

Abstract
In genetic association studies, it is necessary to correct for population structure to avoid inference bias. During the past decade, prevailing corrections often only involved adjustments of global ancestry differences between sampled individuals. Nevertheless, population structure may vary across local genomic regions due to the variability of local ancestries associated with natural selection, migration, or random genetic drift. Adjusting for global ancestry alone may be inadequate when local population structure is an important confounding factor. In contrast, adjusting for local ancestry can more effectively prevent false-positives due to local population structure. To more accurately locate disease genes, we recommend adjusting for local ancestries by interrogating local structure. In practice, locus-specific ancestries are usually unknown and cannot be accurately inferred when ancestral population information is not available. For such scenarios, we propose employing local principal components (PC) to represent local ancestries and adjusting for local PCs when testing for genotype–phenotype association. With an acceptable computation burden, the proposed algorithm successfully eliminates the known spurious association between SNPs in the LCT gene and height due to the population structure in European Americans.
Keywords: Genome-wide association studies, Local ancestries, Local principal components, Migration, Random genetic drift, Natural selection, Genomic inflation factor, Genomic control, Local ancestry principal components correction, Fine mapping
1. Introduction
In association studies, the concerns for population stratification can be dated back to twenty years ago (1). It has been well realized that population stratification—systematic ancestry differences between study subjects—can confound association tests (2–13). Population structure exists in study subjects as a result of distinct ethnic groups or a single pool of admixed individuals. Global population structure can be characterized by individual global ancestries. An individual’s global ancestry can be calculated as the proportions of his/her genome inherited from the underlying ancestral populations. Local population structure in a local genomic region or at a locus can be similarly characterized. During the past decade, statistical methods to control the false-positive rate due to population stratification have mainly involved adjustments for global population structure, which is mainly due to recent migration and random genetic drift. Some prevailing paradigms are genomic control (3), structural association methods (4, 5), and principal component methods (6, 7, 10) that use markers randomly selected across the genome. In genomic control, the variance inflation factor of a test statistic is assumed to be a constant across the entire genome. In a classical principal component (PC) method, the PCs of the genotype score matrix of genome-wide markers (referred to as global PCs) are used as ancestry surrogates in the association analysis for each testing marker. Many studies have shown that the global PCs can effectively represent human demographic history (6, 10–12, 14–16).
Nevertheless, subtle local structures do occur in some small genomic regions, owing to demographic history, natural selection pressure, and random fluctuations of admixture (13, 17, 18). The imprint of natural selection, for example, has recently been identified in many regions across the genome (19, 20) and can create substantial variation in population differentiation, which in turn affects the degree of variance inflation for specific loci (21). Even subtle structure, if ignored, can either inflate type I error or reduce statistical power, especially when the sample size is large (8).
For recently admixed populations with known reference ancestral populations (e.g., African Americans), locus-specific ancestry can be inferred accurately using hidden Markov model-based methods (4, 22–26). However, it is difficult to accurately infer locus-specific ancestries for an admixed individual owing to either the lack of ancestral population information or the high similarity between ancestral populations. For example, population admixture of European Americans occurred within populations of similar origin. For such scenarios, we propose using local PCs of the genotype score matrix of the markers within local genomic regions to represent local ancestries, at the same time using global PCs to represent global ancestries, and interrogating the ancestries in local genomic regions for fine mapping (13). Extensive simulations and applications to the data sets from three genome-wide association studies illustrate the necessity and practical implications of the adjustment of local ancestry principal components. Both European Americans and African Americans demonstrate greater variability in local ancestry than do Nigerians. Adjusting local PCs successfully eliminates the well-known spurious association between the LCT gene and height of European Americans due to the underlying population structure. In this chapter, we illustrate how to run the local ancestry principal components correction (LAPCC).
In LAPCC, we divide each chromosome into 4-Mb adjacent segments (referred to as window cores hereafter) according to the SNP map of base pair positions. Typically, we add an envelope with an 8-Mb margin to each side of a core (Fig. 1) and construct a 20-Mb window. We choose this window width according to the linkage disequilibrium due to recent population admixture (22, 24). The left (right) envelopes of the first (last) 2 windows of some chromosomes might be shorter than 8 Mb. For each local window, we compute the first ℓ = 10 (see Note 1) PCs of the genotypic scores of window-wide SNPs to adjust for the population structure of the SNPs within the 4-Mb core. At each SNP in the core, we compute genotype score residuals g̃i and trait value residuals ỹi by regressing genotype scores gi and trait values yi on the 10 local PCs. We measure the evidence of genotype–phenotype association by s2 = (N − ℓ − 2)r2/(1 − r2), where N is the number of individuals used after excluding individuals with missing genotypes and r is the correlation coefficient between the residuals ỹi and g̃i. Asymptotically, s2 follows a distribution under the null of no genotype–phenotype association if all confounding factors are well adjusted.
Fig 1.
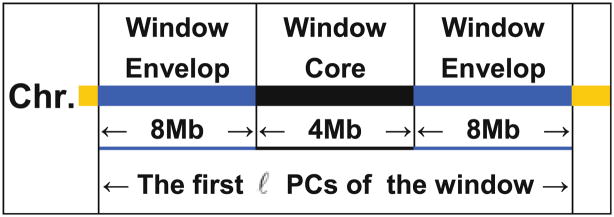
A typical window consists of a 4-Mb core and an envelope with 8-Mb margins on each side of the core. The first ℓ PCs of the genotypic score matrix of the SNPs in the 20-Mb window are employed to adjust for local ancestries of the SNPs within the 4-Mb core.
To be specific, we denote by G = (gij ) the M × N genotypic matrix (of the entire genome or a local window), where gij ∈ {0, 1, 2} is the copy number of a reference allele at the ith marker of the jth subject. We then center each row i of matrix G by the row mean and denote the centered matrix by X = (xij ). We exclude each missing entry gij from the computation of μi and set the corresponding xij to be 0. We denote the eigensystem of the N × N matrix C = X′X by C = VΛV′, where V = [v1, …, vN ], Λ = diag(λ1, …, λN), and λ1> · · · > λN−1> λN are eigenvalues corresponding to eigenvectors v1, …, vN, respectively, and in particular, λN = 0 and vN = N−1/2(1, …, 1)′. Following others’ previous research work (10), we define the kth axis of variation as eigenvector vk = (v1k, …, vNk)′ and select κ eigenvectors (PCs) as the ancestral surrogates. We do not normalize the matrix X (see Note 2) and do not suggest thinning SNPs for calculating the PCs (see Note 3). Instead, we only center the genotypic scores and calculate the PCs from all available genotypic data.
To interrogate local structure, we calculate the coefficients of multiple-determination (R2) and squared coefficients of canonical correlation (λ2) between the global PCs and local PCs. Let N denote the sample size, A = [a1, …, aK ] denote the N × K matrix consisting of the first K global PCs, and B = [b1, …, bK ] denote the N × K matrix consisting of the first K local PCs in a local window. The coefficient of multiple-determination for bj and A is the R2 for the linear regression of bj on A. The j th largest squared coefficient of canonical correlation between A and B is the j th largest coefficient of determination between any linear combination of B’s columns and any linear combination of A’s columns. Mathematically, , and . equals the jth diagonal element, and equals the j th largest eigenvalue of B′AA′B, and , where is the coefficient of determination between bj and ai. Canonical correlation analysis and multiple-determination analysis enable us to evaluate the degree of discrepancies between local and global PCs and facilitate the exploration of population structures. R2 indicates how much variation of the local PCs can be accounted for by the global PCs, and λ2 measures the shared variance between the local and global PCs.
2. Methods
We will illustrate how to use the software package LAPCC.exe with genome-wide genotypic data sets—the Maywood, Nigeria, and Framingham data sets (27, 28). The file LAPCC.exe can be downloaded from http://darwin.cwru.edu/LAPCC/LAPCC.exe. The user needs to put the file LAPCC.exe and the formatted data in the same folder.
2.1. Formatting the Data
First, we will explain how to preprocess and format the data. For example, the Maywood cohort comprises 775 unrelated African-Americans from Maywood, IL, with 909,622 SNPs genotyped on the Affymetrix 6.0 platform. We drop 74 individuals because of possible DNA contamination, false identity, and relatedness. For the 701 retained individuals, we remove 86,800 SNPs whose missing rates >5% and minor allele frequencies <1%. The final analysis data set includes 822,822 SNPs in each of 701 individuals. Similarly, after QC, the Nigeria Affymetrix 6.0 data set contains 759,222 SNPs genotyped for 982 individuals. For the Framingham data set, Mendelian errors are checked, and the corresponding SNPs with Mendelian inconsistence are set missing. SNPs with HWE P values <10−6 are dropped. We select unrelated individuals from each family (i.e., spouses) based on an algorithm that prioritizes individuals with higher genotyping rates, selecting individuals at random when needed. After QC, the Framingham data set contains 415,281 SNPs genotyped for 1,106 unrelated individuals.
For each chromosome of each data set, five plain input files must be prepared like the following example. In the Maywood data set, the first autosome contains 67,242 SNPs. The first file is conf_chr1.txt, which contains three rows as below:
67242 711153 247165315
The first row is the number of SNPs on autosome 1; the second and third rows give the base pair positions of the first and the last SNPs on the autosome, respectively. The second input file is bp_chr1.txt, containing 67,242 rows. Each row contains the base pair position of one SNP, and all the base pair positions are sorted in ascending order:
711153 730720 742429 751595 . . . . . .
The third file is snp_chr1.txt, containing the rs-numbers of all the 67,242 SNPs:
rs12565286 rs12082473 rs3094315 rs2286139 . . . . . .
The fourth file is geno_chr1.txt, containing a 701-by-67,242 white-spaced matrix of genotypic scores. For each SNP (column), a reference allele is randomly assigned, and the copy number of the reference allele in each person is recorded as his/her genotypic score at the SNP. Missing genotypes are recorded as −9. In the file geno_chr1.txt, the genotypic scores of the first five SNPs of the first four individuals are as below:
| 1 | 2 | 2 | 2 | 2 | … |
| 2 | 1 | 2 | 2 | 2 | … |
| 2 | 1 | 0 | 1 | 2 | … |
| 2 | −9 | 2 | 2 | 2 | … |
| … | … | … | … | … | … |
The last file is y_height.txt, listing the trait values (the residuals of height after adjusting out such covariates such as sex, age, and age-squared) one person per row:
0.710633 −0.346286 −1.25865 0.350496 . . . . . .
2.2. Run the Package
After preparing the 5 plain files, the user can run LAPCC.exe to process all or specified autosomes in a parallel way. For each specified autosome, the LAPCC.exe outputs windows, window-wide R2, and λ2-values as well P values of all input SNPs on the autosome. For the three aforementioned populations, we compare local and global PCs to uncover local ancestry patterns. Figure 2 presents the λ2-values between window-wide local PCs and the first 10 global PCs. In general, local PCs and global PCs here only display a small degree of correlation. However, the Maywood and Framingham data sets demonstrate substantially larger λ2-values and larger variation in λ2-values than does the Nigerian data set. These results suggest that there is relatively little population structure in the Nigerian sample, whereas Maywood and Framingham display relatively more population structure. Somewhat surprisingly, the Framingham data set demonstrates much more complex local population structure than do the other two data sets.
Fig 2.

The distributions of the λ2-values of the local windows in three GWAS data sets. For each window in a given genotype data set, λ2 is the largest squared coefficient of canonical correlation between the first 10 local PCs and the first 10 global PCs. Relatively, the Maywood participants demonstrate more population structure, the Nigerian samples demonstrate little population structure, whereas the Framingham participants demonstrate a much more complex local population structure than do the other two samples.
Fig 3.

PCs with (a) and without (b) normalizing the GAW17 genotypic data set of 697 unrelated individuals. Clearly, the PCs without normalization provide better discrimination, although both PCs roughly classify the 697 individuals into 3 large groups: CEPI and Tuscan, Luhya and Yoruba, as well as Denver Chinese, Han Chinese, and Japanese. The PCs without normalization appear more robust to outliers.
Fig 4.

Pearson correlation coefficients between the standard first global PC coordinates of distinct subsets of 1,969,739 SNPs and individual global ancestries of the 2,000 individuals. The data set is generated by the GenoAnceBase0 program (13) applied to the CEU and YRI haplotypes of the HapMap data (Phase II) to simulate African-American genomes. The standard first global PC coordinates of the 3,029 unlinked AIMs across the genome are highly correlated with true individual global ancestries. The first standard global PC coordinates using more random markers represent the true global ancestries even better, regardless of there being more abundant LD.
Acknowledgments
We thank the members in Dr. Zhu’s lab for their helpful comments. This work was supported by NIH grants HL074166, HL086718, and HG003054 to XZ.
Footnotes
Setting ℓ = 10 is somewhat arbitrary. Patterson et al. (29) developed a formal procedure to determine ℓ using the genotypic data. This procedure is based on the Tracy–Widom theory (29, 30) and proves to be conservative if the normalized genotypic data matrix is a true Wishart matrix (29). Improvements may be possible since the first ℓ PCs may not be the most informative.
According to our analysis of real data, normalization may hurt the representativeness of the global (local) PCs for the true ancestry when the data involve more than two ancestral populations (Fig. 3). For the case of only two ancestral populations, the results without normalization are mathematically valid. Thus, we do not suggest normalization even if it may yield better results under certain conditions (29), which may be violated in practice.
In our simulations, random correlations between markers have little impact on global PCs of tens of thousands to millions of genome-wide SNPs (Fig. 4). The first global PC coordinates of a large number of unlinked ancestral informative markers (AIMs) across the genome are highly correlated with true individual global ancestries. The first global PC coordinates using more random markers represent the true global ancestries even better, regardless of the abundant LD. This suggests that global PCs of all available genotypic data would well represent global ancestries. In the same vein, we do not suggest thinning SNPs to calculate the local PCs.
The local regions are often empirically created and thus may be away from the optimal. Standard local PCs may be fragile to apparent random inter-SNP correlations, especially for local genomic regions without sufficient ancestral informative markers. Thus, for local PCs, one may weigh genotypic scores of the SNPs within a local window (31). It is reasonable to incorporate the adjustment of global PCs to prevent false-positives due to other sources of confounding.
The LAPCC was designed for data sets comprising unrelated individuals and would fail to account for the other types of sample structure, i.e., cryptic relatedness and family structure. Cryptic relatedness may occur in a wide range of data sets, and it is necessary to model family structure in family-based association studies with sample ascertainment (32). It may be instructive to incorporate the full covariance structure across individuals using mixed models.
References
- 1.Knowler WC, Williams RC, Pettitt DJ, Steinberg AG. Gm3;5,13,14 and type 2 diabetes mellitus: an association in American Indians with genetic admixture. Am J Hum Genet. 1988;43:520–526. [PMC free article] [PubMed] [Google Scholar]
- 2.Lander ES, Schork NJ. Genetic dissection of complex traits. Science. 1994;265:2037–2048. doi: 10.1126/science.8091226. [DOI] [PubMed] [Google Scholar]
- 3.Devlin B, Roeder K. Genomic control for association studies. Biometrics. 1999;55:997–1004. doi: 10.1111/j.0006-341x.1999.00997.x. [DOI] [PubMed] [Google Scholar]
- 4.Pritchard JK, Stephens M, Rosenberg NA, Donnelly P. Association mapping in structured populations. Am J Hum Genet. 2000;67:170–181. doi: 10.1086/302959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Satten GA, Flanders WD, Yang Q. Accounting for unmeasured population substructure in case–control studies of genetic association using a novel latent-class model. Am J Hum Genet. 2001;68:466–477. doi: 10.1086/318195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhu X, Zhang S, Zhao H, Cooper RS. Association mapping, using a mixture model for complex traits. Genet Epidemiol. 2002;23:181–196. doi: 10.1002/gepi.210. [DOI] [PubMed] [Google Scholar]
- 7.Zhang S, Zhu X, Zhao H. On a semi-parametric test to detect associations between quantitative traits and candidate genes using unrelated individuals. Genet Epidemiol. 2003;24:44–56. doi: 10.1002/gepi.10196. [DOI] [PubMed] [Google Scholar]
- 8.Marchini J, Cardon LR, Phillips MS, Donnelly P. The effects of human population structure on large genetic association studies. Nat Genet. 2004;36:512–517. doi: 10.1038/ng1337. [DOI] [PubMed] [Google Scholar]
- 9.Campbell CD, et al. Demonstrating stratification in a European American population. Nat Genet. 2005;37:868–872. doi: 10.1038/ng1607. [DOI] [PubMed] [Google Scholar]
- 10.Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006;38:904–909. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- 11.Zhu X, et al. A unified association analysis approach for family and unrelated samples correcting for stratification. Am J Hum Genet. 2008a;82:352–365. doi: 10.1016/j.ajhg.2007.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhu X, et al. Admixture mapping and the role of population structure for localizing disease genes. Adv Genet. 2008b;60:547–569. doi: 10.1016/S0065-2660(07)00419-1. [DOI] [PubMed] [Google Scholar]
- 13.Qin, et al. Interrogating local population structure for fine mapping in genome-wide association studies. Bioinformatics. 2010;26 (23):2961–2968. doi: 10.1093/bioinformatics/btq560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cavalli-Sforza LL, Bodmer WF. The genetics of human populations. Dover Publications; Mineola, New York: 1999. [Google Scholar]
- 15.Epstein MP, et al. A simple and improved correction for population stratification in case–control studies. Am J Hum Genet. 2007;80:921–930. doi: 10.1086/516842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Novembre J, Stephens M. Interpreting principal component analyses of spatial population genetic variation. Nat Genet. 2008;40:646–649. doi: 10.1038/ng.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tang H, et al. Recent genetic selection in the ancestral admixture of Puerto Ricans. Am J Hum Genet. 2007;81(3):626–633. doi: 10.1086/520769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang X, et al. Adjustment for local ancestry in genetic association analysis of admixed populations. Bioinformatics. 2011;27(5):670–677. doi: 10.1093/bioinformatics/btq709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Voight BF, et al. A map of recent positive selection in the human genome. PLoS Biol. 2006;4:e72. doi: 10.1371/journal.pbio.0040072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sabeti PC, et al. Genome-wide detection and characterization of positive selection in human populations. Nature. 2007;449:913–918. doi: 10.1038/nature06250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Crow JF, Kimura M. An introduction to population genetics theory. Harper & Row; New York: 1970. pp. 469–478. [Google Scholar]
- 22.Patterson N, et al. Methods for high-density admixture mapping of disease genes. Am J Hum Genet. 2004;74:979–1000. doi: 10.1086/420871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tang H, et al. Reconstructing genetic ancestry blocks in admixed individuals. Am J Hum Genet. 2006;79:1–12. doi: 10.1086/504302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhu X, et al. A classical likelihood based approach for admixture mapping using EM algorithm. Hum Genet. 2006;120:431–445. doi: 10.1007/s00439-006-0224-z. [DOI] [PubMed] [Google Scholar]
- 25.Sankararaman S, et al. Estimating local ancestry in admixed populations. Am J Hum Genet. 2008;82:290–303. doi: 10.1016/j.ajhg.2007.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Price AL, et al. Sensitive detection of chromosomal segments of distinct ancestry in admixed populations. PLoS Genet. 2009;5:e1000519. doi: 10.1371/journal.pgen.1000519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kang SJ, et al. Genome wide association of anthropometric traits in African and African derived populations. Human Molecular Genetics. 2010;19 (13):2725–2738. doi: 10.1093/hmg/ddq154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Levy D, et al. Genome-wide association study of blood pressure and hypertension. Nat Genet. 2009;41:677–687. doi: 10.1038/ng.384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Patterson N, Price AL, Reich D. Population structure and eigenanalysis. PLoS Genet. 2006;2(12):2074–2093. e190. doi: 10.1371/journal.pgen.0020190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Johnstone I. On the distribution of the largest eigenvalue in principal components analysis. Ann Stat. 2001;29:295–327. [Google Scholar]
- 31.Zou F, et al. Quantification of population structure using correlated SNPs by shrinkage principal components. Human Heredity. 2010;70:9–22. doi: 10.1159/000288706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Price AL, et al. New approaches to population stratification in genome-wide association studies. Nature Reviews. 2010;11:459–463. doi: 10.1038/nrg2813. [DOI] [PMC free article] [PubMed] [Google Scholar]