that are not conserved among the parental enzymes. As the TmBglA and TrBgl2 structures vary considerably, a SCHEMA contact map was built for each parent, and a final average SCHEMA contact map weighted each contact depending on the number of parents in which it was present (0.5 if in a single parent, 1 if in both parents). PDB structures 2WBG.pdb chain A and 3AHY.pdb chain A were used to create the TmBglA and TrBgl2 SCHEMA contact maps, respectively.
The SCHEMA contact map was abstracted as a graph. Each nonconserved residue represented a node, and each edge represented an average weighted SCHEMA contact between two residues. Finding crossover locations that minimize the average number of SCHEMA contacts in the chimeras was reformulated as a problem of minimizing the cut edges when partitioning a graph. The hMETIS graph partitioning suite14,15 was used to find two-way partitions of the SCHEMA contact map—these partitions gave designs for two-block chimeragenesis of TmBglA and TrBgl2. A design was selected that would produce a chimera with a SCHEMA energy (number of disrupted contacts) of 27.5 and 144 mutations from the closest parent.
Random chimeragenesis designs
This analysis was carried out with PDB structure 2WBG chain A. The structure was partitioned into two blocks by a randomly generated cut plane through the protein's center. Each residue was assigned to one of the two blocks based on the coordinates of its alpha carbon. Swapping the residues of the blocks among the parents TmBglA and TrBgl2 created two possible chimeras with equal SCHEMA energies. The chimera SCHEMA energies were calculated using the SCHEMA contact map from 2WBG chain A.
Gene synthesis
The NcrBgl gene (Supporting Information Table 1) was optimized for expression in E. coli and synthesized by DNA2.0, Menlo Park, CA.
Protein preparation and crystallization
A 1-L baffled flask of Luria broth (LB) with 100 mg/L ampicillin was inoculated with 5 mL of an overnight culture of E. coli BL21 DE3 cells containing the NcrBgl gene with an N-terminal his6 tag on a pET-22(+) vector. The flask was grown for 4 h at 37°C, 250 rpm before being induced with isopropyl β-d-1-thiogalactopyranoside to a final concentration of 10 μM and incubated for 16 h at 16°C and 250 rpm. The cells were pelleted by centrifugation at 5000 g and frozen at −20°C. The cells were resuspended in 10 mM Tris, pH 7.4 and lysed by sonication. The lysate was spun at 60,000 g for 20 min, and the supernatant filtered with a Nalgene 0.2-μm aPES filter. The supernatant was loaded onto a 5-mL Ni-NTA His-trap HP column (GE Healthcare) and purified by washing with 1% elution buffer (20 mM Tris, pH 7.4, 100 mM NaCl, 300 mM imidazole) for 15 column volumes (CV), followed by a gradient elution (increase by 80% elution buffer in 10 CV). Fractions containing the NcrBgl protein were buffer exchanged to 20 mM Tris, pH 7.4 and loaded onto a 5-mL HiTrap Q HP column (GE healthcare). The column was washed with 1% elution buffer (20 mM Tris, 1M NaCl, pH 7.4) for 15 CV, and the protein purified by a gradient elution (increase by 80% elution buffer in 10 CV). Fractions containing the NcrBgl protein were pooled and concentrated using 30,000 molecular weight cut off protein concentrators with cellulose-free membranes (Vivaspin). Buffer was exchanged to 10 mM Tris, pH 8.0 by repeated refills, and the protein flash frozen and stored at −20°C. The protein was crystallized by vapor diffusion of a 4:3 mixture of 20 g/L protein in 10 mM Tris, pH 8.0 and 20% polyethylene glycol 3350, 0.4M sodium malonate, pH 7.0 in 24-well sitting drop plates (Hampton Research). Crystal growth occurred over a period of 2–3 days and larger, higher-resolution crystals were obtained by microseeding with pieces of sonicated crystals. Crystals were frozen in 25% glycerol for structure determination.
Structure determination and refinement
X-ray diffraction data were collected on a Dectris Pilatus 6M detector at 100K at the Stanford Synchrotron Radiation Lightsource, beamline 12-2. The wavelength of the beam was 0.9795 Å. Diffraction data were integrated using XDS25 and scaled using SCALA.26 A homology model of the NcrBgl was constructed in Modeller27 using 2WBG.pdb, chain A and 3AHY.pdb, chain C. This model was used by MOLREP,20 a molecular replacement tool that is part of the CCP4 crystallography software,28 to determine the initial phases of the X-ray data. The structure was refined with several rounds of manual model building within Coot29 and automated refinement using REFMAC521 within CCP4. Data refinement and collection statistics are given in Supporting Information Table 2.
Error-prone PCR library construction
For expression in E. coli TOP10 cells, the NcrBgl gene and N-terminal his6 tag was subcloned into the arabinose-inducible pBAD vector using Gibson assembly.30 A library of mutants with 3.4 nucleotide mutations per gene was generated by error-prone PCR using 50 μM MnCl2 and Applied Biosystems AmpliTaq polymerase. The pBAD backbone was amplified by regular PCR. Both PCR products were digested for 30 min by Dpn1 (New England Biolabs), purified on an agarose gel, and ligated together using Gibson assembly. The library was transformed into electrocompetent E. coli TOP10 cells and plated on LB-agar media with 100 mg/L ampicillin.
Library expression in 96-well plates
Individual mutant colonies from the library plates were picked into 96-well plates containing 300-μL LB with 100-mg/L ampicillin and grown at 37°C, 250 rpm, and 80% humidity. Each plate contained four null-control wells with an empty pBAD plasmid, four wells with the NcrBgl gene and four wells with the parent gene from the previous round of directed evolution. After 16 h, 50 μL of each culture was expanded into 96-well plates containing 900-μL LB with 100 mg/L and grown at 37°C for a further 4 h. The plates were then induced with 50 μL of 0.8% arabinose to give a final concentration of 0.04% arabinose. The plates were incubated for 16 h at 16°C and 250 rpm, and the cells pelleted by centrifugation at 4000 g and frozen at −20°C.
Enzyme activity screen
The cell pellets were lysed by adding 300 μL of 10 mM HEPES pH 8.0, 10 mM MgCl2, 0.7 mg/L lysozyme, and 0.1 units of DNAase I (Sigma) to each well and incubating at 37°C for 1 h. Lysate (50 μL) was transferred to a PCR plate containing 150 μL of 10 mM pNPG and incubated at 37°C for 1 h. The reaction was stopped by adding 20 μL of 1M sodium hydroxide and absorbance was read at 410 nm. Twenty plates were screened in each round. The best mutants were streaked onto an LB plate with 100 mg/L ampicillin and individual colonies used to rescreen in quadruplicate.
Acknowledgments
The authors thank A. Wang of the Academia Sinica, Taiwan, for providing the TrBgl2 gene and T. Gloster and G. Davies of the University of York, UK, for providing the TmBglA gene. The authors also acknowledge the Molecular Observatory at Caltech for their support with X-ray crystallography. The content of the information in this document does not necessarily reflect the position or the policy of the sponsoring agencies, and no official endorsement should be inferred.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
References
- 1.Crameri A, Raillard SA, Bermudez E, Stemmer WPC. DNA shuffling of a family of genes from diverse species accelerates directed evolution. Nature. 1998;391:288–291. doi: 10.1038/34663. [DOI] [PubMed] [Google Scholar]
- 2.Dueber JE, Yeh BJ, Chak K, Lim WA. Reprogramming control of an allosteric signaling switch through modular recombination. Science. 2003;301:1904–1908. doi: 10.1126/science.1085945. [DOI] [PubMed] [Google Scholar]
- 3.Otey CR, Landwehr M, Endelman JB, Hiraga K, Bloom JD, Arnold FH. Structure-guided recombination creates an artificial family of cytochromes P450. PLoS Biol. 2006;4:e112. doi: 10.1371/journal.pbio.0040112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li Y, Drummond DA, Sawayama AM, Snow CD, Bloom JD, Arnold FH. A diverse family of thermostable cytochrome P450s created by recombination of stabilizing fragments. Nat Biotechnol. 2007;25:1051–1056. doi: 10.1038/nbt1333. [DOI] [PubMed] [Google Scholar]
- 5.Romero PA, Arnold FH. Random field model reveals structure of the protein recombinational landscape. PLoS Comput Biol. 2012;8:e1002713. doi: 10.1371/journal.pcbi.1002713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Voigt CA, Martinez C, Wang Z-G, Mayo SL, Arnold FH. Protein building blocks preserved by recombination. Nat Struct Biol. 2002;9:553–558. doi: 10.1038/nsb805. [DOI] [PubMed] [Google Scholar]
- 7.Meyer M, Hochrein L, Arnold FH. Structure-guided SCHEMA recombination of distantly related β-lactamases. Protein Eng Des Sel. 2006;19:563–570. doi: 10.1093/protein/gzl045. [DOI] [PubMed] [Google Scholar]
- 8.Heinzelman P, Snow CD, Wu I, Nguyen C, Villalobos A, Govindarajan S, Minshull J, Arnold FH. A family of thermostable fungal cellulases created by structure-guided recombination. Proc Natl Acad Sci USA. 2009;106:5610–5615. doi: 10.1073/pnas.0901417106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Romero P, Stone E, Lamb C, Chantranupong L, Krause A, Miklos A, Hughes R, Fechtel B, Ellington AD, Arnold FH. SCHEMA-designed variants of human arginase I and II reveal sequence elements important to stability and catalysis. ACS Synth Biol. 2012;1:221–228. doi: 10.1021/sb300014t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Romero PA, Krause A, Arnold FH. Navigating the protein fitness landscape with Gaussian processes. Proc Natl Acad Sci USA. 2013 doi: 10.1073/pnas.1215251110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pantazes RJ, Saraf MC, Maranas CD. Optimal protein library design using recombination or point mutations based on sequence-based scoring functions. Protein Eng Des Sel. 2007;20:361–373. doi: 10.1093/protein/gzm030. [DOI] [PubMed] [Google Scholar]
- 12.Garey MR, Johnson DS, Stockmeyer L. Some simplified NP-complete graph problems. Theor Comput Sci. 1976;1:237–267. [Google Scholar]
- 13.Kernighan BW, Lin S. An efficient heuristic procedure for partitioning graphs. Bell Syst Tech J. 1970;49:291–307. [Google Scholar]
- 14.Karypis G, Aggarwal R, Kumar V, Shekhar S. In: Proc. 34th IEEE Design Automation Conference: 1997. pp. 526–529. [Google Scholar]
- 15.Karypis G, Kumar V. Multilevel k-way hypergraph partitioning. VLSI Des. 2000;11:285–300. [Google Scholar]
- 16.Gabelsberger J, Liebl W, Schleifer K-H. Purification and properties of recombinant β-glucosidase of the hyperthermophilic bacterium Thermotoga maritima. Appl Microbiol Biotechnol. 1993;40:44–52. [Google Scholar]
- 17.Zechel D, Boraston A, Gloster T, Boraston CM, Macdonald JM, Tilbrook DMG, Stick RV, Davies GJ. Iminosugar glycosidase inhibitors: structural and thermodynamic dissection of the binding of isofagomine and 1-deoxynojirimycin to β-glucosidases. J Am Chem Soc. 2003;125:14313–14323. doi: 10.1021/ja036833h. [DOI] [PubMed] [Google Scholar]
- 18.Takashima S, Nakamura A, Hidaka M, Masaki H, Uozumi T. Molecular cloning and expression of the novel fungal β-glucosidase genes from Humicola grisea and Trichoderma reesei. J Biochem. 1999;125:728–736. doi: 10.1093/oxfordjournals.jbchem.a022343. [DOI] [PubMed] [Google Scholar]
- 19.Jeng WY, Wang NC, Lin MH, Lin CT, Liaw YC, Chang WJ, Liu CI, Liang PH, Wang AHJ. Structural and functional analysis of three β-glucosidases from bacterium Clostridium cellulovorans, fungus Trichoderma reesei and termite Neotermes koshunensis. J Struct Biol. 2011;173:46–56. doi: 10.1016/j.jsb.2010.07.008. [DOI] [PubMed] [Google Scholar]
- 20.Vagin A, Teplyakov A. MOLREP: an automated program for molecular replacement. J Appl Crystallogr. 1997;30:1022–1025. [Google Scholar]
- 21.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 22.Duret G, Van Renterghem C, Weng Y, Prevost M, Moraga-Cid G, Huon C, Sonner JM, Corringer P-J. Functional prokaryotic-eukaryotic chimera from the pentameric ligand-gated ion channel family. Proc Natl Acad Sci USA. 2011;108:12143–12148. doi: 10.1073/pnas.1104494108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shimoji M, Yin H, Higgins L, Jones JP. Design of a novel P450: a functional bacterial-human cytochrome P450 chimera. Biochemistry. 1998;37:8848–8852. doi: 10.1021/bi972775z. [DOI] [PubMed] [Google Scholar]
- 24.Pei J, Kim B-H, Grishin NV. PROMALS3D: a tool for multiple protein sequence and structure alignments. Nucleic Acids Res. 2008;36:2295–2300. doi: 10.1093/nar/gkn072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kabsch W. XDS. Acta Crystallogr D Biol Crystallogr 66. 2010:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Evans P. Scaling and assessment of data quality. Acta Crystallogr D Biol Crystallogr. 2005;62:72–82. doi: 10.1107/S0907444905036693. [DOI] [PubMed] [Google Scholar]
- 27.Eswar N, Webb B, Marti-Renom MA, Madhusudhan M, Eramian D, Shen MY, Pieper U, Sali A. Comparative protein structure modeling using Modeller. Curr Protoc Protein Sci. 2007;2:15–32. doi: 10.1002/0471140864.ps0209s50. [DOI] [PubMed] [Google Scholar]
- 28.Bailey S. The CCP4 suite programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 29.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 30.Gibson DG, Young L, Chuang R-Y, Venter JC, Hutchison CA, Smith HO. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods. 2009;6:343–345. doi: 10.1038/nmeth.1318. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.