

Abstract
The aim of this study was to determine the role of AMP-activated protein kinase (AMPK) in lipopolysaccharide (LPS)-induced lung endothelial barrier dysfunction and lung injury in vivo. Both cultured human pulmonary artery endothelial cells (HPAECs) and experimental animals [AMPK subunit α–deficient mice and wild-type (WT) control mice (C57BL/6J)] were used. In cultured HPAECs, LPS increased endothelial permeability in parallel with a decrease in AMPK activity. Consistent with this observation, AMPK activation with the potent AMPK activator 5-aminoimidazole-4-carboxamide-1-d-ribofuranoside (AICAR) attenuated LPS-induced endothelial hyperpermeability in vitro. Intratracheal administration of LPS (1 mg/kg) in WT mice reduced AMPK phosphorylation at Thr172 in lung tissue extracts, increased protein content and cell count in bronchial alveolar lavage fluid, and increased Evans Blue dye infiltration into the lung. These same attributes were similarly enhanced in AMPKα-knockout mice, compared with WT mice. Pretreatment with AICAR reduced these lung injury indicators in LPS-treated WT mice. AMPK activation with AICAR attenuated LPS-induced endothelial hyperpermeability by activating the Rac/Cdc42/PAK pathway, with concomitant inhibition of the Rho pathway, and decreased VE-cadherin phosphorylation at Tyr658. We conclude that AMPK activity supports normal endothelial barrier function and that LPS exposure inhibits AMPK, thereby contributing to endothelial barrier dysfunction and lung injury.
Vascular endothelial permeability plays a pivotal role in regulating many physiological and pathological processes, including angiogenesis, immunity, and inflammation.1 In lungs, endothelial cells form a semipermeable barrier between the vessel lumen and underlying alveoli, thereby mediating the transmigration of blood cells and maintaining fluid homeostasis. The integrity of the endothelial cell (EC) monolayer therefore directly determines lung vascular permeability. For example, EC barrier dysfunction can lead to an increase in permeation of fluid and macromolecules into the interstitium and alveolar space, resulting in pulmonary edema, a major characteristic of acute lung injury.
RhoA, Rac1, and Cdc42 are key members of the Rho GTPase family and are activated on binding GTP at the membrane.2 These proteins are intimately involved in regulating cell adhesion and cytoskeletal dynamics, both of which play an important role in endothelial barrier function.3–5 For example, Rac1 and Cdc42 are important in maintaining, stabilizing, and restoring the endothelial barrier.3 More specifically, Baumer et al6 demonstrated that Rac1 is involved in mitigating endothelial hyperpermeability by a subset of agonists, including thrombin and lipopolysaccharide (LPS). In addition, activation of the Rho GTPases Cdc42 and Rac1 restores endothelium integrity after lung injury.7 Although the contributions of Rho GTPases in maintaining endothelial barrier function are well established, how Rho GTPase is regulated in endothelial cells is largely unknown.
AMP-activated protein kinase (AMPK) is a heterotrimeric serine/threonine kinase; a catalytic α subunit and regulatory β and γ subunits are important in maintaining the stability of the complex. AMPK belongs to a family of energy-sensing enzymes that function as fuel gauges, monitoring changes in the energy status of the cell.8 AMPK is activated in response to a variety of stressors that increase the intracellular ratio of AMP to ATP. On activation, AMPK phosphorylates a number of downstream targets, thereby affecting glucose metabolism, fatty acid oxidation, hepatic lipogenesis, and cholesterol synthesis.9 In addition to its regulatory role in metabolism, recent studies have demonstrated a role for AMPK in maintaining normal endothelial function.10 For example, AMPK subunit α2 exerts protective effects against atherosclerosis by inhibiting the endoplasmic reticulum stress response in ECs.11 Thus, agents that enhance EC barrier function are of potential therapeutic value in a variety of pathological settings, including inflammatory disease, atherosclerosis, and tumor angiogenesis. The effects of AMPK on endothelial barrier function and vascular permeability have not been investigated previously. Thus, the aim of the present study was to investigate whether AMPK protects lung endothelial barrier function and mitigates acute lung injury in response to LPS.
Materials and Methods
Materials
5-Aminoimidazole-4-carboxamide-1-d-ribofuranoside (AICAR; also known as acadesine) was purchased from Toronto Chemicals (Toronto, ON, Canada). The Rac1 inhibitor NSC 23766 was purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Antibodies against Thr172-phosphorylated AMPKα, AMPKα, Ser307-phosphorylated LKB1, Ser79-phosphorylated acetyl coenzyme A carboxylase (ACC), vascular endothelial cadherin (VE-cadherin), and diphosphorylated myosin light chain (pp-MLC Thr18/Ser19) were purchased from Cell Signaling Technology (Danvers, MA). Antibody against LKB1 was purchased from Santa Cruz Biotechnology. Tyr658-phosphorylated VE-cadherin was purchased from Calbiochem (San Diego, CA). Texas Red phalloidin–conjugated and Alexa Fluor 488–conjugated secondary antibodies were purchased from Invitrogen (Carlsbad, CA). Bacterial LPS (Escherichia coli O55:B5) was purchased from Sigma-Aldrich (St. Louis, MO). Unless otherwise specified, all biochemical reagents were purchased from Sigma-Aldrich.
Animals
Knockout (KO) mice lacking AMPK subunit α1 (α1-KO)12 or AMPK subunit α2 (α2-KO)13 and their wild-type (WT) genetic controls were used. The two subunits are encoded by Prkaa1 and Prkaa2 (synonyms: Ampka1 and Ampka2). The mice were housed in temperature-controlled cages under a 12-hour light–dark cycle and were given free access to water and normal chow. Mice aged 8 to 10 weeks were used for subsequent experiments. A solution of LPS (1 mg/kg) in PBS was delivered intratracheally; PBS alone was used as a control. After 16 hours, bronchoalveolar lavage was performed by intratracheal injection of 1 mL of PBS solution, followed by gentle aspiration. The recovered fluids were processed for determination of total protein concentration and white blood cell counts. Lungs from challenged mice were collected for histological evaluation by H&E staining or were frozen at −80°C.
The animal protocol was approved by the University of Oklahoma Health Sciences Center Institutional Animal Care and Use Committee.
Evans Blue Staining in Lungs
Accumulation of Evans Blue dye in lung tissue was evaluated according to a protocol described previously.14 In brief, Evans Blue dye (30 mL/kg) was injected into the external jugular vein 2 hours before sacrifice. At the end of the experiment, a thoracotomy was performed and the lungs were perfused with PBS containing 5 mmol/L EDTA, to remove blood. Both left and right lobes of the lung were excised. Evans Blue accumulation in the lung tissue was measured by spectrofluorometric analysis of lung tissue lysates according to a protocol described previously.15
Cell Culture
Human pulmonary artery endothelial cells (HPAECs) and n bovine pulmonary artery endothelial cells (BPAECs) were purchased from Cell Applications (San Diego, CA). For subsequent experiments, cell lines were used at passages 5 to 9.
Rac1/Cdc42 Activity Assay
Cell lysates were immunoprecipitated with PAK-1 PBD (p21/Cdc42/Rac1-activated kinase p21-binding domain). Rac1/Cdc42 activation was evaluated using a Rac1/Cdc42 activation assay kit (Millipore, Billerica, MA) according to the manufacturer’s instructions.
Immunofluorescence Staining
BPAEC monolayers were fixed in 4% paraformaldehyde solution for 10 minutes, followed by permeabilization with 0.2% Triton X-100 surfactant. After blocking with 1% sodium borohydrite, cell monolayers were incubated with antibodies in blocking solution (2% bovine serum albumin in PBS) for 1 hour, followed by staining with Alexa Fluor 488–conjugated secondary antibody (Invitrogen, Grand Island, NY). Actin filaments were stained with Texas Red–conjugated phalloidin for 1 hour. All of these steps were performed at room temperature. After the immunostaining, glass slides were prepared using mounting medium (Santa Cruz Biotechnology, Santa Cruz, CA) and analyzed using a video-imaging system.
AMPK Activity Assay
Total AMPK was immunoprecipitated from 500 μg of protein using an antibody against AMPKα. AMPK activity in immunoprecipitates was assessed by determining the incorporation of 32P into the synthetic SAMs peptide, as described previously.9,16
Western Blot Analysis
Western blotting was performed as described previously.17
siRNA Transfection in Endothelial Cells
Transient transfection of siRNA was performed according to the Santa Cruz Biotechnology protocol. Briefly, the siRNAs were dissolved in RNAse-free H2O to prepare a 10 μmol/L stock solution. HPAECs grown in six-well plates were transfected with siRNA in Gibco transfection medium containing RNAiMax (Invitrogen). For each transfection, 4 μL siRNA in 100 μL transfection medium was gently mixed with 3 μL of transfection reagent in 100 μL transfection medium. After 30 minutes of incubation at room temperature, the siRNA–lipid complexes were added to the cells in 0.8 mL transfection medium. The cells were incubated with this mixture for 6 hours at 37°C. After incubation, the transfection medium was replaced with normal medium, and cells were cultured for 48 hours.
Adenoviral Preparation and Infection
Adenoviral vector AMPK-CA (a constitutively active AMPK mutant) was generated as described previously.18 Adenoviral vector AMPK-DN (a dominant-negative mutant of AMPK) was generated by subcloning the cDNA encoding AMPKα1-DN-(D159A) into adenoviral vector pAdEasy-1. Confluent cells were infected with adenoviruses as described previously.18 Adenovirus encoding GFP was used as control. Under these conditions, efficiency of GFP expression was >80%.
Statistical Analysis
Data are expressed as means ± SEM. Statistical analyses were performed using Student’s t-test (two groups) or one-way analysis of variance followed by the Bonferroni procedure for multiple comparison tests (three or more groups), using GraphPad Prism software version 4 (GraphPad Software, San Diego, CA). A P value of ≤0.05 was considered significant.
Results
LPS Inhibits AMPK in Vivo
Because AMPK is important in maintaining normal endothelial function and because LPS treatment results in endothelial barrier dysfunction, we investigated whether LPS increases endothelial permeability through modulating AMPK. WT mice treated with LPS had significantly reduced levels of Thr172-phosphorylated AMPK (activated) in lung tissue, compared with untreated mice (Figure 1A). This suggests a role for LPS as an inhibitor of AMPK activity in vivo.
Figure 1.

AMPK activation by AICAR attenuates LPS-induced lung vascular leakage. AICAR (500 mg/kg i.p.) was injected 4 hours before injection of LPS (1 mg/kg intratracheal); BALF and lung tissue extracts were collected 16 hours later. A: Phosphorylation of AMPK at Thr172 in lungs. B and C: Cell counts and protein concentration in BALF. D: Immunochemical evaluation of lung tissue by H&E staining (original magnification ×40). E: Lung vascular permeability was assessed by accumulation of Evans Blue dye in the lungs. Lungs were excised after perfusion and imaged. Each panel shows the right and left lungs (at right and left, respectively). Spectrophotometric analysis of Evans Blue–stained albumin content in the lung tissues was quantified. Data are expressed as means ± SEM. n = 4 to 6 per group. *P < 0.05 versus control; †P < 0.05 versus LPS. Con, control.
LPS Causes Endothelial Barrier Dysfunction in Vivo
We then investigated the effect of LPS treatment on endothelial barrier function in WT mice. Lung EC barrier dysfunction is characterized by increased permeation of fluid and macromolecules into the interstitium and alveolar space. Compared with that of control mice, LPS treatment induced a fivefold increase in the white blood cell count within the bronchoalveolar lavage fluid (BALF), which is indicative of an acute inflammatory response in the lungs (Figure 1B). LPS consistently induced a significant increase in the total protein concentration in BALF (Figure 1C), indicating barrier disruption and lung injury. Histological examination of lung tissue from LPS-treated mice revealed enhanced alveolar wall thickening and infiltration of white blood cells into the lung interstitium and alveolar spaces (Figure 1D). LPS injection caused infiltration of Evans Blue–stained albumin from the vessel into the lung tissue, further substantiating the effect of LPS on EC barrier dysfunction (Figure 1E).
AMPK Depletion Accentuates LPS-Induced Acute Lung Injury in Vivo
Because LPS suppressed AMPK activity in vivo (Figure 1A), we next determined whether AMPK deletion accentuates LPS-induced hyperpermeability and lung injury. Lung injury was analyzed in both WT and AMPKα-KO mice challenged with LPS. The predominant AMPKα subunit found in lungs was α1 (Figure 2A). After LPS treatment, BALF protein levels and cell counts were higher in AMPKα-KO mice than in WT mice (Figure 2, B and C). Consistent with this finding, infiltration of inflammatory cells into lung tissue, lung congestion, and edema were also more severe, especially in α1-KO mice (Figure 2, B–D).
Figure 2.

AMPK deletion increases LPS-induced lung vascular leakage and inflammation. WT, α1-KO (α1−/−), and α2-KO (α2−/−) mice were injected with LPS (1 mg/kg intratracheal); lung tissue and BALF were collected 16 hours later. A: Levels of total AMPKα, AMPKα1, and AMPKα2 in lungs. B and C: Protein concentrations and BALF cell counts D: Immunochemical evaluation of lung tissue by H&E staining (original magnification ×10). E and F: WT, α1-KO, and α2-KO mice were injected with AICAR (500 mg/kg i.p.); after 4 hours, the mice were injected with LPS (1 mg/kg intratracheal); lung tissue and BALF were collected 16 hours later. Data are expressed as means ± SEM. *P < 0.05 versus WT. AIC, AICAR. n = 6 per group.
AMPK Activation Attenuates LPS-Induced Endothelial Barrier Dysfunction in Vivo
We next investigated the effects of AICAR in AMPKα-deficient mice, to exclude potential off-target effects of this drug. AICAR alone did not alter the basal level of Evans Blue–stained albumin extravasation and inflammatory cell infiltration in control WT mice (Figure 1, D and E); however, AICAR treatment significantly attenuated LPS-induced microvascular lung leakage (Figure 1E) and decreased BALF protein concentration (Figure 1C). Furthermore, AICAR did not attenuate the decrease of BALF protein concentration and cell count in LPS-treated α1-KO or α2-KO mice (Figure 2, E and F).
LPS Disrupts EC Barrier Function and Increases Endothelial Hyperpermeability in Cultured BPAECs
EC cytoskeletal rearrangement causes formation of gaps between cells and increases endothelial permeability. We therefore examined whether LPS alters cytoskeletal rearrangement in cultured BPAECs. In unstimulated cells, F-actin was primarily organized into actin bundles and cortical actin structures were more diffuse (Figure 3A). After stimulation with 200 ng/mL LPS, F-actin reorganized into thicker stress fibers in the center of cells, whereas peripheral actin bands were weakened (Figure 3A). These changes were associated with the appearance of paracellular gaps, indicating EC barrier disruption by LPS.
Figure 3.

AMPK activation with AICAR attenuates LPS-induced endothelial barrier disruption. A and B: Effects of AICAR on LPS-induced F-actin and VE-cadherin. BPAECs grown on glass coverslips were treated with 1 mmol/L AICAR for 1 hour before 200 ng/mL LPS challenge. Cytoskeletal remodeling was analyzed by double-immunofluorescence staining with VE-cadherin (green) and Texas Red phalloidin. C: BPAECs were infected with adenovirus AMPK-DN or AMPK-CA for 36 hours; adenovirus GFP served as control. Infected BPAECs were then treated with 200 ng/mL LPS for 6 hours. Paracellular gaps are indicated by arrows. Original magnification ×100.
Activation of AMPK Attenuates LPS-Induced Endothelial Hyperpermeability in Cultured HPAECs and BPAECs
We next determined the effects of AICAR on LPS-induced disruption of endothelial barrier function in cultured HPAECs. AICAR did not affect cytoskeletal rearrangement in unstimulated HPAECs; however, AICAR pretreatment attenuated LPS-induced stress fiber and gap formation (Figure 3, A and B). AMPK-CA attenuated LPS-induced cytoskeletal rearrangement, compared with control (Figure 3C). Conversely, AMPK-DN-infected BPAECs exhibited increased stress fiber and gap formation, compared with control (Figure 3C). Consistent with this finding, AICAR decreased the phosphorylation of VE-cadherin at Tyr658 in a dose- and time-dependent manner (Figure 4, A and B). Inhibition of AMPKα1 or AMPKα2 increased the phosphorylation of VE-cadherin at Tyr658 (Figure 4C).
Figure 4.

Activation of AMPK by AICAR decreases phosphorylation of both VE-cadherin (at Tyr658) and MLC. A and B: Dose- and time-dependent effects of AICAR on VE-cadherin phosphorylation at Tyr658. HPAECs were treated with 0.1 to 2.0 mmol/L AICAR for 1 hour or were treated with AICAR at 1 mmol/L for 5 to 120 minutes. C: Immunoblot analysis of VE-cadherin phosphorylation at Tyr658 in HPAECs transfected with AMPKα1 or AMPKα2 siRNA. HPAECs were transfected with control siRNA, AMPKα1 siRNA, or AMPKα2 siRNA for 48 hours, then treated with AICAR for 1 hour. Cell lysates were subjected to Western blot analysis using antibodies against p-VE-cadherin, VE-cadherin, AMPKα1, and AMPKα2. D: Effects of AICAR on the phosphorylation of MLC in HPAECs. HPAECs were pretreated with 1 mmol/L AICAR for 1 hour and then challenged with 200 ng/mL LPS for 1 or 6 hours. Cell extracts were analyzed for levels of pp-MLC (phosphorylated at both Thr18 and Ser19). Blots are representative of three independent experiments.
AICAR Inhibits LPS-Induced MLC Phosphorylation
The balance between Rho and Rac pathways controls endothelial permeability. To determine whether AICAR affects the Rho signaling pathway, we first tested MLC, a well-established downstream target of Rho pathway. AICAR inhibited LPS-induced phosphorylation of MLC (Figure 4D).
LPS Inhibits AMPK Phosphorylation at Thr172 and AMPK Activity in HPAECs
Thr172 is located in the activation loop of the AMPK α1 and α2 subunits, and its phosphorylation leads to AMPK activation.19 Our in vivo data (Figure 1A) suggested a role for LPS in AMPK phosphorylation at Thr172. To confirm this hypothesis, we exposed confluent HPAECs (an established EC model) to LPS for 0.2, 0.5, 1, 2, and 6 hours. AMPK phosphorylation at Thr172 was detected by Western blotting. LPS inhibited phosphorylation of both AMPK at Thr172 and its downstream kinase, acetyl coenzyme A carboxylase (ACC) at Ser79 (Figure 5A). Consistent with this finding, LPS decreased AMPK activity as early as 30 minutes after LPS exposure (Figure 5B).
Figure 5.

Effects of LPS on the phosphorylation of AMPK and LKB in HPAECs. A: Effects of LPS on the phosphorylation of AMPK at Thr172 and ACC at Ser79 in HPAECs. HPAECs were treated with 200 ng/mL LPS for 0.2 to 6 hours. Cell extracts were analyzed for levels of Thr172-phosphorylated AMPK and Ser79-phosphorylated ACC by Western blotting B: Effects of LPS on AMPK activity. HPAECs were treated with 200 ng/mL LPS for 0.5 to 6 hours. AMPK activity was assayed as described under Materials and Methods. C: Effects of LPS on LKB1 phosphorylation in HPAECs. HPAECs were treated with 200 ng/mL LPS for 0.2 to 6 hours. Cell extracts were analyzed for LKB1 phosphorylation at Ser307. D and E: Dose- and time-dependent effects of LPS on PP2C expression. HPAECs were treated with LPS at 0.05 to 5 μg/mL for 1 hour or at 1.0 μg/mL for 0.2 to 6 hours. Cell extracts were analyzed for levels of PP2C by Western blotting. Data are expressed as means ± SEM. n = 3 per group. *P < 0.05 treated versus untreated control. Blots are representative of three independent experiments.
LPS Increases LKB1 Phosphorylation at Ser307
We next determined whether LPS inhibits AMPK by altering phosphorylation of its upstream kinase LKB1 at Ser307. LPS increased LKB1 phosphorylation at Ser307 (Figure 5C), and therefore the inhibitory effects of LPS on AMPK are independent of LKB1.
LPS Has No Effect on PP2C Expression in HPAECs
Because protein phosphatase 2C (PP2C) dephosphorylates AMPK in response to inflammatory mediators such as TNFα, we determined the effects of LPS on PP2C expression in our system. LPS did not affect PP2C expression in either a dose-dependent or a time-dependent manner (Figure 5, D and E).
AICAR Reduces LPS-Induced AMPK Inhibition
Confluent HPAECs were treated with AICAR in a dose- and time-dependent manner, after which AMPK and ACC phosphorylation were detected via Western blotting. The levels of Thr172-phosphorylated AMPK significantly increased in response to AICAR (0.1 to 2 mmol/L) in a dose-dependent manner (Figure 6A). Using 1.0 mmol/L as the optimal concentration of AICAR, we assessed the time course over 120 minutes for AMPK and ACC phosphorylation. Increased phosphorylation of AMPK at Thr172 and of ACC at Ser79 was detected after 15 minutes and peaked at the 60-minute time point (Figure 6B). Importantly, AICAR prevented the LPS-induced reduction in phosphorylation of AMPK at Thr172 and of ACC at Ser79 (Figure 6C).
Figure 6.

AICAR activates AMPK-ACC pathways in HPAECs. A: Dose-dependent phosphorylation of AMPK by AICAR. HPAECs were stimulated with AICAR at 0.1 to 2 mmol/L for 1 hour. B: Time-dependent phosphorylation of AMPK by AICAR. HPAECs were stimulated for 5 to 120 minutes with 1 mmol/L AICAR. C: HPAECs were pretreated with 1 mmol/L AICAR for 30 minutes and then treated with LPS for 1 or 6 hours. Data are expressed as means ± SEM. n = 3 per group. *P < 0.05 versus control; †P < 0.05 versus LPS group. Blots are representative of three independent experiments.
AICAR Activates Rac1/Cdc42
We next determined the mechanism or mechanisms by which AMPK activation by AICAR prevents LPS-induced cytoskeletal rearrangement. Activated Rho family members Cdc42 and Rac1 regulate endothelial barrier function,3–5 by re-establishing EC junction integrity after injury.7 We therefore investigated the effects of AICAR on Rac1/Cdc42 in HPAECs. AICAR activated both Rac1 and Cdc42 in a time-dependent manner (Figure 7, A and B). These results indicate AMPK-dependent activation of Rac1 and Cdc42.
Figure 7.
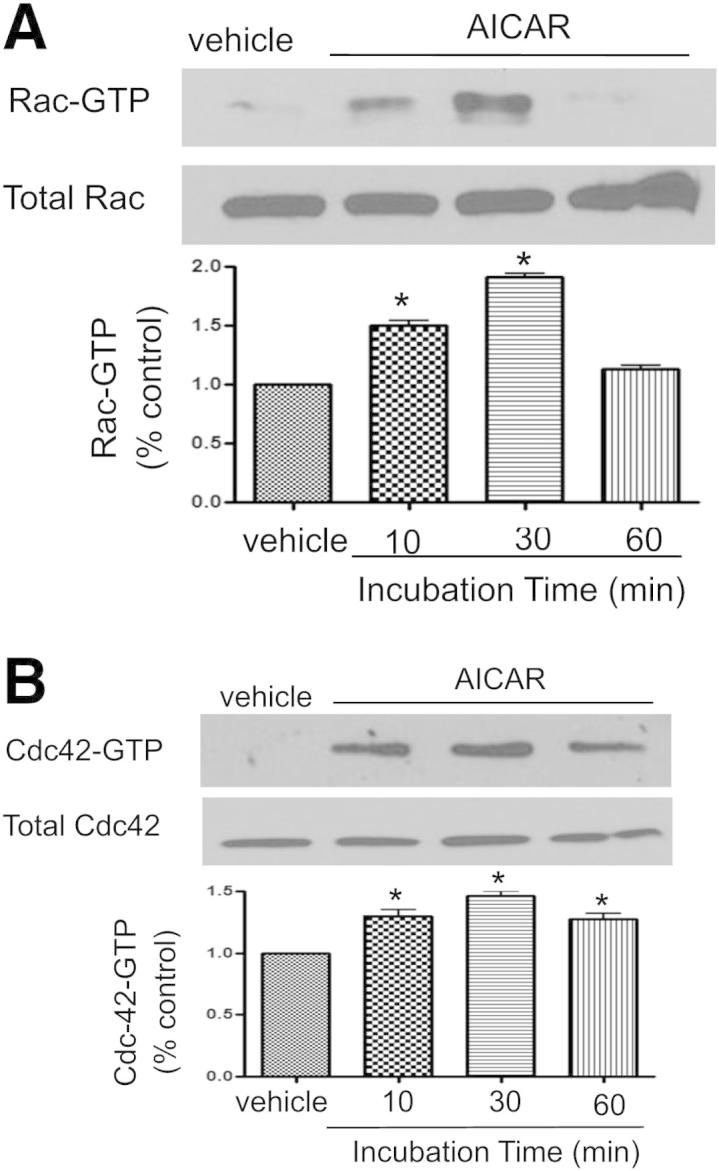
AMPK Regulates RAC/Cd42 signaling. Effects of AICAR on levels of Rac-GTP (A) and Cdc42 (B) in HPAECs. HPAECs were treated with 1 mmol/L AICAR for 10, 30, or 60 minutes. Rac and Cdc42 were analyzed by pull-down assays. Data are expressed as means ± SEM. *P < 0.05 versus control. Blots are representative of three independent experiments.
PAK Is Involved in AICAR-Mediated Rac1/Cdc42 Signaling
To further corroborate the role of AICAR in the Rac1-mediated signaling pathway, we assessed the effects of AMPK on PAK, a downstream target of Rac1 that plays a crucial role in cell motility, cytoskeletal remodeling, cell contraction, and barrier regulation.14 AICAR increased PAK phosphorylation in HPAECs in a dose- and time-dependent manner (Figure 8, A and B). Consistent with this finding, inhibition of AMPKα1 or AMPKα2 with siRNA led to a reduction in p-PAK (Figure 8C). When HPAECs were pretreated with the Rac inhibitor NSC 23766 or with Rac siRNA (Figure 8, D and E), PAK phosphorylation decreased, which confirms PAK as a downstream target for Rac1 in our system.
Figure 8.

Regulation of PAK signaling by AMPK. A: Dose-dependent induction of PAK phosphorylation by AICAR. HPAECs were stimulated with 0.05 to 2.0 mmol/L AICAR for 1 hour. B: Time-dependent increase in PAK phosphorylation in HPAECs by 1 mmol/L AICAR treatment. HPAECs were stimulated for 5 to 120 minutes with 1 mmol/L AICAR. Protein cell lysates (20 μg) were analyzed by immunoblotting using anti–p-PAK and anti-PAK antibodies. C: PAK phosphorylation in HPAECs transfected with AMPKα1 or AMPKα2 siRNA. HPAECs were transfected with control siRNA, AMPKα1 siRNA, or AMPKα2 siRNA for 48 hours, and then treated with 1 mmol/L AICAR for 1 hour. Cell lysates were subjected to Western blot analysis using antibodies against p-PAK, AMPKα1, or AMPKα2. D and E: Inhibition of Rac on phosphorylation of PAK in HPAECs. HPAECs were treated with 1 mmol/L AICAR, with or without 100 μmol/L NSC 23766, for 1 hour. HPAECs were transfected with control siRNA, Rac siRNA for 48 hours, and then treated with 1 mmol/L AICAR for 1 hour. Cell lysates were subjected to Western blot analysis using antibodies against p-PAK. Data are expressed as means ± SEM. n = 3 per group. *P < 0.05 versus control; †P < 0.05 versus AICAR. Blots are representative of three independent experiments.
Discussion
The major finding of the present study is that AMPK inhibition contributes to LPS-induced endothelial barrier dysfunction and lung injury. LPS inhibited AMPK activity both in vitro and in vivo. Importantly, lung tissue inflammatory cell infiltration, lung congestion, and edema were more severe in AMPKα-deficient mice than in WT mice, especially in α1-KO mice, suggesting that AMPK deletion accentuates lung endothelial barrier dysfunction and lung injury in vivo. AICAR, which activates AMPK in the lungs, decreased pulmonary vascular permeability and diminished the severity of LPS-induced acute lung injury. LPS-induced lung injury was more severe in AMPKα-KO mice, indicating that AMPKα is important in maintaining endothelial integrity. Thus, AMPK activation by AICAR attenuates LPS-induced pulmonary vascular hyperpermeability and lung injury.
The effect of AMPK on maintaining endothelial integrity appears to be mediated by multiple pathways. For example, the ability of AMPK activation to enhance pulmonary vascular barrier function appears to be related to its association with cytoskeletal organization, including enhancement of microtubule and actin polymerization. Our data show that AMPK-induced Rac/Cdc42 activation leads to activation of the Rac downstream target PAK, which plays a crucial role in cell motility, cytoskeletal remodeling, cell contraction, and barrier regulation.14 Also, activation of the Rac pathway indirectly inhibits Rho signaling.20 AICAR activated Rac and Cdc42, but inhibited LPS-induced phosphorylation of MLC, a downstream target of Rho, thus inhibiting LPS-induced disruption of the cell cytoskeleton and cell adhesion. Wu and colleagues21 suggest that the protein stability, localization, and phosphorylation of VE cadherin play critical roles in maintaining cell-to-cell junctions. Our data indicate that AICAR decreases VE-cadherin phosphorylation at Tyr658, a key regulatory site of endothelial barrier function,22 thereby leading to the inhibition of cell barrier function.23,24 Conversely, inhibition of AMPKα1 or AMPKα2 increased VE-cadherin phosphorylation at Tyr658. Alternatively, the protective effects of AMPK on endothelial permeability may be due, in part, to its anti-inflammatory activities and antioxidative stress effects,10,25,26 as suggested previously.27
The mechanism or mechanisms by which LPS inhibits AMPK remain unknown. Evidence suggests that inflammatory mediators such as TNFα up-regulate phosphatase PP2C, which results in dephosphorylation of AMPK, thereby decreasing its activity.26,28 In the present study, however, LPS did not alter the levels of PP2C. Moreover, LPS-induced AMPK inhibition does not appear to be related to LKB1, an upstream kinase of AMPK. In fact, LPS treatment increased the phosphorylation of LKB1 at Ser307, a phosphorylation site known to be important for AMPK activation.29 LKB1 is reported to regulate lung cancer cell polarity by mediating Cdc42 recruitment and activity.30 In addition, PAK phosphorylation is impaired in cell lines (eg, the human lung adenocarcinoma epithelial cell line A549) in which LKB1 function is naturally lost.30,31 Whether this effect of LKB1 is mediated by AMPK warrants further investigation.
In summary, the present study yielded three novel findings: i) activation of AMPK protects against lung barrier dysfunction and neutrophil accumulation induced by intratracheal LPS challenge; ii) protective effects of AMPK against LPS-induced lung dysfunction are mediated by lung vascular endothelium and controlled by Rac GTPase effector PAK1 and tyrosine phosphorylation of VE-cadherin; and iii) LPS inhibits endogenous AMPK expression in the lung. In addition to the endothelial effects described here, AMPK may engage other mechanisms (eg, anti-inflammatory or antioxidant). AMPK activation may serve as a potentially useful therapeutic strategy for treating diseases characterized by high permeability states.
Footnotes
Supported by NIH grants HL079584, HL080499, HL074399, HL089920, HL096032, HL105157, and HL110488 (M.-H.Z.), a research award from the American Diabetes Association (M.-H.Z.), funds from the Warren Chair of the University of Oklahoma Health Sciences Center, and funds from the American Heart Association national established investigator award (all to M.-H.Z.).
References
- 1.Cowan C.E., Kohler E.E., Dugan T.A., Mirza M.K., Malik A.B., Wary K.K. Kruppel-like factor-4 transcriptionally regulates VE-cadherin expression and endothelial barrier function. Circ Res. 2010;107:959–966. doi: 10.1161/CIRCRESAHA.110.219592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vogel S.W.M., Farhat K., Zieseniss A., Schnelle M., Le-Huu S., von Ahlen M., Malz C., Camenisch G., Katschinski D.M. Prolyl hydroxylase domain (PHD) 2 affects cell migration and F-actin formation via RhoA/rho-associated kinase-dependent cofilin phosphorylation. J Biol Chem. 2010;285:33756–33763. doi: 10.1074/jbc.M110.132985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Spindler V., Schlegel N., Waschke J. Role of GTPases in control of microvascular permeability. Cardiovasc Res. 2010;87:243–253. doi: 10.1093/cvr/cvq086. [DOI] [PubMed] [Google Scholar]
- 4.Popoff M.R., Geny B. Multifaceted role of Rho, Rac, Cdc42 and Ras in intercellular junctions, lessons from toxins. Biochim Biophys Acta. 2009;1788:797–812. doi: 10.1016/j.bbamem.2009.01.011. [DOI] [PubMed] [Google Scholar]
- 5.Watanabe T., Sato K., Kaibuchi K. Cadherin-mediated intercellular adhesion and signaling cascades involving small GTPases. Cold Spring Harb Perspect Biol. 2009;1:a003020. doi: 10.1101/cshperspect.a003020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baumer Y., Spindler V., Werthmann R.C., Bünemann M., Waschke J. Role of Rac 1 and cAMP in endothelial barrier stabilization and thrombin-induced barrier breakdown. J Cell Physiol. 2009;220:716–726. doi: 10.1002/jcp.21819. [DOI] [PubMed] [Google Scholar]
- 7.Zhao Y.D., Ohkawara H., Rehman J., Wary K.K., Vogel S.M., Minshall R.D., Zhao Y.Y., Malik A.B. Bone marrow progenitor cells induce endothelial adherens junction integrity by sphingosine-1-phosphate-mediated Rac1 and Cdc42 signaling. Circ Res. 2009;105:696–704. doi: 10.1161/CIRCRESAHA.109.199778. 8 p following 704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wu Y., Song P., Xu J., Zhang M., Zou M.H. Activation of protein phosphatase 2A by palmitate inhibits AMP-activated protein kinase. J Biol Chem. 2007;282:9777–9788. doi: 10.1074/jbc.M608310200. [DOI] [PubMed] [Google Scholar]
- 9.Xie Z., Dong Y., Zhang M., Cui M.Z., Cohen R.A., Riek U., Neumann D., Schlattner U., Zou M.H. Activation of protein kinase C zeta by peroxynitrite regulates LKB1-dependent AMP-activated protein kinase in cultured endothelial cells. J Biol Chem. 2006;281:6366–6375. doi: 10.1074/jbc.M511178200. [DOI] [PubMed] [Google Scholar]
- 10.Choi H.C., Song P., Xie Z., Wu Y., Xu J., Zhang M., Dong Y., Wang S., Lau K., Zou M.H. Reactive nitrogen species is required for the activation of the AMP-activated protein kinase by statin in vivo. J Biol Chem. 2008;283:20186–20197. doi: 10.1074/jbc.M803020200. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 11.Dong Y., Zhang M., Liang B., Xie Z., Zhao Z., Asfa S., Choi H.C., Zou M.H. Reduction of AMP-activated protein kinase alpha2 increases endoplasmic reticulum stress and atherosclerosis in vivo. Circulation. 2010;121:792–803. doi: 10.1161/CIRCULATIONAHA.109.900928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jørgensen S.B., Viollet B., Andreelli F., Frøsig C., Birk J.B., Schjerling P., Vaulont S., Richter E.A., Wojtaszewski J.F. Knockout of the alpha2 but not alpha1 5’-AMP-activated protein kinase isoform abolishes 5-aminoimidazole-4-carboxamide-1-beta-4-ribofuranoside but not contraction-induced glucose uptake in skeletal muscle. J Biol Chem. 2004;279:1070–1079. doi: 10.1074/jbc.M306205200. [DOI] [PubMed] [Google Scholar]
- 13.Viollet B., Andreelli F., Jørgensen S.B., Perrin C., Geloen A., Flamez D., Mu J., Lenzner C., Baud O., Bennoun M., Gomas E., Nicolas G., Wojtaszewski J.F., Kahn A., Carling D., Schuit F.C., Birnbaum M.J., Richter E.A., Burcelin R., Vaulont S. The AMP-activated protein kinase alpha2 catalytic subunit controls whole-body insulin sensitivity. J Clin Invest. 2003;111:91–98. doi: 10.1172/JCI16567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Birukova A.A., Xing J., Fu P., Yakubov B., Dubrovskyi O., Fortune J.A., Klibanov A.M., Birukov K.G. Atrial natriuretic peptide attenuates LPS-induced lung vascular leak: role of PAK1. Am J Physiol Lung Cell Mol Physiol. 2010;299:L652–L663. doi: 10.1152/ajplung.00202.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Birukov G.K., Cokic I., Fu P., Junjie X., Birukova A.A. Protective effects of iloprost in cellular and animal models of ventilator-induced lung injury (abstract) Virchows Archiv. 2009;455(Suppl 1):S84. [Google Scholar]
- 16.Zou M.H., Hou X.Y., Shi C.M., Kirkpatrick S., Liu F., Goldman M.H., Cohen R.A. Activation of 5’-AMP-activated kinase is mediated through c-Src and phosphoinositide 3-kinase activity during hypoxia-reoxygenation of bovine aortic endothelial cells. Role of peroxynitrite. J Biol Chem. 2003;278:34003–34010. doi: 10.1074/jbc.M300215200. [DOI] [PubMed] [Google Scholar]
- 17.Zhang M., Dong Y., Xu J., Xie Z., Wu Y., Song P., Guzman M., Wu J., Zou M.H. Thromboxane receptor activates the AMP-activated protein kinase in vascular smooth muscle cells via hydrogen peroxide. Circ Res. 2008;102:328–337. doi: 10.1161/CIRCRESAHA.107.163253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Davis B.J., Xie Z., Viollet B., Zou M.H. Activation of the AMP-activated kinase by antidiabetes drug metformin stimulates nitric oxide synthesis in vivo by promoting the association of heat shock protein 90 and endothelial nitric oxide synthase. Diabetes. 2006;55:496–505. doi: 10.2337/diabetes.55.02.06.db05-1064. [DOI] [PubMed] [Google Scholar]
- 19.Xie Z., Dong Y., Scholz R., Neumann D., Zou M.H. Phosphorylation of LKB1 at serine 428 by protein kinase C-zeta is required for metformin-enhanced activation of the AMP-activated protein kinase in endothelial cells. Circulation. 2008;117:952–962. doi: 10.1161/CIRCULATIONAHA.107.744490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Birukova A.A., Chatchavalvanich S., Oskolkova O., Bochkov V.N., Birukov K.G. Signaling pathways involved in OxPAPC-induced pulmonary endothelial barrier protection. Microvasc Res. 2007;73:173–181. doi: 10.1016/j.mvr.2006.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li R., Ren M., Chen N., Luo M., Zhang Z., Wu J. Vitronectin increases vascular permeability by promoting VE-cadherin internalization at cell junctions. PLoS One. 2012;7:e37195. doi: 10.1371/journal.pone.0037195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cain R.J., Vanhaesebroeck B., Ridley A.J. The PI3K p110alpha isoform regulates endothelial adherens junctions via Pyk2 and Rac1. J Cell Biol. 2010;188:863–876. doi: 10.1083/jcb.200907135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Potter M.D., Barbero S., Cheresh D.A. Tyrosine phosphorylation of VE-cadherin prevents binding of p120- and beta-catenin and maintains the cellular mesenchymal state. J Biol Chem. 2005;280:31906–31912. doi: 10.1074/jbc.M505568200. [DOI] [PubMed] [Google Scholar]
- 24.Starosta V., Wu T., Zimman A., Pham D., Tian X., Oskolkova O., Bochkov V., Berliner J.A., Birukova A.A., Birukov K.G. Differential regulation of endothelial cell permeability by high and low doses of OxPAPC. Am J Respir Cell Mol Biol. 2012;46:331–341. doi: 10.1165/rcmb.2011-0153OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li X.N., Song J., Zhang L., LeMaire S.A., Hou X., Zhang C., Coselli J.S., Chen L., Wang X.L., Zhang Y., Shen Y.H. Activation of the AMPK-FOXO3 pathway reduces fatty acid-induced increase in intracellular reactive oxygen species by upregulating thioredoxin. Diabetes. 2009;58:2246–2257. doi: 10.2337/db08-1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lu J., Wu D.M., Zheng Y.L., Hu B., Zhang Z.F., Shan Q., Zheng Z.H., Liu C.M., Wang Y.J. Quercetin activates AMP-activated protein kinase by reducing PP2C expression protecting old mouse brain against high cholesterol-induced neurotoxicity. J Pathol. 2010;222:199–212. doi: 10.1002/path.2754. [DOI] [PubMed] [Google Scholar]
- 27.Zhao X., Zmijewski J.W., Lorne E., Liu G., Park Y.J., Tsuruta Y., Abraham E. Activation of AMPK attenuates neutrophil proinflammatory activity and decreases the severity of acute lung injury. Am J Physiol Lung Cell Mol Physiol. 2008;295:L497–L504. doi: 10.1152/ajplung.90210.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Steinberg G.R., Michell B.J., van Denderen B.J., Watt M.J., Carey A.L., Fam B.C., Andrikopoulos S., Proietto J., Görgün C.Z., Carling D., Hotamisligil G.S., Febbraio M.A., Kay T.W., Kemp B.E. Tumor necrosis factor alpha-induced skeletal muscle insulin resistance involves suppression of AMP-kinase signaling. Cell Metab. 2006;4:465–474. doi: 10.1016/j.cmet.2006.11.005. [DOI] [PubMed] [Google Scholar]
- 29.Xie Z., Dong Y., Zhang J., Scholz R., Neumann D., Zou M.H. Identification of the serine 307 of LKB1 as a novel phosphorylation site essential for its nucleocytoplasmic transport and endothelial cell angiogenesis. Mol Cell Biol. 2009;29:3582–3596. doi: 10.1128/MCB.01417-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang S., Schafer-Hales K., Khuri F.R., Zhou W., Vertino P.M., Marcus A.I. The tumor suppressor LKB1 regulates lung cancer cell polarity by mediating Cdc42 recruitment and activity. Cancer Res. 2008;68:740–748. doi: 10.1158/0008-5472.CAN-07-2989. [DOI] [PubMed] [Google Scholar]
- 31.Jimenez A.I., Fernandez P., Dominguez O., Dopazo A., Sanchez-Cespedes M. Growth and molecular profile of lung cancer cells expressing ectopic LKB1: down-regulation of the phosphatidylinositol 3’-phosphate kinase/PTEN pathway. Cancer Res. 2003;63:1382–1388. [PubMed] [Google Scholar]