

SUMMARY
c-Jun N-terminal (JNK) family kinases have a common peptide-docking site used by upstream activating kinases, substrates, scaffold proteins, and phosphatases, where the ensemble of bound proteins determines signaling output. Although there are many JNK structures, little is known about mechanisms of allosteric regulation between the catalytic and peptide-binding sites, and the activation loop, whose phosphorylation is required for catalytic activity. Here, we compare three structures of unliganded JNK3 bound to different peptides. These were compared as a class to structures that differ in binding of peptide, small molecule ligand, or conformation of the kinase activation loop. Peptide binding induced an inhibitory interlobe conformer that was reversed by alterations in the activation loop. Structure class analysis revealed the subtle structural mechanisms for allosteric signaling between the peptide-binding site and activation loop. Biochemical data from isothermal calorimetry, fluorescence energy transfer, and enzyme inhibition demonstrated affinity differences among the three peptides that were consistent with structural observations.
INTRODUCTION
The c-Jun N-terminal kinases (JNKs) are members of the mitogen-activated protein kinases (MAPKs), acting as primary mediators of the stress response to regulate insulin signaling, cell fate, DNA repair, and T cell differentiation (Karin and Gallagher, 2005; Weston and Davis, 2007). Differences in the timing and duration of JNK activation can determine whether cells proliferate or undergo programmed cell death (Lin, 2006), highlighting the critical importance of tight regulation of this pathway. The MAPKs, including mammalian JNK, ERK, and p38 families, function as part of a three component signaling module, with the MAPK being activated upon phosphorylation by a MAPK kinase (MKK), which is in turn activated by a MAPK kinase kinase (MAP3K) (Raman et al., 2007). These modules allow for a diverse yet highly tunable set of signaling circuits (Good et al., 2011).
The MAPKs are activated via dual phosphorylation of a TPY motif found in the activation loop (A-loop), which connects the two lobes common to the kinase domain (Zhang et al., 1994) (Figure 1A). The ATP binding site lies between the lobes, bracketed by the A-loop and a peptide-docking site, which are on opposing sides of the interlobe interface. MKKs that phosphorylate the A-loop, along with phosphatases, substrates, inhibitors, and scaffold proteins, share the docking site via a common docking motif (D-motif) (Weston and Davis, 2007). Thus JNK activity derives from the ensemble of interacting proteins that compete for docking at this site.
Figure 1. Formation of the A-Loop into an Inhibitory Helix.
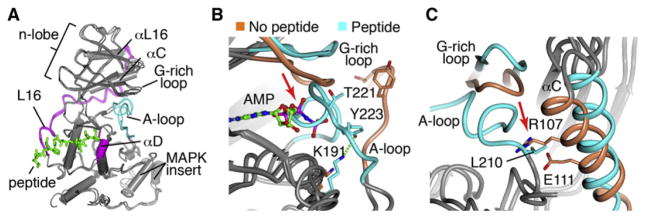
(A) The structures of JNK3 bound to ATF2, SAB, or JIP1 peptides were superimposed, and the JIP1 peptide is shown as green. The A-loops form a helix that docks into catalytic site.
(B) The SAB-bound structure (blue) was superimposed with a liganded, peptide-free structure (JNK1.pdb, colored coral) showing the differences in the positioning of the A-loop. AMP-PNP is shown as a ball and stick.
(C) Same as (B) but showing how relocation of αC is required for the A-loop conformer. The A-loop from the peptide-free structure is not shown for clarity.
The structural features that determine JNK specificity preferences for different D-motifs are currently not known. The motif contains basic residues, a short linker, and a more C-terminal Φ-x-Φ hydrophobic motif, with the specific sequence determining specificity for the different MAPK signaling modules (Bardwell et al., 2009). Here, we compare the structures of JNK3 bound to the higher affinity D-motif from JIP1 with lower affinity motifs from ATF2 and SAB. ATF2 is a JNK substrate in the AP1 family of transcription factors that can heterodimerize with another JNK target, c-Jun, to modulate survival signals (Salameh et al., 2010). JIP1 is a cytoplasmic scaffold protein that interacts with the upstream activating kinases for JNK, and is required for activity in a number of different contexts, including regulation of obesity-induced insulin resistance (Jaeschke et al., 2004; Morel et al., 2010; Weston and Davis, 2007; Whitmarsh et al., 2001). SAB is a more recently identified scaffold protein localized to the mitochondria, accounting for JNK-mediated ROS generation (Chambers and LoGrasso, 2011), and acetaminophen-induced liver injury (Win et al., 2011), and is also a JNK substrate. Thus interaction with these three types of D-motif will drive JNK to different cellular locations, directly affecting JNK signaling specificity.
Allostery among the catalytic site, A-loop, and peptide-docking site of ERK and p38 MAPKs has been well documented (Chang et al., 2002; Goldsmith, 2011; Lee et al., 2004; Wang et al., 1997; Zhang et al., 1994; Zhou et al., 2006a, 2006b), but much less is known about these processes for JNK. The structure of JNK1 bound to a peptide from JIP1 protein showed that the peptide induced a rotation of the two lobes that distorted the active site, which was proposed as a mechanism for JIP1-mediated inhibition of JNK signaling (Heo et al., 2004). However, it is now clear from more physiologically relevant studies that JIP1 is required for activation of JNK (Jaeschke et al., 2004; Morel et al., 2010; Whitmarsh et al., 2001), and is only inhibitory when overexpressed. Thus the function of the peptide-induced conformation is not clear.
In this work we use an approach that we call structure class analysis, which postulates that examining groups of structures reveals subtle differences that are not apparent in an individual structure. It allows for statistical analysis of structural differences, and overcomes the difficulty in interpreting the role of crystal packing by comparing different space groups (Schulze-Gahmen et al., 1993). We previously used this approach to discern differences between the two estrogen receptor subtypes, to identify structural mechanisms for partial agonist activity, and to define a role for ligand dynamics in controlling allosteric signaling (Bruning et al., 2010; Nettles et al., 2004, 2008). There are more than 40 JNK structures in the Protein Data Bank (PDB), the vast majority of which are only minimally described as part of medicinal chemistry campaigns. These include a number of different classes for comparison, including those with an ordered versus disordered A-loop, with or without bound ligands and peptides, and a set with mutation of the phosphoacceptor residues to glutamate, which are bound to peptide and ligand. Here, we add, to our knowledge, a new class: three structures with different bound peptides, but no small molecule ligands. Our structure class analysis identifies allosteric interactions between the catalytic and peptide-binding sites and the A-loop. These define two distinct autoinhibitory mechanisms in JNK that block formation of the active catalytic site in the inactive kinase. We show that A-loop conformer can reverse these autoinhibitory mechanisms. We further identified a structural mechanism for bidirectional signaling between the A-loop and peptide-binding site. Differences between the peptide-binding modes also indicate the structural basis for selectivity of different docking motifs.
RESULTS
Peptide-Induced Autoinhibition of JNK3
We solved crystal structures for JNK3 bound to three different 11-mer D-motif peptides (Table S1A available online), representing two different scaffold proteins, JIP1 (PDB number 4H39) (2.0 Å) and SAB (PDB number 4H3B) (2.1 Å), and a substrate target, ATF2 (PDB number 4H36) (3.0 Å) (Table 1). These structures were obtained without any small molecule ligands, and in three different space groups (Table S1). When these three peptide structures were superimposed, they showed that the A-loop coiled into a helix, docked into the ATP binding site (Figure 1A), and did not participate in crystal packing. An overlay of the JNK3/SAB peptide structure with the structure of JNK3 bound to the nonhydrolyzable analog AMP-PNP shows that the peptide caused the A-loop helix to bind directly in the ATP binding site, shown in blue for peptide and orange for AMP bound structures (Figure 1B). This inhibitory conformation was associated with a substantial shift in the G-rich loop to accommodate the docking of the helix. The inhibitory helix oriented the phosphoacceptor Thr221 toward the peptide-docking site, while Tyr223 H-bonds to Lys191 (Figure 1B). The formation of the A-loop into an inhibitory helix also required an interlobe rotation that is common to peptide bound JNK, with or without ligand. Specifically, the αC helix rotated away from the catalytic site to allow space for the inhibitory helix, because A-loop L210 would clash with αC residues Arg107 and Glu111 (Figure 1C). This suggests a potential mechanism to prevent catalytic activity toward a docked substrate in the absence of phosphorylation of the A-loop.
Table 1.
Isothermal Titration Calorimetry of Peptide Binding to JNK3
| Peptide | Sequence | Kd (μM) | ΔH (cal/mol) | ΔS (cal/mol) | Fold pepJIP Kd |
|---|---|---|---|---|---|
| pepJIPhu_157–167 | RPKRPTTLNLF | 0.8 ± 0.1 | −7,654 | 644 | 1 |
| pepSABhu_40–350 | AVVRPGSLDLP | 17.2 ± 0.6 | −4,019 | 2,479 | 21 |
| pepATF2Hu_46–56 | KHKHEMTLKFG | >10 | ND | ND | ND |
| JlP-SAB | RPKRPGSLDLF | 2.7 ± 0.3 | −6,295 | 1,609 | 5 |
| SAB-JIP | AVVRPTTLNLP | 3.9 ± 0.5 | −5,216 | 2,161 | 5 |
| JIP1 R160H | RPKHPTTLNLF | 11.7 ± 1.0 | −6,713 | 12 | 14 |
| JIP1 R160Q | RPKQPTTLNLF | 32.1 ± 3.2 | −5,455 | 676 | 39 |
| JIP1 T162G | RPKRPGTLNLF | 4.9 ± 0.5 | −7,107 | 138 | 6 |
The structure of JNK with the JIP1 peptide showed that peptide binding to the carboxy-terminal C-lobe induced a rotation of the amino-terminal N-lobe that disrupts the catalytic site and this was suggested to be the mechanism through which overexpression of JIP1 inhibits JNK activity. (Heo et al., 2004). To test if this interlobe rotation is in fact a common feature of binding to different scaffolds and a substrate, we utilized structure class analysis to reveal a substantial interlobe hinge motion for the peptide bound structures, regardless of small molecule ligand. Our three structures of JNK3 were superimposed with the two published structures with peptide + inhibitors, and 24 inhibitor-bound structures (Table S1B; Figure 2A). The analysis showed a substantial hinge motion around the peptide-binding site, such that the MAPK insert region was shifted 6 Å away from αC and αL16 as measured by the distance from Val294 to Thr386 (Figures 2A, 2B, and S2A). In addition to the hinge motion, there was also a clockwise twist of the N-lobe relative to the C-lobe. An examination of the catalytic site shows that the peptide-induced conformer significantly shifted αC Glu111 ~2.5 Å away from N-lobe catalytic site residues, such as Asn194 (Figures 2C and 2D), which disrupted the extensive hydrogen bond network with ordered water and Mg+2 that is required for catalysis. As discussed below, peptide binding to JNK1 induced an identical interlobe hinge and rotation motion.
Figure 2. Interlobe Rotation Induced by Peptide Binding.
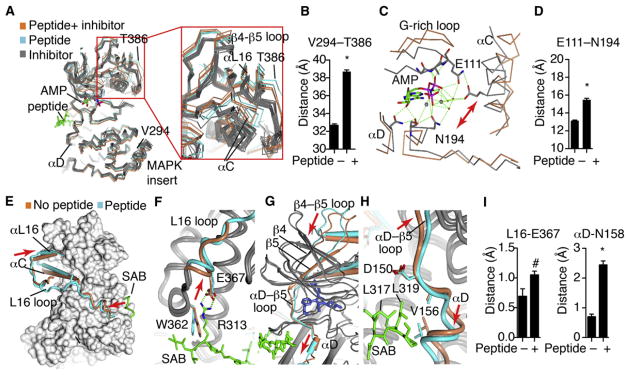
Statistical significance was measured with Student’s t test. *p < 0.0001; #p < 0.08.
(A) All published JNK3 structures (Tables S1A and S1B) were globally superimposed against JNK1.pdb (AMP bound with no peptide) and shown as α-carbon trace, including 24 structures with different inhibitors (gray), two structures with inhibitors+ peptide (cyan), and our three structures with peptides and no inhibitors (coral). Close-ups are also shown in Figure S2.
(B) The distance between the indicated residues in the MAPK insert or αL16 was measured for each structure, showing an opening of the lobes induced by peptide binding.
(C) The structure of JNK3 bound to AMP-PNP (gray, JNK1.pdb) was superimposed with our structure of SAB-bound JNK3 (coral). Shown are select residues in the catalytic site.
(D) The distance between the indicated catalytic site residues was measured for the set of peptide-bound versus peptide-free JNK3 structures, showing significant peptide-induced distortion of the catalytic site. Error bars represent SEM.
(E–H) The SAB/JNK3 structure (gray and cyan) was superimposed with the inhibitor bound structure, 1PMN.pdb (gray and coral). The peptide is shown as a green cylinder, and the inhibitor as spheres. (E) JNK3 is shown as surface, except for the indicated regions. The L16 loop (blue tube) connects the peptide with the altered position of αL16. In the peptide-free structure, (coral), the L16 loop is partially disordered. The peptide is green. Arrows indicate where the peptide pushes on the L16 loop, and where this is transferred to L16. (F) Detail of how the peptide pushes L16 to accommodate a salt bridge. (G) The peptides induce a “pull” in αD that is transmitted to β5 and the β4-β5 loop in the C-lobe. (H) Detail of the peptide-induced shift in αD and the αD-β5 loop.
(I) All the JNK3 structures were superimposed with JNK1.pdb as a reference structure. The distance of the α-carbon from the corresponding residue in JNK1 was measured. Error bars represent SEM.
See also Figure S2.
Our structure class analysis approach revealed a common set of structural mechanisms through which peptide binding induces interlobe motion, regardless of inhibitor binding. One mechanism is via the peptide pushing the L16 loop (colored blue in the peptide bound structure, Figure 2E), which connects the peptide-binding site in the C-lobe to αL16 in the N-lobe. Importantly, the L16 loop was fully ordered in all of the peptide bound structures, and none of the peptide-free structures, demonstrating a class-specific structural phenotype. There was no crystal contact for the ordered region of the L16 loop found in all the peptide bound structures, and it occurred in two different space groups, demonstrating that the ordering and shift in position of the L16 loop was peptide induced. In the peptide, SAB Arg343 (JIP Arg160, ATF2 His49) forms an electrostatic interaction with JNK3 Glu367 that induced a shift in the L16 loop (Figures 2F and S2B), mediating the shift in αL16 and associated shift of the N-lobe (Figure 2E). Statistical analysis showed a trend for the significance of the class differences in L16 positioning (Figure 2I). These analyses confirm that the N terminus of the D-motif controls the interlobe conformer via L16 pushing on α16 and the adjacent αC.
There is also a second mechanism of peptide-induced allo-steric control of interlobe conformation. At the C-terminal end of the peptides, the hydrophobic residues that dock against JNK induced a rotation of helix αD to form Van der Waals (VDW) contacts (Figures 2G, 2H, and S2C), which were then transmitted via β5 to the N-lobe as a pull motion as indicated by the red arrow. The shift in αD as measured by the distance from N158 was ~1.6 Å and was highly significant (p < 0.01) (Figures 2I and S2C). Thus peptide binding to unphosphorylated JNK induced two distinct mechanisms of autoinhibition including interlobe hinge motion and the formation of the A-loop into an inhibitory helix.
Structural Features of Peptide Specificity
A critical feature of MAPK signaling is the use of a single docking site for upstream activators, phosphatases, substrates, as well as scaffold proteins that direct the kinase to specific locations and assemble other interacting proteins. This means that small changes in stoichiometry or differences in peptide affinity can profoundly impact signaling, including cell fate decisions. For example, reducing gene dosage of a single MAPK substrate in Drosophila leads to decreased amounts of active MAPK, due to loss of competition and increased binding to phosphatases (Kim et al., 2011). This highlights the importance of understanding the structural differences between JNK interacting motifs, and how these differences might affect allosteric control mechanisms.
We used a number of approaches to measure the affinity of the D-motif peptides. Utilizing isothermal calorimetry (ITC), we were able to demonstrate a 21-fold affinity preference of JNK3 for JIP peptide compared to SAB peptide (Table 1). ATF2 peptide did not saturate the ITC curves, and the thermodynamic parameters were not attained. We also used two different methods to measure IC50 values for the peptides. In the FRET assay (Table 2; Figure S1A), JIP had a lower IC50 than the others by at least 140-fold. We also performed kinase assays utilizing protein substrates of ATF2 and SAB (Table 3; Figure S1B). Here, the peptides inhibited incorporation of 33P into the protein substrates better than they did when competing against their peptide counterparts in the FRET assay for each peptide, but showed similar higher affinity for JIP1. Lastly, we measured the Km (0.77 ± 0.09 μM) of SAB (1–390) for JNK3α1, compared to Km for ATF2 = 0.16 ± 0.04 μM from our previous work (Ember et al., 2008). We conclude that JIP1 had higher affinity for JNK3 than ATF2 or SAB.
Table 2.
FRET IC50 for Displacement of Labeled JIP1 Peptide
| Kinase | pepJIP | pepATF2 | pepSAB |
|---|---|---|---|
| Inactive JNK1β1 | 2.5 ± 0.1 | 350 ± 12 | 612 ± 21 |
| Active JNK 1β1 | 0.68 ± 0.02 | 169 ± 2 | 386 ± 10 |
| Inactive JNK 3αl | 1.7 ± 0.4 | 257 ± 62 | 834 ± 105 |
| Active JNK 3αl | 0.5 ± 0.02 | 128 ± 6 | 1,018 ± 75 |
FRET IC50 is displayed in μM.
See also Figure S1.
Table 3.
IC50 Values for Peptide Inhibition JNK Kinase Activity
| Protein Substrate | pepJIP | pepATF2 | pepSAB |
|---|---|---|---|
| His-SAB (1–390) | 0.21 ± 0.02 | 53.4 ± 5 | 6.76 ± 0.94 |
| FLAG-ATF2 (2–115) | 0.30 ± 0.07 | 136.9 ± 41 | 7.56 ± 1.74 |
IC50 is displayed in μM. Inhibition of radioactive 33P incorporation by JNK3α1 (39–422) into either His-SAB (1–390) or FLAG-ATF2 (2–115) by pepJIP, pepATF2, or pepSAB.
See also Figure S1.
The crystal structures showed a number of structural features that explain the higher affinity of JIP1 compared to the other peptides (Figure S3). His49 in pepATF2 binds in the same general site as pepJIP1 Arg160 (Figure 3A), but there are numerous differences in the interaction that suggest this is a significant determinant of the lower affinity of pepATF2. The 2.7 Å salt bridge of pepJIP1 Arg160 with JNK3 Glu367 was replaced by a more distant, 3.6 Å H-bond with pepATF2 His49 (Figure 3A). pepJIP1 Arg160 also formed H-bonds with JNK3 Tyr168, the backbone of Trp362 and a network of ordered water. There were also VDW interactions with JNK3 Tyr171 and Asp364 that were not seen with pepATF2. The critical importance of pepJIP1 Arg160 for high affinity binding was verified with ITC, as mutation to His or Gln (a residue found in c-Jun, another JNK substrate) significantly lowered affinity (Table 1). Indeed, the c-Jun 11-mer peptide did not bind to JNK3 at all (data not shown). In the N terminus of the ordered part of the peptides, SAB interacts identically to JIP1 (Figure 3A).
Figure 3. Structural Features of Peptide Specificity.

(A) Hydrogen bonding in the n terminus of the peptides. JNK3 is gray and the peptides, JIP1, SAB, and ATF2 are differently colored.
(B) Differences in binding to JNK3 in the middle of the peptides.
(C) Comparison of the c terminus of JIP1 and ATF2 peptide binding. The Phe residue in ATF2 extends further toward αD and shifts its positioning relative to the JIP1-bound structure.
(D) Comparison of the c terminus of JIP1 and SAB peptide binding. The c-terminal P350 in SAB enforces an altered position of the peptide backbone for the adjacent L349, which is transmitted to a shift in αD.
(E) The shift in αD induced by SAB is transmitted to the adjacent P222 in the A-loop, similar to the ATF2 induced shift (not shown).
See also Figure S3.
The middle sections of the peptides bind very differently. Pro161 in pepJIP1 corresponds to Glu50 in pepATF2, which was flipped out into the solvent, allowing the side chain of the adjacent Met51 to dock against JNK3 in place of JIP1 Pro161 (Figure 3B). In pepJIP1, the backbone and side chains of Thr162 and Thr163 form extensive hydrogen bond networks with JNK3 Arg165 (Figure 3B). The corresponding Gly345 and Ser346 in pepSAB showed only a subset of these H-bonds to JNK3 R165. The altered backbone positioning of ATF2 also repositions Thr52 such that there were no H-bonds with JNK3 R165 (Figure 3B). ITC data showed that the pepSAB glycine in the middle of the sequence was an important difference, as the Thr162Gly mutant pepJIP1 peptide showed 6-fold lower affinity (Table 1). Similarly, mutating the pepSAB residues GSLD to TTLN (the residues found in JIP) increased affinity 4-fold compared to SAB (SAB-JIP peptide, Table 1).
The C termini of the peptides also showed differences. The location of Phe55 in pepATF2 in place of pepJIP1 Leu166 induced significant differences in the peptide backbones (Figure 3C), which caused differences in the docking of ATF2/JIP1 Leu53/Leu164. In pepJIP1, Leu164 makes additional VDW contacts with JNK3 αD at Val197 and Val156 that were not observed with pepATF2.
The SAB peptide is similar to the JIP1 sequence, but still has 21-fold lower affinity (Figure 3D). The C terminus of the peptide showed differences in how L349 was oriented toward JNK3 compared to the corresponding residue in JIP1. This appears to be due to the adjacent SAB P350, which differentially positioned the backbone of L349. SAB induced a similar shift in αD as seen with ATF2, which again was transmitted to P222 in the A-loop of JNK3, stabilizing the well-ordered inhibitory helix (Figure 3E).
Differences in peptide binding were propagated to the A-loop via repositioning of the αD helix (Figures 3C–3E). The peptide-induced shift in αD positions it for VDW interactions with Pro222 in the A-loop. With JIP1 bound JNK3, this slight shift in Pro222 toward αD was associated with a partial disordering of the inactivation helix, suggesting that differences in the docking motifs may affect the positioning or dynamics of the A-loop.
A-Loop Control of the Peptide-Binding Site
To assess whether the A-loop could change the peptide-binding site, we compared peptide binding to active versus inactive JNKs. We prepared recombinant active JNK3α1 and JNK1β1 by incubation with active MKK4 and MKK7 (Chambers et al., 2011b; Figuera-Losada and LoGrasso, 2012b; Kamenecka et al., 2009, 2010; Lisnock et al., 2000). We measured FRET with TAMRA-labeled JIP1 peptide bound to active or inactive JNKs, and assayed inhibition by unlabeled peptides, JIP1, SAB, and ATF2 (Table 2; Figure S1A). MKK4/MKK7 (red) are shown as a control to indicate no binding to these proteins. Except for SAB binding to JNK3α1 (where the binding was very weak), in all other cases the peptides displayed lower IC50s, suggesting tighter binding to the active JNKs. In all cases, there was also a dramatic increase in the FRET signal with the active enzymes, which either represents a conformational change between the FRET pairs, or an increase in the population of JNK molecules that are in a conformation that allows peptide binding. Thus phosphorylation of the A-loop allosterically controls the peptide-binding site.
A-Loop Control of Interlobe Rotation
We used our structure class analysis of all the previously solved crystal structures of JNK3 and JNK1 in the PDB to test a hypothesis about A-loop control of interlobe rotation, and to test if our findings with JNK3 were predictive for a similar analysis of JNK1 structures. To begin this analysis, we reasoned that if peptide binding to inactive JNK inhibits catalysis, which implies that phosphorylation of the A-loop must reverse the autoinhibitory allostery. Though active JNK has not been crystallized, there are a series of JNK1 structures with mutations of the phosphoacceptor residues to glutamate. While these mutations do not yield constitutive activity (Zheng et al., 1999), they induce a different conformer for the A-loop, allowing us to ask how changing the A-loop conformer impacts the catalytic and peptide-binding sites. For JNK1, there are two structures with ligand but no peptides, two peptide bound structures, and six mutant structures with JIP1 peptide (Table S1C).
Our findings with JNK3 were predictive of the differences observed with the three types of JNK1 structures. The peptide-induced interlobe opening and rotation of JNK1 (Figures 4A and 4B), and remodeled L16 and αC to effect this change. Remarkably, mutation of the phosphoacceptor residues to glutamate reverses the peptide-induced hinge motion, but not the interlobe rotation (Figures 4A and 4B), explaining why these mutations are not activating. The mutants show remarkable consistency in αC position compared to the wild-type structures.
Figure 4. Activation Loop Control of Inter-lobe Rotation.

(A) All of the published JNK1 structures (Table S1C) were superimposed and shown as α-carbon trace, including wild-type (2 structures, coral), wild-type with JIP1 peptide (2 structures, cyan), and mutants of the phosphoacceptor residues in the A-loop to Glu (12 structures, blue).
(B) One JNK1 structure in each class was superimposed, showing how the A-loop mutations partially restore the peptide-induced interlobe rotation of αC and αL16.
(C) T183E in the mutant JNK1 structure H-bonds with residues in αC, drawing it closer to the wild-type, peptide-free positioning.
(D) Comparison of phosphoERK1 structure (2ERK. pdb) with phosphoacceptor mutant JNK1 (3O2M. pdb) structure, showing high structural homology, and suggesting that the mutant T183E likely binds differently to αC than phospho-T183 would.
(E) The JNK3 structures with inhibitors and no peptide were superimposed against 1JNK.pdb, of which 12 show an ordered A-loop, and 12 do not. Show is how the ordered A-loop enforces a single common conformer of αC.
(F) Backbone RMSD from 1JNK.pdb was calculated for the structures superimposed in (D). The RMSD for α-carbon in αC His104 was also calculated. *p < 0.0001 using Student’s t test for significance. Error bars represent SEM.
(G) Model of peptide-induced autoinhibition, and restoration of the active conformer by phosphorylation of the active loop.
See also Figure S4.
To understand why the mutations block hinge, but not rotation motion, we compared the active, phosphorylated ERK structure (Canagarajah et al., 1997) with the mutant JNK1 structures. The JNK1 Thr183Glu mutant is positioned for electrostatic interaction with several residues in the N-terminus of αC, including Arg69 and His66 (Figure 4C). This interaction pulls αC but does not reverse the peptide-induced interlobe rotation. Comparison with the active ERK structure suggests that the wild-type active conformer of JNK is similar to that of ERK. The phospho-Thr183 of ERK binds under the αC to a highly positively charged pocket in the N-lobe (Figure 4D). ERK phospho-Tyr185 binds in a positively charged pocket in the C-lobe, where it appears to act as an anchor to limit flexibility of the A-loop (Canagarajah et al., 1997). In contrast, JNK1 Thr183Glu H-bonded on the other side of αC compared to ERK phospho-Thr183, where it reversed the peptide-induced lobe opening, but mis-positioned αC for catalysis (Figures 4C and 4D). The conservation of the control of αC by the activation loop was shown by the presence of sulfate ions in the JNK1 structures in the identical locations as the phosphates in the ERK structure (red arrows, Figure 4D). These observations support the hypothesis that activation of JNK3 and JNK1, which is primarily via phosphorylation of Thr183 (Fleming et al., 2000; Lisnock et al., 2000), stabilizes the active conformation via positioning of αC, counteracting peptide-induced changes.
As a further test of the idea that the A-loop enforces a specific conformation, we examined 24 structures of JNK3 (Table S1B) bound to different inhibitors, of which 12 show a well-ordered A-loop conformer. The A-loop packed up against αC, but also docked into the peptide-binding site of the adjacent symmetry related molecule (Figure S4A), which was blocked in the peptide bound structure (Figure S4B), and thus likely not rendered disordered by the peptide per se. It is not clear why only some peptide-free structures show an ordered A-loop, as it is not due to differences in crystal packing. Here, we use this artificial stabilization of the A-loop to ask how it impacts the catalytic and peptide-docking sites. Visual inspection of the 24 structures suggested that the ordered A-loop reduced conformational heterogeneity (Figure 4E). This was reflected in significant lowering (p < 0.05) of overall differences between the structures in the ordered loop set, as measured by the root mean square deviation (RMSD) between the superimposed models. Less structural heterogeneity was also apparent in αC of the ordered set, as measured by the RMSD of αC at His104 (Figure 4F), indicating that this A-loop conformation is associated with a specific αC conformer. While the role of A-loop phosphorylation could not be directly confirmed, structure class analysis of both JNK1 and JNK3 suggests a model where A-loop phosphorylation reverses the peptide-induced autoinhibitory mechanisms, and locks the two lobes into a catalytically active conformation (Figure 4G).
The mutant and wild-type JNK1 structures show a structural mechanism for how the A-loop alters the peptide-docking site. This can be visualized by the peptide-induced shift in αD, and the “counter-shift” induced by the altered positioning of the A-loop in the mutant structures (Figure 5A). While this could occur via broad impact of the A-loop on the interlobe conformation, the direct role of allosteric communication via the A-loop is more apparent. The counter-shift in αD was caused by the differential positioning of A-loop Tyr191, which formed a tight hydrogen bond with Glu217 in αF, which in turn maintained a close VDW contact with Ile119 in αD (Figure 5B). Thus small changes in the positioning of A-loop Y191 could be directly transmitted to the peptide. Importantly, Leu166 in the JIP1 peptide bound differently between the two sets of structures, due to the shift in V118 in αD (Figure 5B). This demonstrates that subtle changes in the A-loop can allosterically alter how the peptide binds.
Figure 5. Activation Loop Control of Peptide Binding.

(A) All of the JNK1 structures were superimposed, as in Figure 3A. Shown is the α-carbon trace of αD region, and bound JIP1 peptide (green). Error bars represent SEM.
(B) Comparison of JNK1 wild-type and phosphoacceptor mutant structure. The mutations alter the position of the A-loop, shifting Y191, which is transmitted to αD. The shift in αD induced a shift in how the JIP1 peptide binds.
(C) The JNK3 structures with inhibitors and no peptides were superimposed and colored as in Figure 3D. The structures with ordered A-loop show a single, common conformer of αD.
(D) The RMSD of the structures in (D) from JNK1.pdb were calculated. Shown is the data for the α-carbon of αD M159. *p < 0.0001 using Student’s t test for significance. Error bars represent SEM.
We also examined the set of peptide-free JNK3 structures (Table S1B). Among these structures, those with an ordered A-loop showed less conformational heterogeneity in αD than those with a disordered A-loop (Figure 5C), and this difference was significant (Figure 5D). Both the JNK1 and JNK3 sets showed that the A-loop affected the conformation of αD, while the JNK1 data also showed a direct impact on peptide binding. Thus αD functions as a key allosteric sensor that links the A-loop to the peptide-binding site, and mediates interlobe communication. Finally, this communication is bidirectional, because αD also transmits subtle structural information from different peptides to affect A-loop conformation.
DISCUSSION
The three-component signaling module of MAPKs enable a wide range of signaling behavior, including crosstalk between parallel modules, as well as targeted activation of a single pathway (Good et al., 2011). Signaling can be switch-like, or show graded responses, with small changes in stoichiometry altering signaling characteristics (O’Shaughnessy et al., 2011; Takahashi and Pryciak, 2008). As the protein motif responsible for docking onto the MAPK, the D-motif is the primary determinant of signaling outcomes, as it is found in the N-terminus of upstream activating kinases, where it is required for binding and activation of MAPKs. The D-motif is also found in phosphatases, substrates, and scaffold proteins (Raman et al., 2007). A counter-intuitive feature of D-motif binding is called retroactivity, which is that signaling is bidirectional as sequestration of the enzyme alters the ensemble of interacting proteins (Del Vecchio et al., 2008). This has been shown dramatically in Drosophila, where 2-fold reduction in gene dosage of a single substrate leads to a global reduction in activated MAPK, due to greater access of a phosphatase (Kim et al., 2011). Thus subtle differences in D-motifs determine the ensemble of interacting proteins, and the nature of the signal.
In this work we describe two mechanisms of autoinhibition unique to JNK family kinases. JNK signaling needs to be tightly regulated, given its role in cell death. In fact, subtle changes in timing and duration of JNK signaling can determine cell fate decisions (Lin, 2006), highlighting the critical importance of tight regulation. Our results suggest that peptide-induced autoinhibition prevented spurious activity toward substrates. Peptide binding induced a substantial interlobe opening, and a rotation as well, which moved αC out of the position required for catalysis (Figures 1 and 2). In the absence of bound inhibitor, this interlobe motion further allowed the A-loop to bind in the catalytic site, forming an inhibitory helix (Figure 1). The peptides control interlobe motion via a subtle push on L16 (0.5–1 Å), and a more substantial pull on αD (1–1.5 Å) (Figures 2E–2I), which are then transmitted to the N-lobe. These conclusions are supported by the findings of Yan et al., who showed utilizing molecular dynamic simulations that peptide binding caused interdomain motions in the A-loop (Yan et al., 2011). In the initial publication of JIP1-bound JNK (Heo et al., 2004), the interlobe rotation was apparent, but not the more substantial lobe opening, nor the mechanism of allosteric signal transduction, due to the inherent limitations of examining a single structure. Furthermore, comparison of substrate and scaffold protein D-motifs, solved in different space groups as done in this work, demonstrates that this is likely a common mechanism and not an artifact of crystal packing. To get around limitations of single structure analysis we utilized structure class analysis which revealed the subtle allosteric mechanism through which peptide binding can control the JNK catalytic site, as both ends of the peptide modulate N-lobe position like reins on a horse.
We show here that JIP1 makes a number of unique interactions that explain its much higher affinity compared to SAB and ATF2, including both unique hydrogen bond networks, and differential interaction with αD (Figure 3). To our knowledge, these observations represent the first understanding of how JIP1 achieves such high affinity interactions compared to other known D-motif proteins, which can be used to direct structure based design efforts targeting the peptide-binding site (Stebbins et al., 2008). We are currently using these insights to develop both pure D-site substrate competitive inhibitors and bidentate competitive inhibitors that span both the ATP pocket and the D-site. Comparison of the different D-motif peptides also showed that they can directly control the A-loop via positioning of αD (Figures 3C–3E), demonstrating how small changes in D-motif sequence can contribute to allosteric control.
Our structure class analysis suggests that the function of A-loop phosphorylation is to stabilize the αC helix in the conformer required for catalysis, and in doing so reverse the peptide-induced autoinhibitory mechanisms. Though there is no structure of active JNK, the series of phosphoacceptor mutant JNK1 structures show a closing of the peptide-induced interlobe motion, but not a restoration of the rotation motion, explaining why these mutants are not constitutively active (Figures 4A–4C). Furthermore, we compared a series of JNK3 structures with the A-loop ordered versus disordered, showing that the A-loop enforced a single conformer of αC (Figures 4E and 4F). The structural similarity with the active ERK structure reinforces the interpretation of these observations that phosphorylation limits conformational heterogeneity between the kinase lobes (Figure 4D). This occurs largely via stabilization of N-lobe αC by the phosphoThr residue. Phosphorylation of Tyr adds an additional 25% activity by binding to the C-lobe and further locking in the active interlobe conformer. Indeed, this has been shown biochemically for JNK3 because it was suggested that once Thr is phosphorylated, a movement occurred so that Tyr is now accessible for phosphorylation by MKK7 (Lisnock et al., 2000). Thus, a primary mechanism of allosteric control in JNK family kinases is the opposing actions of peptide and A-loop phosphorylation on the interlobe conformation, providing an exquisite control mechanism over the catalytic site, which lies between the lobes (Figure 4G).
Activation of JNK also allosterically regulates the peptide-binding site. Using a FRET assay with recombinant JNK1 or JNK3, we showed that in most, but not all cases, activation increased affinity for the D-motif peptides (Table 2; Figure S1A). These results are consistent with those seen by Ngoei et al. who also observed greater affinity for pepJIP with JNK1α1 (Ngoei et al., 2011). This finding is likely due to the fact that in the current study and the Ngoei et al. study, peptide ligands were utilized whereas in Figuera-Losada and LoGrasso full length protein substrates were used. Given that the ensemble of interacting proteins shows different affinities for JNK (Tables 1, 2, and 3; Figure S1), and different functions and cellular localizations, this suggests that active and inactive JNK will show different ensembles of interacting proteins via this allosteric mechanism. Indeed it is interesting to wonder whether A-loop-induced changes from peptide binding would also occur at the presumed high intracellular ATP concentrations that exist. One clue to this answer may be in the first structure solved for JNK3 with AMPPNP. Here, the Gly-rich loop folds down over the AMPPNP pushing the A-loop out of the pocket. This potentially would likely be the case in the cellular environment as well when ATP levels are high. It will require a ternary complex of JNK-ATP-and peptide to experimentally test this hypothesis. In addition to A-loop control of interlobe conformation, our structural analyses suggest that the A-loop can more directly control the positioning of αD, and alter the peptide-binding site (Figure 5). Further, this interaction was bidirectional, as we show in our structures that differences in JIP1 versus SAB or ATF2 docking against αD influence the subtle positioning of the A-loop (Figures 3C–3E).
In summary, several observations and conclusions can be drawn from our findings. Importantly, to our knowledge, this is the first structural determination of peptides for JNK binding proteins other than JIP, namely SAB and ATF2. As previously mentioned, the structures reveal why JIP binds more potently than the other peptides, and provides insights into how this binding may regulate signaling to influence cell death and survival. Second, the biochemical observations form both inactive and active forms of JNK support the structural findings and again have implications on how signaling is regulated through active and inactive kinases. Finally, we used structure class analysis to define allosteric signaling mechanisms between peptide binding, catalytic, and phosphoacceptor sites in the JNK family of kinases.
Comparison of classes of structures enabled identification of common features across many structures and space groups, and allowed statistical analysis and characterization of subtle features within the noise of a single structure, as we previously used for dissecting estrogen receptor signaling (Bruning et al., 2010; Nettles et al., 2004, 2008). It is noteworthy that most of these structures were not described in the scientific literature other than a single image of the inhibitor binding, as is increasingly common with the high-throughput approaches used in structure base -design and structural genomics campaigns. Advances in all aspects of X-ray crystallography are enabling its evolution to mature science, focusing on data analysis rather than technique. We suggest that the application of techniques to looking at classes of structures will enable X-ray crystallography to be used as a system biology tool. Moreover, these studies lay the foundation for structure-based design of substrate competitive inhibitors for JNK.
EXPERIMENTAL PROCEDURES
Reagents and Protein Production
Recombinant ATF2 and SAB for kinase assays were cloned and purified as previously described (Chambers et al., 2011a). JNK1β1 production for FRET assays was as previously described (Figuera-Losada and Lograsso, 2012a) and full length JNK3α1 was produced similarly, using a 6xHis tag for affinity purification from BL21(DES) E. coli. In vitro activation of JNK1β1 and JNK3α1 for FRET assays was prepared by incubation with active MKK4 and MKK7 (Millipore) (Figuera-Losada and Lograsso, 2012a). Peptides were ordered from Peptidogenics.
Peptide Inhibition Assays for JNK Kinase Activity
To determine the ability of different peptides derived from the D-motifs of JIP1 (pepJIP), ATF2 (pepATF2), or SAB (pepSAB) to inhibit JNK3α1 (39–422) phosphorylation of substrates His-SAB (1–390) or FLAG-ATF2 (2–115), we followed the incorporation of 33P from γ33P-ATP into the substrates using a filtration assay (0.45 μm Immobilon-P MultiScreen HTS plates, Millipore). Reactions were done in 25 mM HEPES pH 7.4, 10 mM MgCl2, 1 mM DTT, 20 mM β-glycerophosphate, 0.1 mM Na3VO4, and 0.5 mg/ml bovine serum albumin buffer in the presence of 0.9–1.2 μCi γ33P-ATP. Constant concentrations of substrates were used: 15 μM ATP, 2.5 μM His-SAB or 0.1 μM FLAG-ATF2, and 2 nM active JNK3α1. Seven different concentrations of the peptides (0.01–1000 μM) were tested and reactions were carried out for 45 min at 30 °C. To stop reactions, 100 mM of H3PO4 was used and 50 μl samples were transferred to the MultiScreen plates in quadruplicate for filtration. After extensive washes with 10 mM HEPES pH 7.4, 100 mM NaCl, 25 mM EDTA, filters were washed with 50% ethanol solution, completely air-dried and read in a Top Count scintillation counter (Packard Instrument) after 100 μl of Ultima Gold XR liquid scintillation counting cocktail (Perkin Elmer) was added in each well. Data analysis and IC50 calculations were accomplished with GraphPad Prism version 5.04. Reported IC50 values represent a single experiment ± standard error; however, each experiment was repeated at least three times and similar results were observed.
Isothermal Titration Calorimetry
ITC studies were performed using a MicroCal ITC200 by titration of each peptide into a solution of purified JNK3α1 (39–422). JNK3α1 (39–422) was expressed and purified in the same manner as JNK3α1 (39–402) and concentrated to 30 mg/ml. The protein was then diluted to working concentration in 50 mM HEPES, pH 7.4, 150 mM NaCl, 10 mM MgCl2, and 2 mM TCEP that had been thoroughly degassed. Each peptide was then diluted in the same buffer as the protein. Since the peptides displayed widely varying affinities and amount of heat given off upon binding to JNK3α1 (39–422), the concentration of JNK3α1 (39–422) and peptide used was optimized for each ligand to obtain a full binding curve. The concentrations of JNK3α1 (39–422) used are given here, with the concentration of the corresponding peptide in parentheses: pepJIP: 50 μM (500 μM); JIP_SAB: 50 μM (500 μM); SAB_JIP: 120 μM (1,200 μM); pepATF2: 150 μM (1,500 μM); pepSAB: 150 μM (1,500 μM); T158G: 108 μM (1,000 μM); R156Q: 108 μM (1,200 μM); R156H: 108 μM (1,200 μM). The titrations were performed at 25°C. Each experiment consisted of 18 injections of peptide, with each injection consisting of 2.2 μl of peptide solution injected at a rate of 1 μl/sec, with injections spaced 180 s apart. The rotation of the stirrer was set to 500 rpm. The reference power was set at 5 cal. Binding curves were fit to a single-site binding model in the program Origin (MicroCal). The aberrant first injection of each experiment was not included in the curve fitting.
Fluorescence Resonance Energy Transfer Assay
Substrate molecules binding to phopho-JNK (p-JNK) or unphosphorylated JNK were measured in a Fluorescence Resonance Energy Transfer (FRET) assay. Under standard assay conditions, 5 μl of peptide (protein or compound) in buffer (10 mM HEPES/KOH (pH 7.4), 150 mM NaCl, 10 mM MgCl2, 0.005% Brij-35, 0.1% 2-mercaptoethanol, and 0.05% BSA) was dispensed into a 384-well low volume black Greiner 784076 microtiter plate. Final peptide concentrations ranged from 125 to 0 μM. Then, 15 μl of solution containing N-terminal His-JNK3 39–422, Tb-labeled anti-His antibody (Invitrogen), and FITC-labeled JIP 11-mer peptide was added to the microtiter wells to give a final JNK concentration of 10 nM, a final Tb-labeled anti-His antibody concentration of 2 nM, and a final FITC-labeled JIP 11-mer peptide concentration of 100 nM. Plates were read on a Perkin Elmer Envision 2104 multilabel plate reader with a filter set for Tb excitation at 340 ± 60 nm and emission at 492 ± 8 nm and FITC emission at 520 ± 8 nm. The signal is presented as an emission ratio of 520/492. Data were analyzed in Origin to determine the EC50 for each peptide by fitting to a logistic equation.
X-Ray Crystallography
JNK3α1 (39–402) was produced in E. coli as previously described (Kamenecka et al., 2009). The protein was concentrated to 8 mg/ml in 50 mM HEPES pH 7.0, 100 mM NaCl, 5% glycerol, 2 mM DTT. For crystallization trials each peptide (pepJIP, pepSAB, or pepATF2) was diluted in DMSO and added to the protein solution to a 5:1 (pepJIP and pepSAB) or 10:1 (pepATF) peptide:JNK molar ratio. The mixtures were incubated on ice for at least one hour before crystal trays were set up. Crystals of the pepJIP: JNK3α1(39-402) complex formed in the conditions previously described for the pepJIP:JNK1 complex (Heo et al., 2004). However, complexes of JNK3α1 (39–402) with pep-SAB and pepATF2 did not crystallize in these conditions, and so screening and optimization of crystallization conditions was undertaken. Crystallization conditions were identified using the Protein Complex Screen (QIAGEN) and optimized using the Additive Screen (Hampton Research). Crystals for diffraction experiments were grown by mixing the protein solution in ratios from 0.8:1.2 to 1.2:0.8 with 15% PEG6000, 100 mM sodium citrate, pH 5.5, 2% tertbutanol. Crystals were grown via hanging drop over a reservoir of the precipitant solution at 4°C. Before picking crystals the drops containing crystals were equilibrated with a mixture of the mother liquor containing 40% ethylene glycol to a final concentration of approximately 10% ethylene glycol in the drop. The picked crystals were further dipped into perfluoropolyether oil (Hampton Research) before freezing to remove surface water. X-ray diffraction data were collected at Stanford Synchrotron Radiation Light-source (SSRL) beam line 11-1, and scaled using HKL-2000 software (HKL Research, Charlottesville, VA). Structures of the peptide-bound JNK3 complexes were solved by molecular replacement using the PHENIX software suite (Adams et al., 2010) and the catalytic domain of JNK3 (PDB number 1JNK; Xie et al., 1998) without waters, bound peptides, and inhibitors, as the starting model. After initial model building, the peptides were docked in using Coot (Emsley and Cowtan, 2004). Subsequently, the JNK3+peptide models were refined in PHENIX with ExCoR (Extensive combinatorial Refinement) using 256 unique refinement strategies (unpublished data). Elements of several of the best models were combined and validated using PHENIX and MolProbity (Chen et al., 2010). All other crystal structures were obtained from the Protein Data Bank (http://www.pdb.org) (Berman et al., 2000). Structures were superposed globally in CCP4MG using 1JNK.pdb as a reference model for JNK3, or 3PTG.pdb for JNK1 structures. Structural figures were prepared with CCP4MG (McNicholas et al., 2011).
Supplementary Material
Acknowledgments
This work was supported by NIH Grant NS057153 awarded to P.VL. P.V.L. serves as a consultant to OPKO Health.
Footnotes
ACCESSION NUMBERS
The Protein Data Bank accession numbers for the JNK3/pepJIP1, JNK3 pepSAB, and JKN3/pepATF2 structures are 4H39, 4H3B, and 4H36, respectively.
Supplemental Information includes four figures and one table and can be found with this article online at http://dx.doi.org/10.1016/j.str.2012.09.021.
References
- Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardwell AJ, Frankson E, Bardwell L. Selectivity of docking sites in MAPK kinases. J Biol Chem. 2009;284:13165–13173. doi: 10.1074/jbc.M900080200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The Protein Data Bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruning JB, Parent AA, Gil G, Zhao M, Nowak J, Pace MC, Smith CL, Afonine PV, Adams PD, Katzenellenbogen JA, Nettles KW. Coupling of receptor conformation and ligand orientation determine graded activity. Nat Chem Biol. 2010;6:837–843. doi: 10.1038/nchembio.451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canagarajah BJ, Khokhlatchev A, Cobb MH, Goldsmith EJ. Activation mechanism of the MAP kinase ERK2 by dual phosphorylation. Cell. 1997;90:859–869. doi: 10.1016/s0092-8674(00)80351-7. [DOI] [PubMed] [Google Scholar]
- Chambers JW, LoGrasso PV. Mitochondrial c-Jun N-terminal kinase (JNK) signaling initiates physiological changes resulting in amplification of reactive oxygen species generation. J Biol Chem. 2011;286:16052–16062. doi: 10.1074/jbc.M111.223602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers JW, Cherry L, Laughlin JD, Figuera-Losada M, Lograsso PV. Selective inhibition of mitochondrial JNK signaling achieved using peptide mimicry of the Sab kinase interacting motif-1 (KIM1) ACS Chem Biol. 2011a;6:808–818. doi: 10.1021/cb200062a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers JW, Pachori A, Howard S, Ganno M, Hansen D, Jr, Kamenecka T, Song X, Duckett D, Chen W, Ling YY, et al. Small Molecule c-jun-N-terminal Kinase (JNK) Inhibitors Protect Dopaminergic Neurons in a Model of Parkinson’s Disease. ACS Chem Neurosci. 2011b;2:198–206. doi: 10.1021/cn100109k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang CI, Xu BE, Akella R, Cobb MH, Goldsmith EJ. Crystal structures of MAP kinase p38 complexed to the docking sites on its nuclear substrate MEF2A and activator MKK3b. Mol Cell. 2002;9:1241–1249. doi: 10.1016/s1097-2765(02)00525-7. [DOI] [PubMed] [Google Scholar]
- Chen VB, Arendall WB, 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Vecchio D, Ninfa AJ, Sontag ED. Modular cell biology: retroactivity and insulation. Mol Syst Biol. 2008;4:161. doi: 10.1038/msb4100204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ember B, Kamenecka T, LoGrasso P. Kinetic mechanism and inhibitor characterization for c-jun-N-terminal kinase 3alpha1. Biochemistry. 2008;47:3076–3084. doi: 10.1021/bi701852z. [DOI] [PubMed] [Google Scholar]
- Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Figuera-Losada M, Lograsso PV. Enzyme kinetics and interaction studies for human JNK1[beta]1 and substrates ATF2 and cJun. J Biol Chem. 2012a;287:13291–13302. doi: 10.1074/jbc.M111.323766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figuera-Losada M, LoGrasso PV. Enzyme kinetics and interaction studies for human JNK1β1 and substrates activating transcription factor 2 (ATF2) and c-Jun N-terminal kinase (c-Jun) J Biol Chem. 2012b;287:13291–13302. doi: 10.1074/jbc.M111.323766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming Y, Armstrong CG, Morrice N, Paterson A, Goedert M, Cohen P. Synergistic activation of stress-activated protein kinase 1/c-Jun N-terminal kinase (SAPK1/JNK) isoforms by mitogen-activated protein kinase kinase 4 (MKK4) and MKK7. Biochem J. 2000;352:145–154. [PMC free article] [PubMed] [Google Scholar]
- Goldsmith EJ. Three-dimensional docking in the MAPK p38α. Sci Signal. 2011;4:pe47. doi: 10.1126/scisignal.2002697. [DOI] [PubMed] [Google Scholar]
- Good MC, Zalatan JG, Lim WA. Scaffold proteins: hubs for controlling the flow of cellular information. Science. 2011;332:680–686. doi: 10.1126/science.1198701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heo YS, Kim SK, Seo CI, Kim YK, Sung BJ, Lee HS, Lee JI, Park SY, Kim JH, Hwang KY, et al. Structural basis for the selective inhibition of JNK1 by the scaffolding protein JIP1 and SP600125. EMBO J. 2004;23:2185–2195. doi: 10.1038/sj.emboj.7600212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaeschke A, Czech MP, Davis RJ. An essential role of the JIP1 scaffold protein for JNK activation in adipose tissue. Genes Dev. 2004;18:1976–1980. doi: 10.1101/gad.1216504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamenecka T, Habel J, Duckett D, Chen W, Ling YY, Frackowiak B, Jiang R, Shin Y, Song X, LoGrasso P. Structure-activity relationships and X-ray structures describing the selectivity of aminopyrazole inhibitors for c-Jun N-terminal kinase 3 (JNK3) over p38. J Biol Chem. 2009;284:12853–12861. doi: 10.1074/jbc.M809430200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamenecka T, Jiang R, Song X, Duckett D, Chen W, Ling YY, Habel J, Laughlin JD, Chambers J, Figuera-Losada M, et al. Synthesis, biological evaluation, X-ray structure, and pharmacokinetics of aminopyrimidine c-jun-N-terminal kinase (JNK) inhibitors. J Med Chem. 2010;53:419–431. doi: 10.1021/jm901351f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karin M, Gallagher E. From JNK to pay dirt: jun kinases, their biochemistry, physiology and clinical importance. IUBMB Life. 2005;57:283–295. doi: 10.1080/15216540500097111. [DOI] [PubMed] [Google Scholar]
- Kim Y, Paroush Z, Nairz K, Hafen E, Jiménez G, Shvartsman SY. Substrate-dependent control of MAPK phosphorylation in vivo. Mol Syst Biol. 2011;7:467. doi: 10.1038/msb.2010.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee T, Hoofnagle AN, Kabuyama Y, Stroud J, Min X, Goldsmith EJ, Chen L, Resing KA, Ahn NG. Docking motif interactions in MAP kinases revealed by hydrogen exchange mass spectrometry. Mol Cell. 2004;14:43–55. doi: 10.1016/s1097-2765(04)00161-3. [DOI] [PubMed] [Google Scholar]
- Lin A. A five-year itch in TNF-alpha cytotoxicity: the time factor determines JNK action. Dev Cell. 2006;10:277–278. doi: 10.1016/j.devcel.2006.02.006. [DOI] [PubMed] [Google Scholar]
- Lisnock J, Griffin P, Calaycay J, Frantz B, Parsons J, O’Keefe SJ, LoGrasso P. Activation of JNK3 alpha 1 requires both MKK4 and MKK7: kinetic characterization of in vitro phosphorylated JNK3 alpha 1. Biochemistry. 2000;39:3141–3148. doi: 10.1021/bi992410+. [DOI] [PubMed] [Google Scholar]
- McNicholas S, Potterton E, Wilson KS, Noble ME. Presenting your structures: the CCP4mg molecular-graphics software. Acta Crystallogr D Biol Crystallogr. 2011;67:386–394. doi: 10.1107/S0907444911007281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morel C, Standen CL, Jung DY, Gray S, Ong H, Flavell RA, Kim JK, Davis RJ. Requirement of JIP1-mediated c-Jun N-terminal kinase activation for obesity-induced insulin resistance. Mol Cell Biol. 2010;30:4616–4625. doi: 10.1128/MCB.00585-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nettles KW, Sun J, Radek JT, Sheng S, Rodriguez AL, Katzenellenbogen JA, Katzenellenbogen BS, Greene GL. Allosteric control of ligand selectivity between estrogen receptors alpha and beta: implications for other nuclear receptors. Mol Cell. 2004;13:317–327. doi: 10.1016/s1097-2765(04)00054-1. [DOI] [PubMed] [Google Scholar]
- Nettles KW, Bruning JB, Gil G, Nowak J, Sharma SK, Hahm JB, Kulp K, Hochberg RB, Zhou H, Katzenellenbogen JA, et al. NFkappaB selectivity of estrogen receptor ligands revealed by comparative crystallographic analyses. Nat Chem Biol. 2008;4:241–247. doi: 10.1038/nchembio.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ngoei KR, Catimel B, Church N, Lio DS, Dogovski C, Perugini MA, Watt PM, Cheng HC, Ng DC, Bogoyevitch MA. Characterization of a novel JNK (c-Jun N-terminal kinase) inhibitory peptide. Biochem J. 2011;434:399–413. doi: 10.1042/BJ20101244. [DOI] [PubMed] [Google Scholar]
- O’Shaughnessy EC, Palani S, Collins JJ, Sarkar CA. Tunable signal processing in synthetic MAP kinase cascades. Cell. 2011;144:119–131. doi: 10.1016/j.cell.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raman M, Chen W, Cobb MH. Differential regulation and properties of MAPKs. Oncogene. 2007;26:3100–3112. doi: 10.1038/sj.onc.1210392. [DOI] [PubMed] [Google Scholar]
- Salameh A, Galvagni F, Anselmi F, De Clemente C, Orlandini M, Oliviero S. Growth factor stimulation induces cell survival by c-Jun. ATF2-dependent activation of Bcl-XL. J Biol Chem. 2010;285:23096–23104. doi: 10.1074/jbc.M109.087221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulze-Gahmen U, Rini JM, Wilson IA. Detailed analysis of the free and bound conformations of an antibody. X-ray structures of Fab 17/9 and three different Fab-peptide complexes. J Mol Biol. 1993;234:1098–1118. doi: 10.1006/jmbi.1993.1663. [DOI] [PubMed] [Google Scholar]
- Stebbins JL, De SK, Machleidt T, Becattini B, Vazquez J, Kuntzen C, Chen LH, Cellitti JF, Riel-Mehan M, Emdadi A, et al. Identification of a new JNK inhibitor targeting the JNK-JIP interaction site. Proc Natl Acad Sci USA. 2008;105:16809–16813. doi: 10.1073/pnas.0805677105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi S, Pryciak PM. Membrane localization of scaffold proteins promotes graded signaling in the yeast MAP kinase cascade. Current Biol. 2008;18:1184–1191. doi: 10.1016/j.cub.2008.07.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Harkins PC, Ulevitch RJ, Han J, Cobb MH, Goldsmith EJ. The structure of mitogen-activated protein kinase p38 at 2.1-A resolution. Proc Natl Acad Sci USA. 1997;94:2327–2332. doi: 10.1073/pnas.94.6.2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weston CR, Davis RJ. The JNK signal transduction pathway. Curr Opin Cell Biol. 2007;19:142–149. doi: 10.1016/j.ceb.2007.02.001. [DOI] [PubMed] [Google Scholar]
- Whitmarsh AJ, Kuan CY, Kennedy NJ, Kelkar N, Haydar TF, Mordes JP, Appel M, Rossini AA, Jones SN, Flavell RA, et al. Requirement of the JIP1 scaffold protein for stress-induced JNK activation. Genes Dev. 2001;15:2421–2432. doi: 10.1101/gad.922801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Win S, Than TA, Han D, Petrovic LM, Kaplowitz N. c-Jun N-terminal kinase (JNK)-dependent acute liver injury from acetaminophen or tumor necrosis factor (TNF) requires mitochondrial Sab protein expression in mice. J Biol Chem. 2011;286:35071–35078. doi: 10.1074/jbc.M111.276089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie X, Gu Y, Fox T, Coll JT, Fleming MA, Markland W, Caron PR, Wilson KP, Su MS. Crystal structure of JNK3: a kinase implicated in neuronal apoptosis. Structure. 1998;6:983–991. doi: 10.1016/s0969-2126(98)00100-2. [DOI] [PubMed] [Google Scholar]
- Yan C, Kaoud T, Lee S, Dalby KN, Ren P. Understanding the specificity of a docking interaction between JNK1 and the scaffolding protein JIP1. J Phys Chem B. 2011;115:1491–1502. doi: 10.1021/jp1073522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Strand A, Robbins D, Cobb MH, Goldsmith EJ. Atomic structure of the MAP kinase ERK2 at 2.3 A resolution. Nature. 1994;367:704–711. doi: 10.1038/367704a0. [DOI] [PubMed] [Google Scholar]
- Zheng C, Xiang J, Hunter T, Lin A. The JNKK2-JNK1 fusion protein acts as a constitutively active c-Jun kinase that stimulates c-Jun transcription activity. J Biol Chem. 1999;274:28966–28971. doi: 10.1074/jbc.274.41.28966. [DOI] [PubMed] [Google Scholar]
- Zhou H, Zheng M, Chen J, Xie C, Kolatkar AR, Zarubin T, Ye Z, Akella R, Lin S, Goldsmith EJ, Han J. Determinants that control the specific interactions between TAB1 and p38alpha. Mol Cell Biol. 2006a;26:3824–3834. doi: 10.1128/MCB.26.10.3824-3834.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou T, Sun L, Humphreys J, Goldsmith EJ. Docking interactions induce exposure of activation loop in the MAP kinase ERK2. Structure. 2006b;14:1011–1019. doi: 10.1016/j.str.2006.04.006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.