

Abstract
The process of cellular eating, or the phagocytic swallowing of one cell by another, is an ancient manifestation of the struggle for life itself. Following the endosymbiotic origin of eukaryotic cells, increased cellular and then multicellular complexity was accompanied by the emergence of autophagic mechanisms for self-digestion. Heterophagy and autophagy function not only to protect the nutritive status of cells, but also as defensive responses against microbial pathogens externally or the ill effects of damaged proteins and organelles within. Because of the key roles played by phagocytosis and autophagy in a wide range of acute and chronic human diseases, pathologists have played similarly key roles in elucidating basic regulatory phases for both processes. Studies in diverse organ systems (including the brain, liver, kidney, lung, and muscle) have defined key roles for these lysosomal pathways in infection control, cell death, inflammation, cancer, neurodegeneration, and mitochondrial homeostasis. The literature reviewed here exemplifies the role of pathology in defining leading-edge questions for continued molecular and pathophysiological investigations into all forms of cellular digestion.
“Life is cell activity; its uniqueness is the uniqueness of the cell.” –Rudolf Virchow1
Invitation to a Cellular Banquet
In the 19th century, Rudolph Virchow’s observation that diseases originate with changes in individual cells gave rise to the principles of cellular pathology, which remain as cornerstones of diagnostic medicine to this day. Although Virchow disagreed with his contemporary Louis Pasteur concerning the germ theory, he recognized that no single authority should be considered infallible, and advocated for active involvement of physicians in experimental pathology. Indeed, we now know that diseases can be caused both by invasion of microorganisms and by inappropriate host responses to them.
The ingestion of one cell by another undoubtedly arose as a feeding mechanism in unicellular organisms. In a twist of fate, proteobacteria and cyanobacteria engulfed by larger archaebacteria managed to survive, giving rise to symbiotic relationships that form the basis of eukaryotic cells.2 In our bodies, professional phagocytes function as a first line of defense against external pathogens, adapting ancient engulfment mechanisms from unicellular ameboid organisms. (An example of normal phagocytosis of bacteria is shown in Figure 1.) Likewise, as cells became more complex, self-cannibalism by autophagy evolved from a starvation response to fulfill additional roles in cellular homeostasis. These include the defense of the cell against mitochondrial endosymbionts gone bad or in response to disorders of proteostasis that threaten the cell from within. Moreover, autophagy forms another line of defense against microbial pathogens that can subvert the endocytic/phagocytic system to gain entry into the cell.3
Figure 1.
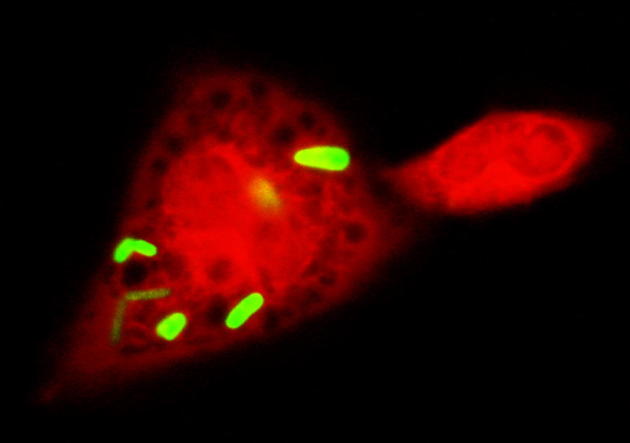
Normal phagocytosis of bacteria, demonstrated with immunofluorescence. A wild-type, EC-SOD–expressing, peritoneal macrophage (red) (Invitrogen CellTracker CMRA; Life Technologies, Carlsbad, CA) exposed to EGFP-expressing E. coli (green) rapidly internalizes several bacteria.
It is not surprising, then, to find that a wide range of human diseases are associated with dysfunctional phagocytosis or autophagy. Given that both phagocytosis and autophagy represent integral cellular responses to pathological stimuli, experimental pathologists have played key roles in defining central themes for investigation since the 1890s, and their findings often appeared in The American Journal of Pathology (AJP) or its precursor journals. Using biochemical, ultrastructural, and fluorescence methods, pathologists defined key stages in these dynamic processes, for which detailed molecular mechanisms are still being unraveled today. More recent studies use molecular approaches to manipulate disease models, revealing that either inadequate or excessive cellular eating can contribute to the pathophysiology of diseases. These studies foreshadow current interests in defining specific macromolecular signaling and adaptor mechanisms that underlie the physical recognition and sequestration of cargo, the completeness of their disposal, and future implications for disease therapy.
Phagocytosis Throughout the Last Century
Setting the Table: Early Studies in Phagocytosis
Phagocytosis is an evolutionarily conserved mechanism by which certain cells engulf and digest other cells or foreign particles. Ilya (also Élie) Metchnikoff, a Russian zoologist, first described phagocytosis in 1882 after observing that specific cells in starfish larvae absorbed thorns from his tangerine tree.4,5 Over the next few years, Metchnikoff established that phagocytosis is a conserved defense mechanism among species, and that it relies on the function of specialized cells (phagocytes).4,5 His landmark work earned him the Nobel Prize for Physiology or Medicine in 1908 (shared with the German scientist Paul Ehrlich) and sparked more than a century of research on the subject.
The earliest articles on phagocytosis sought to define the phenomenon: Which cells are active? Where in the body are they acting? What can these cells ingest? What conditions trigger phagocytic activity? In 1897, J.L. Goodale6 reported a study in the Journal of the Boston Society of Medical Sciences (which later became the Journal of Medical Research and then the AJP), showing that polynuclear leukocytes in the tonsillar mucus membrane absorb foreign matter and proving that specialized cells in the body possess phagocytic activity against bacteria. In 1906, Buxton and Torrey7 reported for the first time that macrophages can engulf foreign animal cells, in addition to bacteria. In that study they also examined migration patterns of phagocytes after ingestion of a bacterial load, observing that most of these cells end up in lymph nodes. Buxton and Torrey found that resident tissue mononuclear phagocytes (macrophages) are the initial responders to infection, and that polynuclear cells (neutrophils) arrive later, to complete the removal of the invading organism. Finally, they found that nondigestible particles can remain in phagocytes for months before eventually being fragmented and released.
The next wave of research followed the landmark 1903 article by Wright and Douglas,8 in which they described opsonins (molecules in the bloodstream that bind to bacteria to promote phagocytosis by host cells). Antibodies, opsonins, and other substances in body fluids became the focus of the work of many pathologists in the 1900s and 1910s, as they worked to understand what regulates phagocytosis. Destruction of opsonins through heating of blood serum was found to markedly inhibit the ability of phagocytes to digest bacteria.9 These findings also led to studies examining the role of opsonins in vaccine development.10
In 1917, Bartlett and Ozaki11 reported additional in vivo studies of phagocytosis. They found that bacteria are rapidly removed from the blood within minutes after inoculation, usually by mobile leukocytes. During heavy infection, however, uptake by mobile phagocytes is insufficient, and the majority of invading bacteria enter the tissues to be engulfed by phagocytic cells within the tissue. As foreshadowed by the study of Buxton and Torrey,7 a large body of research focused on the migratory patterns of phagocytes in various organs after ingestion of bacteria, including lung,12 liver,13 spleen,13,14 and digestive tract.7 In the setting of acute pneumonia, Permar12,15 studied the migration of leukocytes from tissues into interstitial spaces and eventually into the lymphatic system, where the cells can reside in lymph nodes for an extended period.
In the 1920s and 1930s, a debate about the cellular origin of mononuclear phagocytes emerged in the literature. Scientists struggled to determine whether phagocytic cells originated from vascular endothelium, epithelium, or blood monocytes.16–18 Howard Permar19,20 at the University of Pittsburgh argued that mononuclear phagocytes are formed from vascular endothelium in amounts proportional to the number of invading bacteria, and that these numbers are especially high in blood vessels adjacent to infected and inflamed tissue. Although Permar was correct in noting that infection triggers increased phagocytic cell production and phagocyte migration to the site of infection, he was incorrect in his theory about the origin of mononuclear phagocytes. In 1922, Simpson21 proposed that macrophages originate from monocytes that are released into the blood in bursts, or showers, during infection. This proposal withstood the test of time. In 1927, Nathan Chandler Foot22 stated, in an article in the AJP, that what were then called dust cells in the lung originated from blood monocytes, not from epithelial tissue. By the mid-20th century, it was established that mononuclear phagocytes originate as myeloid cells in the bone marrow, and that infection triggers release of monocytes into the bloodstream from the bone marrow.23 The exact cytokine signals causing this release are not yet well defined. Monocytes extravasate through vascular endothelial cells (which may account for Permar’s conclusions) into tissue, where they then differentiate into macrophages and dendritic cells.24
Phagocyte Activation: A Knife That Cuts Both Ways
In the mid-20th century, researchers began to focus on defining the regulatory crosstalk between phagocytes and their environment. The complement system was already known to promote neutrophil chemotaxis, but in 1968 it was discovered that bacteria themselves secrete substances that draw neutrophils to the site of infection.25 In a 1976 review in the AJP, Unanue26 examined interactions between phagocytes and their environment during acute and chronic inflammation, wound healing, and normal immune responses. He not only discussed several chemotactic stimuli, but also highlighted the importance of surface receptors on the phagocyte that are necessary for the binding, ingestion, and degradation of foreign particulates (Figure 2). The following year, it was found that disorders of recognition and ingestion of foreign matter by these phagocytic receptors can lead to clinical pathology.27 Recent studies have confirmed that an inability to properly complete phagocytosis contributes to a variety of diseases, including atherosclerosis and autoimmune diseases.28,29
Figure 2.

Major steps in phagocytosis and autophagy. The proper induction (A and B), cargo targeting (C), maturation (D and E), and completion of lysosomal clearance (F) are all important for both phagocytosis and autophagy. For phagocytosis, the recognition of danger signals from dead cells or microbial pathogens stimulates phagocyte migration. A number of different receptors are involved in capturing and internalizing bacterial, apoptotic, or particulate cargoes through a process enhanced by Atg proteins and dependent on a proper local redox environment maintained by EC-SOD. Once the phagocytic cargo is inside the phagosome, concentrated production of ROS or degradative enzymes are stimulated, depending on the ingested contents. Similarly, either general or localized insults within cells serve to trigger the membrane deposition of Atg 5—Atg 12 and of LC3 (through Beclin 1–dependent or independent mechanisms), which are essential for the extension of autophagic membranes. Whereas nonselective sequestration of cytoplasm is induced by starvation, damaged and potentially harmful cargoes need to be appropriately targeted to LC3-bound membranes. Autophagy is also induced by phagosome-derived signals to sequester pathogens that escape or damage the phagosome membrane. In turn, induction of the autophagic pathway serves to further promote phagosome–lysosomal fusion. Thus, these two complementary systems cooperate in removing exogenous and endogenous danger signals to limit proinflammatory, carcinogenic, and prodeath stimuli. For both phagocytic and autophagic pathways, inefficient lysosomal fusion or digestion can contribute to many classes of disease (red-shaded boxes). Accumulation of undigested lysosomal cargo is seen in aging and in chronic granulomatous disease (CGD), neurodegenerative diseases, and many other diseases or disorders. Inefficient utilization of digested products for energy production or in the regeneration of mitochondria and other essential cellular structures may also contribute to cell death and neurodegeneration.
That phagocytes are necessary for proper wound repair in the absence of infection was also elucidated in the 1970s. Phagocytes were found to be required not only for removal of dead tissue, but also for communication with fibroblasts, to stimulate fibrous scarring.30 Indeed, as Unanue26 noted, phagocytes secrete a wide array of substances into the environment, including enzymes (eg, lysozyme, plasminogen activator, collagenase, and elastase), signals for immune modulation (eg, lymphocyte recruitment, colony-stimulating factor, and inhibitory and lytic signals), and complement proteins. Unanue26 also made the astute observation that some of these secretions could be harmful to host tissue—an important concept for chronic inflammatory conditions. The pathological role of phagocytes is perhaps best illustrated in a series of AJP articles on atherosclerosis that appeared in the 1980s.31–33 Monocytes were identified as the precursors to foam cells in plaques, actively recruiting more inflammatory cells to the lesion and thus promoting growth of atherosclerotic lesions.
In recent decades, much of the work on phagocytosis has been at the molecular level, with focus on specific receptors and molecules that dictate phagocyte behavior.34–37 Identifying the various roles of phagocytic cells in a variety of diseases has remained an important area of active investigation, including studies of neurodegeneration,38–41 wound repair,42 and cancer.43 Indeed, more recent studies have focused on different classes of macrophages involved in inflammatory responses [currently, these are simplified as classically activated M1 (proinflammatory) and alternatively activated M2 (tissue remodeling) responses]. A growing number of recent studies, published in the AJP and elsewhere, have shown that inappropriate regulation of the M1/M2 balance may contribute to the pathogenesis of a number of diseases, including spinal cord injury,44 sarcoidosis,45 glomerulonephritis,46 and pulmonary fibrosis.47
Frustrated Phagocytosis and Indigestion: Role of Reactive Oxygen Species
Once a phagocyte has engulfed a pathogen, the immune cell must be able to kill the bacteria (Figure 2). One method by which neutrophils and macrophages kill ingested bacteria is through the respiratory burst, which is initiated via the NADPH oxidase system. A 1982 review in the AJP underscores the role of free radicals in phagocyte bactericidal activity.48 Superoxide ions released by NADPH oxidase produce hydrogen peroxide after spontaneous or enzymatic dismutation. Myeloperoxidase then produces hypochlorous acid (bleach) from the hydrogen peroxide. This results in concentrated levels of reactive oxygen species (ROS) within the phagolysosome that are toxic to bacteria. The respiratory burst was first reported in 1933, by Baldridge and Gerard,49 but it was not until the 1960s and 1970s that its role in bacterial killing was understood.50 The discovery of impaired activity of the NADPH oxidase enzyme (and thus no oxidative burst) in chronic granulomatous disease highlighted the importance of the respiratory burst in immunity. Patients with chronic granulomatous disease are at a high risk of developing chronic and recurring infections, especially abscesses and pneumonia; even though their phagocytes can ingest invading bacteria, they cannot efficiently complete the killing of these bacteria within their phagolysosomes.
Although ROS production by phagocytic cells plays a crucial role in bacterial killing, overproduction of these molecules during inflammation can also damage host tissues. Oxidant levels must, therefore, be tightly regulated in the body. A vast antioxidant system is present in normal tissues and within phagocytes themselves, to protect against oxidative damage.48
Importantly, phagocytosis itself can be impaired by a failure to appropriately regulate oxidant levels in the body, as shown in a 2011 AJP report of a study in mice lacking the antioxidant enzyme extracellular superoxide dismutase (EC-SOD).51 The data showed that EC-SOD is localized inside macrophage and neutrophil granules. Whereas wild-type EC-SOD expressing macrophages undergo efficient bacterial phagocytosis (Figure 1), phagocytosis is markedly impaired in EC-SOD knockout macrophages (not illustrated).3 This suggests that ROS production by phagocytic cells can disrupt normal phagocytosis if EC-SOD is not present to protect the phagocyte itself from its own production of ROS. This finding further illustrates the importance of regulating ROS production, even in settings where large quantities of ROS are physiologically produced for bactericidal activity. The targets through which excess superoxide or downstream ROS interfere with phagocytosis are unclear, but this study highlights an important role for EC-SOD in phagocytic cells in promoting appropriate innate immune responses to bacterial pathogens.51
The antioxidant regulation of ROS production is also important for maintaining effective phagocytic function at later stages of bacterial killing, as highlighted in a study of the intracellular pathogen Mycobacterium abscessus.52 This study found that antioxidants promote killing of the mycobacteria by promoting phagosome–lysosome fusion. Thus, regulation of ROS production in phagocytic cells appears to be essential not only in the maintenance of normal phagocytic uptake of bacteria51 but also in promoting the phagosome–lysosome fusion needed for completion of bacterial killing.52
The redox environment within the phagosome is important also for adaptive immune responses. Proteolysis within the phagosome allows macrophages to degrade pathogen proteins in preparation for antigen presentation. NADPH oxidase activity during the respiratory burst was recently shown to reversibly oxidize cysteine residues in some cathepsins, which the authors propose may alter the repertoire of antigenic peptides produced.53 Additionally, ROS release from phagocytes has been shown to activate NF-κB,54 a transcription factor involved in promoting both innate and adaptive immune responses. These findings indicate that, even during a respiratory burst generated for bacterial killing, tight regulation by antioxidants is important for enhancing the overall efficiency of phagocytic functions.
Macroautophagy: Six Decades of Progress
Hors d’Oeuvre: Early Studies in Autophagy
The biochemical discovery of lysosomes (specialized intracellular compartments devoted to hydrolytic digestion) by Christian de Duve and colleagues55 coincided with pathological studies of cytoplasmic inclusions reported by H.W. Altmann in 1955. Altmann (as cited by Hruban et al56) speculated that certain inclusions represent a stage in the cellular reaction to injury in which damaged portions of cytoplasm are sequestered for digestion or extrusion. This process of localized cellular digestion represents the defining feature of macroautophagy. Moreover, mechanisms underlying autophagy-related cellular exocytosis, as predicted by this study more than 50 years ago, have recently been described.57,58
In 1963, in one of the first experimental studies of autophagy reported in the AJP, Z. Hruban et al56 recognized a continuum of structures reflective of what they defined as a single dynamic process, outlining three progressive stages for focal cytoplasmic degradation: i) sequestration, ii) formation of complex bodies, and iii) formation of lysosome-like bodies (Figure 2). This study, which involved systemic treatment of rats with an array of stressors followed by ultrastructural examination of liver cells revealed that the contents inside of autophagosomes depends on the nature of the insult. Furthermore, the authors noted that focal cytoplasmic degradation is not limited to pathological states, but occurs also under physiological conditions “for disposal of organelles when the cell changes its functional state.” These observations, made before the term autophagy came into use, laid the foundation for current studies of autophagic flux and selective autophagy in differentiation and disease.
In 1968, Antti Arstila and Benjamin Trump59 reported another landmark study, which remains the second most cited autophagy study, after that of Hruban, published in the AJP (Thompson Reuters Web of Science version 5.9, last accessed January 29, 2013). In this study, they addressed a set of important questions for cellular autophagocytosis. These questions relate to the origin or origins of the hydrolytic enzymes, the origin or origins of the enclosing autophagic membrane, and “the control mechanisms and metabolic requirements involved in autophagy.” Using density gradient fractions and histochemical techniques to visualize enzyme activity by transmission electron microscopy, they showed that the double membrane phase of autophagosomes lacked hydrolytic enzymes, but exhibited features of smooth endoplasmic reticulum. Fusion of the autophagosome outer membrane with vesicles containing hydrolytic enzymes resulted in delivery of enzyme to the space between the double limiting membranes, which was followed by loss of the inner membrane. The fusion process required “high energy compounds,” but not protein synthesis. This was followed by a series of studies in the 1970s by Shelburne, Arstila, and/or Trump defining the roles of cAMP, protein synthesis, nutritive status, and energy metabolism in hepatocyte, renal tubules, and HeLa cells (for example, Shelburne et al60). An early description of selective mitophagy elicited in renal tubules by hyperbaric oxygen exposure was published in the AJP in 1979,61 nearly 30 years before this would re-emerge as a central topic in autosomal recessive Parkinson’s disease.62,63
In a similar time period, the occurrence of autophagy was delineated in several systems of cell death, both developmental and injurious (reviewed by Bursch64). Indeed, three characteristic morphological patterns of cell death were defined: i) shrinkage necrosis, which is now known as apoptosis, ii) the controversial autophagic cell death, or cell death accompanied by autophagosomes, and iii) necrotic cell death, which is characterized by organelle swelling and loss of membrane integrity. Nonetheless, the role or roles of autophagy in cell degeneration and death (whether beneficial, maladaptive, coincidental, or part of an active cell death program) would remain inscrutable until the discovery of autophagy-related (Atg) genes and proteins in the years surrounding the turn of the millennium (reviewed by Klionsky et al65).
Autophagy in Diseases: Cytoprotection versus Gluttony?
In the 1980s and 1990s, autophagolysosomal processes were identified in association with characteristic cytopathologic hallmarks of several human diseases. These include the rimmed vacuoles of certain forms of myopathy,66 and granulovacuolar degeneration in hippocampal neurons of patients with Alzheimer disease.67 These authors proposed that autophagy was activated as a means of protective sequestration against early apoptotic changes, expanding on the concepts advanced by Altmann and by Hruban.56 Indeed, a recent study of autolysosomal pathology in Alzheimer disease, by Ralph Nixon and colleagues, 68 revealed a mechanism by which autophagy protects against apoptosis through degradation of activated caspase-3 in dystrophic neurites. Likewise, clearance of damaged mitochondria by autophagy serves a neuroprotective role in a PTEN-induced kinase 1 (PINK1)–deficient recessive model of Parkinson’s disease.63 Protective or compensatory roles for autophagy were also delineated in acute injury models and sepsis.69,70
In analogy to the field of apoptosis after the identification of Bcl-2 as a survival oncogene in 1989,71 the identification of conserved autophagy genes beginning a decade later triggered a surge of interest in the role of autophagy in physiology and disease, and this interest continues to grow exponentially. In particular, the delineation in 2000 of a ubiquitin-like conjugation event involving Atg8/microtubule associated protein-1 light chain 3 (LC3), which is essential for the extension of autophagic membranes,72,73 provided molecular tools for monitoring autophagosomes and for manipulating autophagy induction. A landmark study in 2004 demonstrated the therapeutic potential for autophagy in animal models of Huntington’s disease.74 This has been followed by reports indicating protective effects of autophagy-enhancing drugs in other protein aggregation diseases.75,76 The concept of autophagy as a key clearance mechanism for pathological protein inclusions in turn inspired investigations into mechanisms underlying selective cargo recognition, resulting in the discovery of p62 and other LC3-interacting proteins as molecular adaptors for ubiquitin-coated protein aggregates.77
The prospect of harnessing autophagy-related clearance mechanisms for various human diseases, and potentially in slowing the process of aging itself, has engendered much excitement. Our understanding of the network of regulatory mechanisms that affect induction and completion of autophagy, not to mention its crosstalk with biosynthetic and other stress responses, has been growing rapidly, but has also stimulated further questions that remain to be answered. For example, the ability to therapeutically upregulate autophagy by rapamycin may be limited by metabolic disturbances associated with the chronic complex I deficiency observed in Parkinson’s patients.78 This and other articles published in the AJP in the first decade of the 21st century have pioneered several key considerations concerning the biology of autophagy in the context of disease.
In 2007, the fourth and fifth most cited autophagy articles in the AJP linked autophagy to the outcome of endoplasmic reticulum stress in cancer cells79 and defined a role for noncanonical beclin 1–independent autophagy in cell death (discussed in the following paragraph).80 In the former study, Xiao-Ming Yin and colleagues79 showed that proteasome inhibition activated autophagy as a compensatory mechanism through the IRE1-JNK signaling arm of the unfolded protein response. Subsequent studies implicate the unfolded protein response in several other disease states to include pretangle neurons in postmortem Alzheimer disease specimens81 and muscle biopsies of sporadic inclusion-body myositis.82 An amplifying cycle is proposed in which endoplasmic reticulum stress impairs cathepsin function,82 causing autophagic stress, defined as an imbalance of autophagy induction and completion.83 Cathepsin deficiency itself causes cellular pathology similar to that observed in human neuronal ceroid lipofuscinoses,84 and induction of autophagy may prove harmful in the context of lysosomal dysfunction.85,86
The context-dependent outcome of autophagy is a central area for disease pathophysiology and therapy. Also in 2007, a novel pathway of autophagy/mitophagy was described in toxin-treated neuronal cells, which used only a subset of the canonical Atg proteins that had been defined for starvation-induced autophagy.80 Beclin 1/PI3K–independent autophagy has since been shown in a growing number of pathological situations.87–89 Given that the prosurvival protein Bcl-2 binds Beclin 1 to limit its autophagy-related function,90 Beclin 1–independent autophagy most likely represents an escape from this important rheostat function. Indeed, siRNA knockdown of autophagy mediators results in reduced cell death.80,87 Like inflammation, autophagy mediates beneficial responses when it is activated transiently with appropriate clearance of intermediates, but may convert to a maladaptive response with excessive or sustained demands that interfere with completion of the autophagic response (Figure 2).
Although it may be difficult for researchers studying physiologically regulated autophagy in otherwise normal cells to envision excessive autophagy induction, data are emerging to put into context examples of these situations in ailing cells. In disease models, impairment in microtubule or lysosomal function results in inefficient clearance of autophagosomes.91,92 In this context, tuning down the capacity for autophagy induction by siRNA knockdown of key proteins would serve to reduce autophagic stress. Aging or diseased cells may also show declining capacities for regenerative biosynthesis. For example, excessive ERK1/2 activation drives mitophagy93 while concurrently suppressing mitochondrial biogenesis.94 The resulting catabolic–anabolic imbalance leads to an uncompensated loss of mitochondria, which is poorly tolerated in neurons and other mitochondria-dependent cell types. Aberrant ERK1/2 activation has also been implicated in the autophagic degeneration of arsenite-injured renal tubular cells.95 For the most part, these examples of harmful autophagy are limited to cell culture systems, which are readily manipulated in a controlled fashion. Exceptions include the murine central nervous system. Although a complete loss of constitutive autophagy is harmful to subsets of neurons,96 suppression of injury-induced autophagy through conditional deletion of Atg7 has been shown to mediate neuroprotection in vivo against neonatal ischemia–reperfusion injury and acute nigrostriatal injuries in mice.97,98
These findings likely reflect a greater demand of neurons for precise bioenergetic homeostasis, compared with other glycolytic or proliferative cell types. In several disease models, autophagosomes are unusually enriched in dendritic and axonal processes, and these changes appear early in the course of injury.99 Although insufficient autophagy induction and completion have each been implicated in axonal degeneration,96,100 autophagy also degrades receptors and other important synaptic components.101,102 The subsequent decrease in function may play into early stages of disease pathophysiology. Mitochondrial distribution is critically important to the development and maintenance of synaptic contacts,103 and a recent article in AJP showing dendritic mitophagy in a model of Parkinson’s disease reveals another mechanism by which autophagy can affect neuronal function. In that study, mitophagy induced as a response to calcium dysregulation, contributes to the degeneration of dendrites in neurons expressing mutant forms of leucine-rich repeat kinase 2.104
Shared Features of Cellular Consumption
Autophagy and phagocytosis probably each began as a method of promoting cellular nutrition; however, both processes have evolved to become key players in cellular homeostasis and defense against pathogens. Thus, both self-eating and the consumption of other cells, living or dead, play key roles in cell survival and tissue homeostasis.
Autophagy and phagocytosis are often upregulated concurrently with apoptosis. In some contexts, autophagy serves to prevent apoptosis,105 as in sequestration of damaged mitochondria that could release death mediators.63,69 On the other hand, autophagy is often associated with tumor-suppressing pathways,106,107 and autophagy (or its key regulatory proteins) may be essential for promoting subsequent cell death.95,97 Likewise, phagocytosis is engaged not only by professional phagocytes to kill living (microbial) cells or to scavenge necrotic tissue, but also comprises a key mechanism by which epithelial cells can eliminate the apoptotic corpses of neighboring cells.108 In this way, danger signals that may trigger sustained inflammation, tissue fibrosis, allergies, or inflammatory carcinogenesis can be eliminated.109–112
Although autophagy and phagocytosis are activated by different mechanisms, their final steps converge on similar pathways that are regulated by shared molecules. For example, phosphatidylinositol-3-phosphate (PI3P) is required for both phagosome and autophagosome generation and subsequent fusion with the lysosome.113,114 Recent studies, discussed below, have further highlighted the crosstalk between autophagy and phagocytosis in the clearance of dead cells (homeostasis) and the destruction of pathogens (defense).
A Well-Balanced Diet: Autophagy and Phagocytosis in Homeostasis
Rapid and efficient clearing of apoptotic and necrotic cells is a crucial housekeeping function in the body. Without clearance of dead cells, there is inadequate room for growth of new cells, and the dead cells release inflammatory danger signals that can be harmful to host tissue. Dead cells that persist in tissue increase the risk for autoimmune diseases such as systemic lupus erythematosus,28,115 as well as other morbidities.29 Recent studies show that autophagy and phagocytosis work together to reduce the volume of dying cells, preventing the harmful release of inflammatory mediators.
Cells that have undergone apoptosis display phosphatidylserine on their surface, flagging them for macrophage ingestion. Macrophages secrete a protein called the milk fat globule epidermal growth factor 8 (MFG-E8), which binds to phosphatidylserine and promotes phagocytosis of apoptotic cells, much like the opsonins first described in 1903 by Wright and Douglas8 in bacterial infections. A recent study by Qu et al116 described the necessity of the autophagy genes ATG5 or BECN1 for the proper display of phosphatidylserine eat-me signals on apoptotic cells. The authors showed that, without proper autophagic function, phagocytes could not efficiently engulf dead cells in embryoid bodies, leading to developmental abnormalities. Likewise, autophagy regulated by the Toll-like receptor 4 may be protective in a model of pulmonary fibrosis by facilitating the clearance of injured or dead cells,117 thereby accelerating the resolution of inflammation.
Uninvited Dinner Guests: Autophagy and Phagocytosis in Pathogen Clearance
When phagocytosis fails to eliminate a pathogen, autophagy can act as a back-up system to fight off infection. Certain bacteria, when phagocytosed, can persist within the phagosome or can damage the phagosome to be released into the intracellular environment. Modifications or damage to the phagosome are thought to activate the autophagic machinery. For example, Listeria monocytogenes bacteria secrete listeriolysin-O, which lyses the phagosome to release phagocytosed bacteria, but can also stimulate autophagy.3 Salmonella enterica enterica, serovar Typhimurium, resides within the phagosome for protection; however, it secretes proteins as part of its type III secretion system, which trigger autophagy of the entire salmonella-containing vesicle.3 Additionally, Mycobacterium tuberculosis inhibits phagosome maturation, but activation of autophagy is sufficient to override these effects, resulting in decreased phagosome pH and restoration of bacterial killing.118 Following Toll-like receptor signaling on macrophages during phagocytosis,119 LC3 recruitment to phagosomes serves to promote phagosome–lysosome fusion to aid in bacterial killing. Despite these dual mechanisms of phagocytic and autophagic immune surveillance, pathogens such as hepatitis C viruses have evolved mechanisms by which they can avoid clearance by interfering with autophagosome–lysosomal fusion, as was demonstrated in a study of human liver biopsies reported in the AJP in 2011.120
Tomorrow’s Menu: Future Themes for Investigation
As we celebrate the Centennial Anniversary of the American Society for Investigative Pathology, it is clear that central advances in defining and answering key questions in basic phagocytosis and autophagy research have been made in the context of tissue injury, repair, and disease by investigative pathologists using biochemical, morphological, and molecular approaches. Future studies in phagocytosis will likely involve continued identification of specific receptor targets for immunomodulation therapy in a variety of diseases involving phagocytosis.
Likewise, given the double-edged nature of autophagic responses, a better understanding of molecular interactions that underlie the selective recognition of damaged cargo may allow development of therapies to clear damaged cellular constituents while avoiding bystander effects. Although the autophagy response is induced in a wide array of developmental, physiological, and pathological situations, the outcome of autophagy induction is far from uniform. Future studies in aging and diseased tissues will serve to more fully delineate the cell type- or stimulus-specific mechanisms that determine cell fate after autophagy. It is likely that the outcome will depend on a balance of several factors.
Finally, given the convergence of autophagic, phagocytic, and endocytic pathways at the lysosome, a more detailed understanding of the cellular signaling events that induce cytoskeletal rearrangements and regulate lysosomal fusion and activity is of primary importance. Impairments in completing the process of autophagic degradation, microbial killing, or clearance of danger signals from damaged cells undoubtedly play major roles in human diseases. Understanding the factors that regulate fusion of phagosomes and autophagosomes with lysosomes may yield insight into mechanisms by which some pathogens evade lysosomal killing. Likewise, efforts to enhance the autophagic degradation of aggregate-prone proteins within the lysosome will likely prove beneficial for treatment of neurodegeneration and other diseases of disordered proteostasis.
Ultimately, disease therapies harnessing the power of phagocytosis or autophagy will likely be dependent on three factors: i) the ability to correctly target cargo by the phagosome or autophagosome, ii) successful intracellular trafficking and lysosomal delivery, and iii) the capacity for generating germicidal ROS bursts or hydrolytic enzyme activity to complete the process of lysosomal clearance. As either a cellular recycling or a defensive response, impairment at any step of the autophagic process—at sequestration and cargo targeting, at lysosomal fusion and degradation, or at reutilization of liberated products to replace digested parts—may represent potential future targets for disease-modulating therapies.
Footnotes
Supported in part by the NIH (AG026389, NS065789, and NS059806 sub-project 6235 to C.T.C.; HL095495 to T.D.O.). E.A.O. is a predoctoral trainee on T32 HL094295. C.T.C. is recipient of a Julie Martin Mid-Career Award in Aging Research funded by the Ellison Medical Foundation and the American Federation for Aging Research.
References
- 1.Virchow R: Disease, life, and man; selected essays. Translated and with an introduction by LJ Rather. Stanford, CA, Stanford University Press, 1958, p 106
- 2.Sagan L. On the origin of mitosing cells. J Theor Biol. 1967;14:255–274. doi: 10.1016/0022-5193(67)90079-3. [DOI] [PubMed] [Google Scholar]
- 3.Sanjuan M.A., Green D.R. Eating for good health: linking autophagy and phagocytosis in host defense. Autophagy. 2008;4:607–611. doi: 10.4161/auto.6397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kaufmann S.H. Immunology’s foundation: the 100-year anniversary of the Nobel Prize to Paul Ehrlich and Elie Metchnikoff. Nat Immunol. 2008;9:705–712. doi: 10.1038/ni0708-705. [DOI] [PubMed] [Google Scholar]
- 5.Mechnikov I. Elsevier; Amsterdam: 1967. Nobel lecture: on the present state of the question of immunity in infectious diseases. Nobel Lectures, Physiology or Medicine 1901–1921.http://www.nobelprize.org/nobel_prizes/medicine/laureates/1908/mechnikov-lecture.html [available from. October 25, 2012] [Google Scholar]
- 6.Goodale J.L. On the absorption of foreign substances by the faucial tonsils in the human subject. J Boston Soc Med Sci. 1897;1:10–17. [PMC free article] [PubMed] [Google Scholar]
- 7.Buxton B.H., Torrey J.C. Studies in absorption. J Med Res. 1906;15:3–88.43. [PMC free article] [PubMed] [Google Scholar]
- 8.Wright A., Douglas S.R. An experimental investigation of the rôle of the blood fluids in connection with phagocytosis. Proc R Soc Lond. 1903;72:357–370. doi: 10.1093/clinids/11.5.827. [DOI] [PubMed] [Google Scholar]
- 9.Walker E.L. The relative influence of the blood fluids and the bacterial toxins on phagocytosis. J Med Res. 1905;14:173–180. [PMC free article] [PubMed] [Google Scholar]
- 10.Sappington S.W. Studies in typhoid vaccines and opsonins. J Med Res. 1910;22:435–460.23. [PMC free article] [PubMed] [Google Scholar]
- 11.Bartlett C.J., Ozaki Y. Phagocytosis in vivo under various conditions. J Med Res. 1917;37:139–160. [PMC free article] [PubMed] [Google Scholar]
- 12.Permar H.H. The migration and fate of the mononuclear phagocyte of the lung. J Med Res. 1921;42:209–225. [PMC free article] [PubMed] [Google Scholar]
- 13.Motohashi S. Fixed-tissue phagocytosis. J Med Res. 1922;43:419–434. [PMC free article] [PubMed] [Google Scholar]
- 14.Ozaki Y. Phagocytosis of bacteria in the excised spleen after perfusion with Locke’s solution. J Med Res. 1917;37:247–258.1. [PMC free article] [PubMed] [Google Scholar]
- 15.Permar H.H. The mononuclear phagocytes in experimental pneumonia. J Med Res. 1923;44:27–50.1. [PMC free article] [PubMed] [Google Scholar]
- 16.Foot N.C. Studies on endothelial reactions: (first paper.) The macrophages of the loose connective tissue. J Med Res. 1919;40:353–370.1. [PMC free article] [PubMed] [Google Scholar]
- 17.Briscoe J.C. An experimental investigation of the phagocytic action of the alveolar cells of the lung. J Pathol Bacteriol. 1908;12:66–100. [Google Scholar]
- 18.Gazayerli M.E. On the nature of the pulmonary alveolar lining and the origin of the alveolar phagocyte. J Pathol Bacteriol. 1936;43:357–366. [Google Scholar]
- 19.Permar H.H. An experimental study of the mononuclear phagocytes of the lung. J Med Res. 1920;42:9–32. [PMC free article] [PubMed] [Google Scholar]
- 20.Permar H.H. The development of the mononuclear phagocyte of the lung. J Med Res. 1921;42:147–162.1. [PMC free article] [PubMed] [Google Scholar]
- 21.Simpson M.E. The experimental production of macrophages in the circulating blood. J Med Res. 1922;43:77–144.1. [PMC free article] [PubMed] [Google Scholar]
- 22.Foot N.C. Studies on endothelial reactions: X. On the origin of the pulmonary “dust cell”. Am J Pathol. 1927;3:413–444.7. [PMC free article] [PubMed] [Google Scholar]
- 23.Kumar S., Jack R. Origin of monocytes and their differentiation to macrophages and dendritic cells. J Endotoxin Res. 2006;12:278–284. doi: 10.1179/096805106X118861. [DOI] [PubMed] [Google Scholar]
- 24.Gordon S., Taylor P.R. Monocyte and macrophage heterogeneity. Nat Rev Immunol. 2005;5:953–964. doi: 10.1038/nri1733. [DOI] [PubMed] [Google Scholar]
- 25.Ward P.A., Lepow I.H., Newman L.J. Bacterial factors chemotactic for polymorphonuclear leukocytes. Am J Pathol. 1968;52:725–736. [PMC free article] [PubMed] [Google Scholar]
- 26.Unanue E.R. Secretory function of mononuclear phagocytes: a review. Am J Pathol. 1976;83:396–418. [PMC free article] [PubMed] [Google Scholar]
- 27.Stossel T.P. Phagocytosis. Clinical disorders of recognition and ingestion. Am J Pathol. 1977;88:741–752. [PMC free article] [PubMed] [Google Scholar]
- 28.Hanayama R., Tanaka M., Miyasaka K., Aozasa K., Koike M., Uchiyama Y., Nagata S. Autoimmune disease and impaired uptake of apoptotic cells in MFG-E8-deficient mice. Science. 2004;304:1147–1150. doi: 10.1126/science.1094359. [DOI] [PubMed] [Google Scholar]
- 29.Ravichandran K.S. Find-me and eat-me signals in apoptotic cell clearance: progress and conundrums. J Exp Med. 2010;207:1807–1817. doi: 10.1084/jem.20101157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Leibovich S.J., Ross R. The role of the macrophage in wound repair. A study with hydrocortisone and antimacrophage serum. Am J Pathol. 1975;78:71–100. [PMC free article] [PubMed] [Google Scholar]
- 31.Schaffner T., Taylor K., Bartucci E.J., Fischer-Dzoga K., Beeson J.H., Glagov S., Wissler R.W. Arterial foam cells with distinctive immunomorphologic and histochemical features of macrophages. Am J Pathol. 1980;100:57–80. [PMC free article] [PubMed] [Google Scholar]
- 32.Gerrity R.G. The role of the monocyte in atherogenesis: I. Transition of blood-borne monocytes into foam cells in fatty lesions. Am J Pathol. 1981;103:181–190. [PMC free article] [PubMed] [Google Scholar]
- 33.Gerrity R.G. The role of the monocyte in atherogenesis: II. Migration of foam cells from atherosclerotic lesions. Am J Pathol. 1981;103:191–200. [PMC free article] [PubMed] [Google Scholar]
- 34.Ganesan S., Faris A.N., Comstock A.T., Sonstein J., Curtis J.L., Sajjan U.S. Elastase/LPS-exposed mice exhibit impaired innate immune responses to bacterial challenge: role of scavenger receptor A. Am J Pathol. 2012;180:61–72. doi: 10.1016/j.ajpath.2011.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gorgani N.N., Thathaisong U., Mukaro V.R., Poungpair O., Tirimacco A., Hii C.S., Ferrante A. Regulation of CRIg expression and phagocytosis in human macrophages by arachidonate, dexamethasone, and cytokines. Am J Pathol. 2011;179:1310–1318. doi: 10.1016/j.ajpath.2011.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chuang P.C., Lin Y.J., Wu M.H., Wing L.Y., Shoji Y., Tsai S.J. Inhibition of CD36-dependent phagocytosis by prostaglandin E2 contributes to the development of endometriosis. Am J Pathol. 2010;176:850–860. doi: 10.2353/ajpath.2010.090551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mittal R., Gonzalez-Gomez I., Goth K.A., Prasadarao N.V. Inhibition of inducible nitric oxide controls pathogen load and brain damage by enhancing phagocytosis of Escherichia coli K1 in neonatal meningitis. Am J Pathol. 2010;176:1292–1305. doi: 10.2353/ajpath.2010.090851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Frautschy S.A., Cole G.M., Baird A. Phagocytosis and deposition of vascular beta-amyloid in rat brains injected with Alzheimer beta-amyloid. Am J Pathol. 1992;140:1389–1399. [PMC free article] [PubMed] [Google Scholar]
- 39.Trebst C., Sørensen T.L., Kivisäkk P., Cathcart M.K., Hesselgesser J., Horuk R., Sellebjerg F., Lassmann H., Ransohoff R.M. CCR1+/CCR5+ mononuclear phagocytes accumulate in the central nervous system of patients with multiple sclerosis. Am J Pathol. 2001;159:1701–1710. doi: 10.1016/s0002-9440(10)63017-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gaikwad S., Larionov S., Wang Y., Dannenberg H., Matozaki T., Monsonego A., Thal D.R., Neumann H. Signal regulatory protein-beta1: a microglial modulator of phagocytosis in Alzheimer’s disease. Am J Pathol. 2009;175:2528–2539. doi: 10.2353/ajpath.2009.090147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sokolowski J.D., Mandell J.W. Phagocytic clearance in neurodegeneration. Am J Pathol. 2011;178:1416–1428. doi: 10.1016/j.ajpath.2010.12.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brancato S.K., Albina J.E. Wound macrophages as key regulators of repair: origin, phenotype, and function. Am J Pathol. 2011;178:19–25. doi: 10.1016/j.ajpath.2010.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schoppmann S.F., Birner P., Stöckl J., Kalt R., Ullrich R., Caucig C., Kriehuber E., Nagy K., Alitalo K., Kerjaschki D. Tumor-associated macrophages express lymphatic endothelial growth factors and are related to peritumoral lymphangiogenesis. Am J Pathol. 2002;161:947–956. doi: 10.1016/S0002-9440(10)64255-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kigerl K.A., Gensel J.C., Ankeny D.P., Alexander J.K., Donnelly D.J., Popovich P.G. Identification of two distinct macrophage subsets with divergent effects causing either neurotoxicity or regeneration in the injured mouse spinal cord. J Neurosci. 2009;29:13435–13444. doi: 10.1523/JNEUROSCI.3257-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Prokop S., Heppner F.L., Goebel H.H., Stenzel W. M2 polarized macrophages and giant cells contribute to myofibrosis in neuromuscular sarcoidosis. Am J Pathol. 2011;178:1279–1286. doi: 10.1016/j.ajpath.2010.11.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fujita E., Shimizu A., Masuda Y., Kuwahara N., Arai T., Nagasaka S., Aki K., Mii A., Natori Y., Iino Y., Katayama Y., Fukuda Y. Statin attenuates experimental anti-glomerular basement membrane glomerulonephritis together with the augmentation of alternatively activated macrophages. Am J Pathol. 2010;177:1143–1154. doi: 10.2353/ajpath.2010.090608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Trujillo G., O’Connor E.C., Kunkel S.L., Hogaboam C.M. A novel mechanism for CCR4 in the regulation of macrophage activation in bleomycin-induced pulmonary fibrosis. Am J Pathol. 2008;172:1209–1221. doi: 10.2353/ajpath.2008.070832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fantone J.C., Ward P.A. Role of oxygen-derived free radicals and metabolites in leukocyte-dependent inflammatory reactions. Am J Pathol. 1982;107:395–418. [PMC free article] [PubMed] [Google Scholar]
- 49.Baldridge C.W., Gerard R.W. The extra respiration of phagocytosis. Am J Physiol. 1933;103:235–236. [Google Scholar]
- 50.Babior B.M. Phagocytes and oxidative stress. Am J Med. 2000;109:33–44. doi: 10.1016/s0002-9343(00)00481-2. [DOI] [PubMed] [Google Scholar]
- 51.Manni M.L., Tomai L.P., Norris C.A., Thomas L.M., Kelley E.E., Salter R.D., Crapo J.D., Chang L.Y., Watkins S.C., Piganelli J.D., Oury T.D. Extracellular superoxide dismutase in macrophages augments bacterial killing by promoting phagocytosis. Am J Pathol. 2011;178:2752–2759. doi: 10.1016/j.ajpath.2011.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Oberley-Deegan R.E., Lee Y.M., Morey G.E., Cook D.M., Chan E.D., Crapo J.D. The antioxidant mimetic, MnTE-2-PyP, reduces intracellular growth of Mycobacterium abscessus. Am J Respir Cell Mol Biol. 2009;41:170–178. doi: 10.1165/rcmb.2008-0138OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rybicka J.M., Balce D.R., Khan M.F., Krohn R.M., Yates R.M. NADPH oxidase activity controls phagosomal proteolysis in macrophages through modulation of the lumenal redox environment of phagosomes. Proc Natl Acad Sci USA. 2010;107:10496–10501. doi: 10.1073/pnas.0914867107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gloire G., Legrand-Poels S., Piette J. NF-kappaB activation by reactive oxygen species: fifteen years later. Biochem Pharmacol. 2006;72:1493–1505. doi: 10.1016/j.bcp.2006.04.011. [DOI] [PubMed] [Google Scholar]
- 55.De Duve C., Pressman B.C., Gianetto R., Wattiaux R., Appelmans F. Tissue fractionation studies. 6. Intracellular distribution patterns of enzymes in rat-liver tissue. Biochem J. 1955;60:604–617. doi: 10.1042/bj0600604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hruban Z., Spargo B., Swift H., Wissler R.W., Kleinfeld R.G. Focal cytoplasmic degradation. Am J Pathol. 1963;42:657–683. [PMC free article] [PubMed] [Google Scholar]
- 57.Manjithaya R., Anjard C., Loomis W.F., Subramani S. Unconventional secretion of Pichia pastoris Acb1 is dependent on GRASP protein, peroxisomal functions, and autophagosome formation. J Cell Biol. 2010;188:537–546. doi: 10.1083/jcb.200911149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang A.L., Lukas T.J., Yuan M., Du N., Tso M.O., Neufeld A.H. Autophagy and exosomes in the aged retinal pigment epithelium: possible relevance to drusen formation and age-related macular degeneration. PLoS One. 2009;4:e4160. doi: 10.1371/journal.pone.0004160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Arstila A.U., Trump B.F. Studies on cellular autophagocytosis. The formation of autophagic vacuoles in the liver after glucagon administration. Am J Pathol. 1968;53:687–733. [PMC free article] [PubMed] [Google Scholar]
- 60.Shelburne J.D., Arstila A.U., Trump B.F. Studies on cellular autophagocytosis. The relationship of autophagocytosis to protein synthesis and to energy metabolism in rat liver and flounder kidney tubules in vitro. Am J Pathol. 1973;73:641–670. [PMC free article] [PubMed] [Google Scholar]
- 61.Greene W.B., Balentine J.D., Hennigar G.R. Selective mitochondrial degeneration in renal tubules following hyperbaric oxygen exposure. Am J Pathol. 1979;96:737–752. [PMC free article] [PubMed] [Google Scholar]
- 62.Narendra D., Tanaka A., Suen D.F., Youle R.J. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol. 2008;183:795–803. doi: 10.1083/jcb.200809125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dagda R.K., Cherra S.J., 3rd, Kulich S.M., Tandon A., Park D., Chu C.T. Loss of PINK1 function promotes mitophagy through effects on oxidative stress and mitochondrial fission. J Biol Chem. 2009;284:13843–13855. doi: 10.1074/jbc.M808515200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bursch W. The autophagosomal-lysosomal compartment in programmed cell death. Cell Death Differ. 2001;8:569–581. doi: 10.1038/sj.cdd.4400852. [DOI] [PubMed] [Google Scholar]
- 65.Klionsky D.J., Cregg J.M., Dunn W.A., Jr., Emr S.D., Sakai Y., Sandoval I.V., Sibirny A., Subramani S., Thumm M., Veenhuis M., Ohsumi Y. A unified nomenclature for yeast autophagy-related genes. Dev Cell. 2003;5:539–545. doi: 10.1016/s1534-5807(03)00296-x. [DOI] [PubMed] [Google Scholar]
- 66.Kominami E., Kunio I., Katunuma N. Activation of the intramyofibral autophagic-lysosomal system in muscular dystrophy. Am J Pathol. 1987;127:461–466. [PMC free article] [PubMed] [Google Scholar]
- 67.Stadelmann C., Deckwerth T.L., Srinivasan A., Bancher C., Brück W., Jellinger K., Lassmann H. Activation of caspase-3 in single neurons and autophagic granules of granulovacuolar degeneration in Alzheimer’s disease. Evidence for apoptotic cell death. Am J Pathol. 1999;155:1459–1466. doi: 10.1016/S0002-9440(10)65460-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yang D.S., Kumar A., Stavrides P., Peterson J., Peterhoff C.M., Pawlik M., Levy E., Cataldo A.M., Nixon R.A. Neuronal apoptosis and autophagy cross talk in aging PS/APP mice, a model of Alzheimer’s disease. Am J Pathol. 2008;173:665–681. doi: 10.2353/ajpath.2008.071176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Takahashi A., Kimura T., Takabatake Y., Namba T., Kaimori J., Kitamura H., Matsui I., Niimura F., Matsusaka T., Fujita N., Yoshimori T., Isaka Y., Rakugi H. Autophagy guards against cisplatin-induced acute kidney injury. Am J Pathol. 2012;180:517–525. doi: 10.1016/j.ajpath.2011.11.001. [DOI] [PubMed] [Google Scholar]
- 70.Li S., Zhou Y., Fan J., Cao S., Cao T., Huang F., Zhuang S., Wang Y., Yu X., Mao H. Heat shock protein 72 enhances autophagy as a protective mechanism in lipopolysaccharide-induced peritonitis in rats. Am J Pathol. 2011;179:2822–2834. doi: 10.1016/j.ajpath.2011.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.McDonnell T.J., Deane N., Platt F.M., Nunez G., Jaeger U., McKearn J.P., Korsmeyer S.J. bcl-2-immunoglobulin transgenic mice demonstrate extended B cell survival and follicular lymphoproliferation. Cell. 1989;57:79–88. doi: 10.1016/0092-8674(89)90174-8. [DOI] [PubMed] [Google Scholar]
- 72.Kabeya Y., Mizushima N., Ueno T., Yamamoto A., Kirisako T., Noda T., Kominami E., Ohsumi Y., Yoshimori T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing [Erratum appeared in EMBO J 2003, 22:4577] EMBO J. 2000;19:5720–5728. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ichimura Y., Kirisako T., Takao T., Satomi Y., Shimonishi Y., Ishihara N., Mizushima N., Tanida I., Kominami E., Ohsumi M., Noda T., Ohsumi Y. A ubiquitin-like system mediates protein lipidation. Nature. 2000;408:488–492. doi: 10.1038/35044114. [DOI] [PubMed] [Google Scholar]
- 74.Ravikumar B., Vacher C., Berger Z., Davies J.E., Luo S., Oroz L.G., Scaravilli F., Easton D.F., Duden R., O’Kane C.J., Rubinsztein D.C. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet. 2004;36:585–595. doi: 10.1038/ng1362. [DOI] [PubMed] [Google Scholar]
- 75.Spencer B., Potkar R., Trejo M., Rockenstein E., Patrick C., Gindi R., Adame A., Wyss-Coray T., Masliah E. Beclin 1 gene transfer activates autophagy and ameliorates the neurodegenerative pathology in alpha-synuclein models of Parkinson’s and Lewy body diseases. J Neurosci. 2009;29:13578–13588. doi: 10.1523/JNEUROSCI.4390-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hidvegi T., Ewing M., Hale P., Dippold C., Beckett C., Kemp C., Maurice N., Mukherjee A., Goldbach C., Watkins S., Michalopoulos G., Perlmutter D.H. An autophagy-enhancing drug promotes degradation of mutant alpha1-antitrypsin Z and reduces hepatic fibrosis. Science. 2010;329:229–232. doi: 10.1126/science.1190354. [DOI] [PubMed] [Google Scholar]
- 77.Bjørkøy G., Lamark T., Brech A., Outzen H., Perander M., Overvatn A., Stenmark H., Johansen T. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol. 2005;171:603–614. doi: 10.1083/jcb.200507002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yu W.H., Dorado B., Figueroa H.Y., Wang L., Planel E., Cookson M.R., Clark L.N., Duff K.E. Metabolic activity determines efficacy of macroautophagic clearance of pathological oligomeric alpha-synuclein. Am J Pathol. 2009;175:736–747. doi: 10.2353/ajpath.2009.080928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ding W.X., Ni H.M., Gao W., Yoshimori T., Stolz D.B., Ron D., Yin X.M. Linking of autophagy to ubiquitin-proteasome system is important for the regulation of endoplasmic reticulum stress and cell viability. Am J Pathol. 2007;171:513–524. doi: 10.2353/ajpath.2007.070188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zhu J.H., Horbinski C., Guo F., Watkins S., Uchiyama Y., Chu C.T. Regulation of autophagy by extracellular signal-regulated protein kinases during 1-methyl-4-phenylpyridinium-induced cell death. Am J Pathol. 2007;170:75–86. doi: 10.2353/ajpath.2007.060524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hoozemans J.J., van Haastert E.S., Nijholt D.A., Rozemuller A.J., Eikelenboom P., Scheper W. The unfolded protein response is activated in pretangle neurons in Alzheimer’s disease hippocampus. Am J Pathol. 2009;174:1241–1251. doi: 10.2353/ajpath.2009.080814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Nogalska A., D’Agostino C., Terracciano C., Engel W.K., Askanas V. Impaired autophagy in sporadic inclusion-body myositis and in endoplasmic reticulum stress-provoked cultured human muscle fibers. Am J Pathol. 2010;177:1377–1387. doi: 10.2353/ajpath.2010.100050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Chu C.T. Autophagic stress in neuronal injury and disease. J Neuropathol Exp Neurol. 2006;65:423–432. doi: 10.1097/01.jnen.0000229233.75253.be. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Koike M., Shibata M., Waguri S., Yoshimura K., Tanida I., Kominami E., Gotow T., Peters C., von Figura K., Mizushima N., Saftig P., Uchiyama Y. Participation of autophagy in storage of lysosomes in neurons from mouse models of neuronal ceroid-lipofuscinoses (Batten disease) Am J Pathol. 2005;167:1713–1728. doi: 10.1016/S0002-9440(10)61253-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Yu W.H., Kumar A., Peterhoff C., Shapiro Kulnane L., Uchiyama Y., Lamb B.T., Cuervo A.M., Nixon R.A. Autophagic vacuoles are enriched in amyloid precursor protein-secretase activities: implications for beta-amyloid peptide over-production and localization in Alzheimer’s disease. Int J Biochem Cell Biol. 2004;36:2531–2540. doi: 10.1016/j.biocel.2004.05.010. [DOI] [PubMed] [Google Scholar]
- 86.Walls K.C., Ghosh A.P., Franklin A.V., Klocke B.J., Ballestas M., Shacka J.J., Zhang J., Roth K.A. Lysosome dysfunction triggers Atg7-dependent neural apoptosis. J Biol Chem. 2010;285:10497–10507. doi: 10.1074/jbc.M110.103747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Scarlatti F., Maffei R., Beau I., Codogno P., Ghidoni R. Role of non-canonical Beclin 1-independent autophagy in cell death induced by resveratrol in human breast cancer cells. Cell Death Differ. 2008;15:1318–1329. doi: 10.1038/cdd.2008.51. [DOI] [PubMed] [Google Scholar]
- 88.Tian S., Lin J., Zhou J., Wang X., Li Y., Ren X., Yu W., Zhong W., Xiao J., Sheng F., Chen Y., Jin C., Li S., Zheng Z., Xia B. Beclin 1-independent autophagy induced by a Bcl-X(L)/Bcl-2 targeting compound, Z18. Autophagy. 2010;6:1032–1041. doi: 10.4161/auto.6.8.13336. [DOI] [PubMed] [Google Scholar]
- 89.Kim I., Lemasters J.J. Mitophagy selectively degrades individual damaged mitochondria after photoirradiation. Antioxid Redox Signal. 2011;14:1919–1928. doi: 10.1089/ars.2010.3768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Pattingre S., Tassa A., Qu X., Garuti R., Liang X.H., Mizushima N., Packer M., Schneider M.D., Levine B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. 2005;122:927–939. doi: 10.1016/j.cell.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 91.Elrick M.J., Yu T., Chung C., Lieberman A.P. Impaired proteolysis underlies autophagic dysfunction in Niemann-Pick type C disease. Hum Mol Genet. 2012;21:4876–4887. doi: 10.1093/hmg/dds324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Arduino D.M., Esteves A.R., Cortes L., Silva D.F., Patel B., Grazina M., Swerdlow R.H., Oliveira C.R., Cardoso S.M. Mitochondrial metabolism in Parkinson’s disease impairs quality control autophagy by hampering microtubule-dependent traffic. Hum Mol Genet. 2012;21:4680–4702. doi: 10.1093/hmg/dds309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Dagda R.K., Zhu J., Kulich S.M., Chu C.T. Mitochondrially localized ERK2 regulates mitophagy and autophagic cell stress: implications for Parkinson’s disease. Autophagy. 2008;4:770–782. doi: 10.4161/auto.6458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Zhu J.H., Gusdon A.M., Cimen H., Van Houten B., Koc E., Chu C.T. Impaired mitochondrial biogenesis contributes to depletion of functional mitochondria in chronic MPP+ toxicity: dual roles for ERK1/2. Cell Death Dis. 2012;3:e312. doi: 10.1038/cddis.2012.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kimura A., Ishida Y., Wada T., Hisaoka T., Morikawa Y., Sugaya T., Mukaida N., Kondo T. The absence of interleukin-6 enhanced arsenite-induced renal injury by promoting autophagy of tubular epithelial cells with aberrant extracellular signal-regulated kinase activation. Am J Pathol. 2010;176:40–50. doi: 10.2353/ajpath.2010.090146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Komatsu M., Wang Q.J., Holstein G.R., Friedrich V.L., Jr., Iwata J., Kominami E., Chait B.T., Tanaka K., Yue Z. Essential role for autophagy protein Atg7 in the maintenance of axonal homeostasis and the prevention of axonal degeneration. Proc Natl Acad Sci USA. 2007;104:14489–14494. doi: 10.1073/pnas.0701311104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Koike M., Shibata M., Tadakoshi M., Gotoh K., Komatsu M., Waguri S., Kawahara N., Kuida K., Nagata S., Kominami E., Tanaka K., Uchiyama Y. Inhibition of autophagy prevents hippocampal pyramidal neuron death after hypoxic-ischemic injury. Am J Pathol. 2008;172:454–469. doi: 10.2353/ajpath.2008.070876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Cheng H.C., Kim S.R., Oo T.F., Kareva T., Yarygina O., Rzhetskaya M., Wang C., During M., Talloczy Z., Tanaka K., Komatsu M., Kobayashi K., Okano H., Kholodilov N., Burke R.E. Akt suppresses retrograde degeneration of dopaminergic axons by inhibition of macroautophagy. J Neurosci. 2011;31:2125–2135. doi: 10.1523/JNEUROSCI.5519-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Jaworski T., Lechat B., Demedts D., Gielis L., Devijver H., Borghgraef P., Duimel H., Verheyen F., Kügler S., Van Leuven F. Dendritic degeneration, neurovascular defects, and inflammation precede neuronal loss in a mouse model for tau-mediated neurodegeneration. Am J Pathol. 2011;179:2001–2015. doi: 10.1016/j.ajpath.2011.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Boland B., Kumar A., Lee S., Platt F.M., Wegiel J., Yu W.H., Nixon R.A. Autophagy induction and autophagosome clearance in neurons: relationship to autophagic pathology in Alzheimer’s disease. J Neurosci. 2008;28:6926–6937. doi: 10.1523/JNEUROSCI.0800-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Rowland A.M., Richmond J.E., Olsen J.G., Hall D.H., Bamber B.A. Presynaptic terminals independently regulate synaptic clustering and autophagy of GABAA receptors in Caenorhabditis elegans. J Neurosci. 2006;26:1711–1720. doi: 10.1523/JNEUROSCI.2279-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Hernandez D., Torres C.A., Setlik W., Cebrián C., Mosharov E.V., Tang G., Cheng H.C., Kholodilov N., Yarygina O., Burke R.E., Gershon M., Sulzer D. Regulation of presynaptic neurotransmission by macroautophagy. Neuron. 2012;74:277–284. doi: 10.1016/j.neuron.2012.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Li Z., Okamoto K., Hayashi Y., Sheng M. The importance of dendritic mitochondria in the morphogenesis and plasticity of spines and synapses. Cell. 2004;119:873–887. doi: 10.1016/j.cell.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 104.Cherra S.J., 3rd, Steer E., Gusdon A.M., Kiselyov K., Chu C.T. Mutant LRRK2 elicits calcium imbalance and depletion of dendritic mitochondria in neurons. Am J Pathol. 2013;182:474–478. doi: 10.1016/j.ajpath.2012.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Boya P., González-Polo R.A., Casares N., Perfettini J.L., Dessen P., Larochette N., Métivier D., Meley D., Souquere S., Yoshimori T., Pierron G., Codogno P., Kroemer G. Inhibition of macroautophagy triggers apoptosis. Mol Cell Biol. 2005;25:1025–1040. doi: 10.1128/MCB.25.3.1025-1040.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Botti J., Djavaheri-Mergny M., Pilatte Y., Codogno P. Autophagy signaling and the cogwheels of cancer. Autophagy. 2006;2:67–73. doi: 10.4161/auto.2.2.2458. [DOI] [PubMed] [Google Scholar]
- 107.Horbinski C., Mojesky C., Kyprianou N. Live free or die: tales of homeless (cells) in cancer. Am J Pathol. 2010;177:1044–1052. doi: 10.2353/ajpath.2010.091270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Viuff B., Tjørnehøj K., Larsen L.E., Røntved C.M., Uttenthal A., Rønsholt L., Alexandersen S. Replication and clearance of respiratory syncytial virus: apoptosis is an important pathway of virus clearance after experimental infection with bovine respiratory syncytial virus. Am J Pathol. 2002;161:2195–2207. doi: 10.1016/S0002-9440(10)64496-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Milutinovic P.S., Alcorn J.F., Englert J.M., Crum L.T., Oury T.D. The receptor for advanced glycation end products is a central mediator of asthma pathogenesis. Am J Pathol. 2012;181:1215–1225. doi: 10.1016/j.ajpath.2012.06.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Koedel U., Merbt U.M., Schmidt C., Angele B., Popp B., Wagner H., Pfister H.W., Kirschning C.J. Acute brain injury triggers MyD88-dependent, TLR2/4-independent inflammatory responses. Am J Pathol. 2007;171:200–213. doi: 10.2353/ajpath.2007.060821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Deng L., Zhou J.F., Sellers R.S., Li J.F., Nguyen A.V., Wang Y., Orlofsky A., Liu Q., Hume D.A., Pollard J.W., Augenlicht L., Lin E.Y. A novel mouse model of inflammatory bowel disease links mammalian target of rapamycin-dependent hyperproliferation of colonic epithelium to inflammation-associated tumorigenesis. Am J Pathol. 2010;176:952–967. doi: 10.2353/ajpath.2010.090622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Rodriguez-Manzanet R., Sanjuan M.A., Wu H.Y., Quintana F.J., Xiao S., Anderson A.C., Weiner H.L., Green D.R., Kuchroo V.K. T and B cell hyperactivity and autoimmunity associated with niche-specific defects in apoptotic body clearance in TIM-4-deficient mice. Proc Natl Acad Sci USA. 2010;107:8706–8711. doi: 10.1073/pnas.0910359107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Deretic V. Autophagy as an immune defense mechanism. Curr Opin Immunol. 2006;18:375–382. doi: 10.1016/j.coi.2006.05.019. [DOI] [PubMed] [Google Scholar]
- 114.Deretic V. Autophagosome and phagosome. Methods Mol Biol. 2008;445:1–10. doi: 10.1007/978-1-59745-157-4_1. [DOI] [PubMed] [Google Scholar]
- 115.Nagata S., Hanayama R., Kawane K. Autoimmunity and the clearance of dead cells. Cell. 2010;140:619–630. doi: 10.1016/j.cell.2010.02.014. [DOI] [PubMed] [Google Scholar]
- 116.Qu X., Zou Z., Sun Q., Luby-Phelps K., Cheng P., Hogan R.N., Gilpin C., Levine B. Autophagy gene-dependent clearance of apoptotic cells during embryonic development. Cell. 2007;128:931–946. doi: 10.1016/j.cell.2006.12.044. [DOI] [PubMed] [Google Scholar]
- 117.Yang H.Z., Wang J.P., Mi S., Liu H.Z., Cui B., Yan H.M., Yan J., Li Z., Liu H., Hua F., Lu W., Hu Z.W. TLR4 activity is required in the resolution of pulmonary inflammation and fibrosis after acute and chronic lung injury. Am J Pathol. 2012;180:275–292. doi: 10.1016/j.ajpath.2011.09.019. [DOI] [PubMed] [Google Scholar]
- 118.Gutierrez M.G., Master S.S., Singh S.B., Taylor G.A., Colombo M.I., Deretic V. Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell. 2004;119:753–766. doi: 10.1016/j.cell.2004.11.038. [DOI] [PubMed] [Google Scholar]
- 119.Sanjuan M.A., Dillon C.P., Tait S.W., Moshiach S., Dorsey F., Connell S., Komatsu M., Tanaka K., Cleveland J.L., Withoff S., Green D.R. Toll-like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature. 2007;450:1253–1257. doi: 10.1038/nature06421. [DOI] [PubMed] [Google Scholar]
- 120.Rautou P.E., Cazals-Hatem D., Feldmann G., Mansouri A., Grodet A., Barge S., Martinot-Peignoux M., Duces A., Bièche I., Lebrec D., Bedossa P., Paradis V., Marcellin P., Valla D., Asselah T., Moreau R. Changes in autophagic response in patients with chronic hepatitis C virus infection. Am J Pathol. 2011;178:2708–2715. doi: 10.1016/j.ajpath.2011.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]