

Abstract
Comparative integrome analysis has revealed that the most neutral integration pattern among retroviruses is attributed to alpharetroviruses. We chose X-linked chronic granulomatous disease (X-CGD) as model to evaluate the potential of self-inactivating (SIN) alpharetroviral vectors for gene therapy of monogenic diseases. Therefore, we combined the alpharetroviral vector backbone with the elongation factor-1α short promoter, both considered to possess a low genotoxic profile, to drive transgene (gp91phox) expression. Following efficient transduction transgene expression was sustained and provided functional correction of the CGD phenotype in a cell line model at low vector copy number. Further analysis in a murine X-CGD transplantation model revealed gene-marking of bone marrow cells and oxidase positive granulocytes in peripheral blood. Transduction of human X-CGD CD34+ cells provided functional correction up to wild-type levels and long-term expression upon transplantation into a humanized mouse model. In contrast to lentiviral vectors, no aberrantly spliced transcripts containing cellular exons fused to alpharetroviral sequences were found in transduced cells, implying that the safety profile of alpharetroviral vectors may extend beyond their neutral integration profile. Taken together, this highlights the potential of this SIN alpharetroviral system as a platform for new candidate vectors for future gene therapy of hematopoietic disorders.
Introduction
Chronic granulomatous disease (CGD) is a rare congenital immunodeficiency disorder characterized by the inability of phagocytes to eliminate ingested pathogens. The underlying cause of the disease is any of several mutations in the NADPH oxidase enzyme complex resulting in deficient antimicrobial activity of phagocytes. This multiprotein complex plays a pivotal role in microbial killing by reducing molecular oxygen to superoxide, which subsequently reacts to form reactive oxygen species (ROS). The genetic defect renders the patient susceptible to severe, recurrent, and life threatening bacterial and fungal infections, eventually leading to therapy-refractory granuloma and abscess formation even under prophylaxis treatment with antibiotics and antimycotics.1 Allogeneic hematopoietic stem cell transplantation is to date the only curative option for patients with an human leukocyte antigen-matched donor, when conventional care and therapy fail.2 In most cases (~65%) the X-chromosomal CYBB gene encoding for gp91phox, a subunit of the NADPH oxidase, is affected (X-CGD).3 Therefore, reconstitution of oxidase activity by gene delivery of an intact gp91phox to autologous hematopoietic stem cells (HSCs) is a reasonable approach for the treatment of gp91phox-deficient patients lacking a suitable matched stem cell donor. In 2004, our group initiated a phase I/II clinical trial using a gammaretroviral, long-terminal repeat (LTR) driven vector encoding for gp91phox. Autologous CD34+ cells were transduced ex vivo and the genetically modified cells were re-infused into two patients.4 At first, a clear clinical benefit was observed as revealed by the resolution of therapy-resistant lesions in lung and liver. However, long-term observations revealed the occurrence of clonal dominance and a steady decline in transgene expression due to vector silencing. Furthermore, insertional activation of EVI1 led to genomic instability and to myelodysplasia with monosomy 7 in both patients.5 Taken together with the outcomes of other clinical trials for primary immunodeficiencies using LTR-driven gammaretroviral vectors (as reviewed elsewhere),6 these serious adverse events emphasize the need for new approaches for therapeutic gene transfer with enhanced safety profile.
We report here on the evaluation of a recently developed replication-deficient alpharetroviral self-inactivating (SIN) vector with a split packaging system7,8 in the context of X-CGD as a paradigmatic approach in hematopoietic gene therapy. Integration site analysis had revealed that infection with avian sarcoma and leukosis virus (ASLV), an alpharetroviral subfamily, results in a relatively neutral integration pattern as compared with lenti- and gammaretroviral vectors which show a high preference to either integrate into the proximity of transcription start sites or into active genes, respectively.9,10,11 Combined with the SIN design, the alpharetroviral vectors are expected to have a reduced risk of insertional mutagenesis. In addition, alpharetroviruses are per se replication-deficient in mammalian cells,12 high titers can be obtained and even transduction of nondividing cells has been reported.13
Previous evaluation of SIN alpharetroviral vectors already met most expectations: first, enhancer activity was abolished by the SIN LTR configuration in a luciferase reporter assay.7 Even more importantly, in an in vitro immortalization assay Suerth et al.8 observed a reduced incidence of immortalized cells upon transduction of lineage negative (Lin−) murine bone marrow cells with a SIN alpharetroviral vector expressing enhanced green fluorescent protein (eGFP). In addition, most of the immortalized cells showed a reduced fitness in comparison with immortalized cells arising from transduction with gamma- and lentiviral vectors. In vivo evaluation of a SIN alpharetroviral vector driving eGFP under the transcriptional control of the spleen focus forming virus promoter (AS.SFFV.eGFP.PRE*) demonstrated not only the long-term repopulation capacity of SIN alpharetroviral vector transduced cells in a mouse transplantation model but also revealed a favorable integration pattern as compared with lenti- and gammaretroviral vectors, with long-term maintenance of multilineage transgene expression.8 Besides the pivotal role of the integration profile in determining the genotoxicity potential of integrating vectors, aberrant splicing of cellular transcripts into the integrated vector backbone has recently emerged as an additional cause of adverse effects. In a β-thalassemia clinical gene therapy trial as well as in a murine model aberrantly spliced products of endogenous transcripts fused to vector sequences led to either proto-oncogene upregulation or haploinsufficiency of a tumor suppressor gene, respectively.14,15,16 Noteworthy, in most cases engagement of splice sites within the leader sequence contributed to abnormally spliced transcripts.14,15 The lack of canonical splice sites in the SIN alpharetroviral vectors' leader sequence may further contribute to the safety profile of this type of vectors.7
Here, we demonstrate that SIN alpharetroviral vectors generate high titers and stable gp91phox expression, sufficient to functionally rescue NADPH oxidase activity in the human X-CGD PLB-985 cell line as well as in primary murine and human X-CGD HSCs, even at low vector copy numbers (VCN). In addition, no induction of aberrantly spliced transcripts upon intronic integration was observed in PLB-XCGD cells. Transplantation of transduced Lin− bone marrow HSCs in a murine model of X-CGD resulted in reconstitution of ROS-producing neutrophils in peripheral blood. Myeloid differentiation of transduced primary human X-CGD CD34+ cells either in vitro or after transplantation into a humanized mouse model led to the reconstitution of gp91phox expression and NADPH oxidase activity in the range of wild-type cells. Taken together, this report supports the application of SIN alpharetroviral vectors to rescue a monogenetic disease phenotype upon transplantation of ex vivo modified HSC.
Results
Vector design and efficient gp91phox expression in a human X-CGD model cell line
To obtain an optimal SIN alpharetroviral construct providing stable and high expression of gp91phox, we combined three previously described elements known to enhance transgene expression in vitro and in vivo: first, we chose a codon-optimized transgene (gp91s), which was shown to improve viral titers and expression levels,17 thus reducing the number of vector copies required for an effective functional rescue of CGD. For the same reasons, an optimized posttranslational regulatory element (oPRE) devoid of any open reading frames and X protein promoter activity was placed downstream of the gp91phox coding region.18 Finally, we chose the human elongation factor-1α promoter in its intronless, 240 bp short version (EFS)19 to drive transgene expression. This regulatory element drives physiological expression levels without perturbing the expression of genes proximal to the vector integration site.20,21,22 Furthermore, the EFS promoter was shown to be less prone to downregulation than other promoters within the context of lentiviral vectors.23 We evaluated the performance of this SIN alpharetroviral vector (AS.EFS.gp91s, Figure 1a) in comparison with its corresponding SIN lentiviral vector analogue LV.EFS.gp91s. Transduction efficiencies as well as vector titers were comparable between both constructs reaching 2.56 ± 0.88 × 108 T.U./ml (n = 5) for AS.EFS.gp91s (LV.EFS.gp91s: 2.53 ± 0.49 × 108 T.U./ml) as estimated after transduction of the human myeloid PLB-XCGD cell line24 (Supplementary Figure S1c online). The levels of gp91phox expression, as estimated from the mean fluorescence intensity (MFI), were similar for both vectors at comparable mean vector copy numbers (0.39 ± 0.23 vs. 0.42 ± 0.05 for AS.EFS.gp91s and LV.EFS.gp91s, respectively) (Figure 1b) with an estimate of more than 90% of the integrated copies expressing the gp91s transgene (AS.EFS.gp91s 90.2 ± 2.0% vs. LV.EFS.gp91s 91.1 ± 5.9%; P = 0.88, Figure 1c and Supplementary Figure S1d online). As expected, the percentage of gp91phox expressing PLB-XCGD cells and vector copy numbers increased with increasing multiplicity of infection (Figure 1d and Supplementary Figure S1a,b online). Noteworthy, the mean fluorescence intensities (MFI) of the gp91phox positive populations and hence the expression levels were in the range of the parental wild-type cell line PLB-985, even at low VCNs (Figure 1d). Analysis of transduced clonal populations obtained by limiting dilution revealed expression levels independent of VCN and cell differentiation state (Figure 1e), suggesting that maximal gp91phox expression can be achieved with a single AS.EFS.gp91s integrant.
Figure 1.

Vector constructs and gp91phox expression in PLB-XCGD cells. (a) Schema of the self-inactivating (SIN) alpharetroviral (SIN-α) and SIN lentiviral (SIN-LV) proviruses used in this study. The transgene cassette contains the elongation factor-1α promoter in its intronless, 240 bp short version (EFS) promoter and the codon-optimized gp91s and is followed by a woodchuck hepatitis virus posttranscriptional regulatory element (oPRE). In addition, indicated are splice donor and acceptor site (SD and SA), Rev-responsive element (RRE), central polypurine tract (cPPT) and PPT, the direct repeat element (DRE) for the alpharetroviral provirus and remaining viral gag-coding sequences (Δgag) for the lentiviral construct. (b) Human myelomonocytic PLB-XCGD cells transduced with either SIN alpharetroviral vector (alpha) or its SIN lentiviral (LV) analogue were subjected to comparative expression analysis by flow cytometry (n = 3; mean + SD). Transduced populations were analyzed for MFI in the gp91phox positive subfraction (MFI (gp91+)) undifferentiated (undiff; CD11b-) and after 8 days of myeloid differentiation (diff; CD11b+) in parallel. Mean percentages of gp91phox positive cells upon differentiation are indicated below the corresponding bar's label. (c) Comparative analysis of effective transcription on proviral level by a SIN alpharetro- or equivalent lentiviral vector. Transduced cell populations (n = 4) with varying percentage of gp91phox expressing cells were FACS-sorted into gp91phox positive and negative populations, and subsequently analyzed for vector copy numbers (VCN). To estimate the fraction of transcriptionally active proviral integrations, the VCN of the fraction expressing gp91phox was set in relation to the corresponding cumulative VCN of positive and negative fractions. (d) PLB-XCGD cells were transduced (td) with the VSV-G pseudotyped AS.EFS.gp91s vector at different multiplicity of infection (MOI) as indicated. Percentage of gp91phox positive cells was determined 5 days post transduction (ptd) by flow cytometry. Representative blots from flow cytometric analysis of transduced PLB-XCGD cells. Mean fluorescence intensities (MFI) of gp91phox positive populations were as indicated. Stained nontransduced (ntd) PLB-XCGD and PLB-985 wild-type (wt) cells served as controls. Mean VCNs were determined at day 7 after transduction by qPCR. (e) Clonal populations (n = 32) of AS.EFS.gp91s transduced cells established by limited dilution were grouped according to VCN. Expression levels compared to PLB-XCGD control were determined as ΔMFI (mean + SD) by flow cytometry of either undifferentiated cells (undiff; CD11b−) or cells subjected to 8 days of differentiation (diff; CD11b+).
Long-term expression of gp91phox and functional rescue of the CGD phenotype
Since a previous report implicated that retroviral delivery of gp91phox was prone to silencing in PLB-XCGD cells,25 we monitored alpharetroviral gp91phox expression in this cell line over 150 days. In order to minimize the risk of masking silencing due to expression from several vector copies per cell, PLB-XCGD cells were transduced at a low multiplicity of infection of 0.1 followed by immunomagnetic sorting of gp91phox-expressing cells resulting in a cell population with an average vector copy number of 1.3. Long-term analysis of this population revealed stable expression levels (MFI) and percentages of gp91phox positive cells (Figure 2a,b), recapitulating similar observations made with an alpharetroviral vector expressing eGFP under the transcriptional control of the EFS promoter.8 Upon differentiation and phorbol myristate acetate stimulation of the transduced and sorted PLB-XCGD cells restoration of NADPH oxidase activity to levels similar to that of the wild-type cell line (P = 0.498) were observed as assessed by superoxide production over time in a quantitative cytochrome C reduction assay (Figure 2c). Even after 150 day culture, no significant changes in superoxide production (P = 0.645) were observed in this population after differentiation and subsequent functional analysis (Figure 2c). Taken together, the AS.EFS.gp91s vector not only provides efficient transduction and long-term stable gp91phox expression but also complete functional rescue of the X-CGD phenotype at low VCN in a model X-CGD cell line in vitro.
Figure 2.
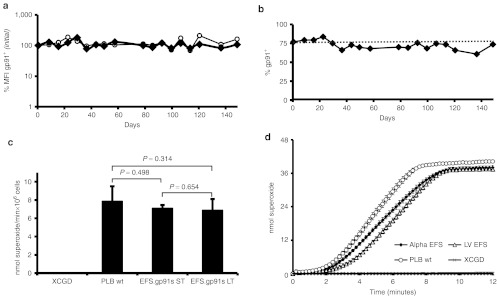
Long-term analysis of gp91phox expression and functional evaluation in PLB-XCGD cells. X-linked chronic granulomatous disease (X-CGD) PLB-985 cells were transduced with AS.EFS.gp91s at a MOI of 0.1 and transgene expressing cells were enriched by magnetic sorting. (a) Long-term analysis of gp91phox expression levels (MFI) in sorted cells (filled symbols) as compared with that of wild-type PLB-985 cells (open symbols). (b) Percentage of gp91phox positive cells in the sorted population over time. (c) Cytochrome C assay comparing superoxide production per minute in transduced cells 5 weeks (ST) and 20 weeks (LT) after transduction. Superoxide production was compared with that of wild-type cells after myeloid differentiation. Three individually differentiated populations were analyzed in duplicates. (d) Kinetics of cytochrome C reduction determining superoxide production upon PMA stimulation after 8 days of myeloid differentiation of PLB-XCGD ntd, PLB-985 and sorted PLB-XCGD td AS.EFS.gp91s or LV.EFS.gp91s as indicated. Error bars = mean + SD. PMA, phorbol myristate acetate.
We also assessed the performance of the SIN alpharetroviral vector in comparison with its lentiviral equivalent in terms of functional activity. Therefore, we chose two sorted gp91phox+ cell populations with similar mean VCN (alpha: 1.27 vs. LV: 1.30; Supplementary Figure S1d online) and subjected them to differentiation and subsequent cytochrome C assay. Both cell populations revealed comparable kinetics for superoxide production close to wild-type levels (Figure 2d). Superoxide production in cells transduced with AS.EFS.gp91s was equivalent to that of LV.EFS.gp91s transduced cells (5.3 vs. 5.5 nmol/min 106 cells−1, respectively). Hence, the vectors were not only comparable in terms of titer and expression levels, but also in terms of functional activity.
Lack of aberrantly spliced transcripts in alpharetrovirally transduced cells
Since aberrantly spliced cellular transcripts have recently been detected caused by lentiviral vector integrations, we addressed the occurrence of aberrant splicing in cells transduced with SIN alpharetroviral vectors. In contrast to lentiviral vectors, SIN alpharetroviral vectors are devoid of canonical splice sites in the leader region,7 suggesting the absence of cellular exon–vector fusion transcripts in cells transduced with these vectors. However, recent reports have revealed that cryptic splice sites are also engaged in splicing leading to aberrant cellular exon–vector fusion transcripts.14,15,26 Therefore, we analyzed the SIN alpharetroviral leader sequence in silico for cryptic splice sites using the NetGene2 software (http://www.cbs.dtu.dk/services/NetGene2/) as this procedure led to the identification of cryptic splice sites in the leader region of lentiviral vectors involved in aberrant splicing.15 In total, no splice donor and seven cryptic splice acceptors with intermediate score (0.2 < score < 0.5) were predicted within the alpharetroviral leader sequence (Table 1). To identify potential aberrantly spliced transcripts in alpharetrovirally transduced PLB-XCGD cells, a clonal population harboring five well-defined intronic integrations was selected for this analysis. A similar procedure was followed for the analysis of aberrant splicing in lentivirally transduced cells (Table 2).
Table 1. Internal vector splice sites.
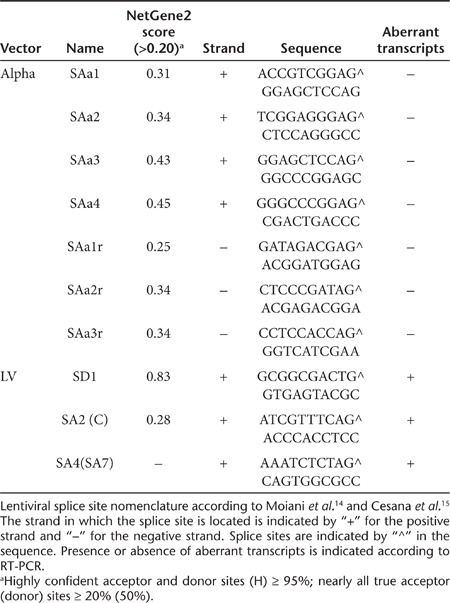
Table 2. Intronic vector integrations in monoclonal PLB-XCGD.
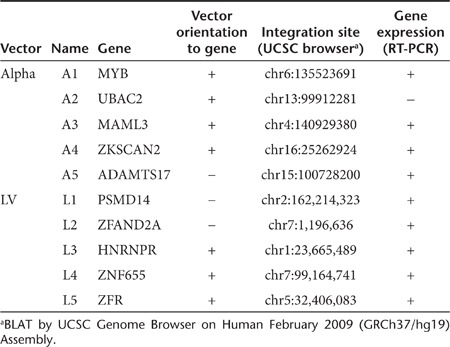
First, we asked if the genes targeted by the integration of alpharetroviral or lentiviral vectors were actively transcribed. Using RT-PCR primers spanning exons flanking the integration sites, transcripts were detected for all targeted loci with exception of A2 (Table 2; Supplementary Figures S2a,b online). For the detection of cellular exon–vector fusion transcripts in transduced cells RT-PCR primers spanning the upstream exon adjacent to the integration sites and the in silico predicted splice acceptor sites for the alpharetroviral vector were selected, whereas previously published primer sets15 were used for the evaluation of aberrant splicing in lentivirally transduced cells (Figure 3a,b; Supplementary Table S1 online). In alpharetrovirally transduced cells no cellular exon–vector fusion transcripts were detectable (Supplemenatry Figure S2c online). Using the same cDNA sample, sustained expression of the targeted genes was confirmed by PCR (Supplementary Figure S2a online), indicating that the lack of exon–vector fusion transcripts was not due to inefficient cDNA synthesis. On the contrary, cellular exon–vector fusion transcripts were detected (and confirmed by sequencing) in all LV integrants orientated in sense to the targeted gene (Figure 3b and Supplementary Figure S2d online).
Figure 3.

Analysis of aberrant transcripts induced by self-inactivating (SIN) alpharetroviral or SIN lentiviral vectors by RT-PCR and 5′RACE. (a and b) Monoclonal PLB-XCGD populations harboring either SIN-α or SIN-lentiviral (LV) integrations in intronic genomic sequences were subjected to RT-PCR for detection of aberrantly spliced transcripts. Schematical representation of proviral intronic integrations in sense orientation to the affected gene for SIN-α (a) or SIN-LV (b). Splice donor (SD) and acceptor sites (SA) mentioned in Table 1 are indicated. The internal promoter is indicated by a thick black arrow whereas primers used for RT-PCR are drawn as narrowed black arrows (compare Supplementary Table S1 online). The open arrow heads denote the primers used for positive control on gDNA together with the alpharetroviral primers used for detection of cellular exon–vector fusion transcripts (compare Supplementary Figure S2c online). Cryptic splice sites as defined in silico by the NetGene2 software are shown above (sense orientation) or below (reverse orientation) of the proviral schemes. (b) Schematic representation of the aberrant splice events between the cellular exon and the vector sequence are shown below the lentiviral provirus. The sequence of the retrieved fusion transcripts is shown at the right (sequence centered at the exon–vector fusion ± 10 bp). Exon sequences are in black, vector sequences are shown in white lettering. Transcript labels according to Supplementary Figure S2d online. (c) RNA of polyclonal PLB-XCGD cells transduced with either AS.EFS.gp91s or LV.EFS.gp91s and positively sorted for gp91phox expression was subjected to 5′RACE. Leader specific primers (alpha, LV) were used according to the respective vector transduced. RNA derived from nontransduced PLB-XCGD served as negative control.
We then extended our analysis to polyclonal PLB-XCGD populations. Therefore, we chose two transduced populations that matched in VCN of either AS.EFS.gp91s or LV.EFS.gp91s (VCN: 2.92 and 2.94, respectively) after FACS sorting of gp91phox positive cells. A 5′RACE was performed to recover aberrantly spliced transcripts originating from endogenous genes spliced to cryptic or canonical splice sites within the proviral leader sequences as described above. Transfected HEK293T cells served as positive control for this approach (Supplementary Figure S2e,f online). In line with our previous findings, no RT-PCR products containing cellular exons fused to alpharetroviral sequences were found in AS.EFS.gp91s transduced cells whereas multiple cellular exon–LV fusion transcripts were identified in LV.EFS.gp91s transduced cells (Figure 3c and Supplementary Figure S2g and Tables S2–S4 online).
The abundance of lentivirally induced aberrant splice products recovered by these approaches as compared with the lack of detectable cellular exon–vector fusion transcripts in SIN alpharetroviral vector transduced cells indicate that the contribution of the alpharetroviral vector backbone to splice-mediated genotoxicity may be lower than that observed for lentiviral vectors.
In vitro and in vivo expression of gp91phox and functional analysis in primary murine X-CGD cells
Next we transduced murine Lin− cells obtained from X-CGD mice with AS.EFS.gp91s at a moderate multiplicity of infection of 15. This resulted in 33% gp91phox positive cells as measured 5 days after transduction by flow cytometry (Figure 4a). Transduction of hematopoietic precursors was confirmed by superoxide positive colonies in an nitroblue tetrazolium (NBT) assay obtained after plating transduced cells in semi-solid media. The percentage of NBT-positive colonies matched with 35 ± 6% (n = 4) the initial gp91phox expression in Lin− cells (Figure 4a,b). Moreover, five out of seven (71.4%) provirus positive colonies showed functional reconstitution of superoxide activity (Figure 4c). The lack of superoxide detection in the two vector positive colonies can be explained by incomplete myeloid differentiation and/or low gp91phox expression. Please note that only 30% of the hematopoietic colonies derived from wild-type mice showed superoxide activity in this particular assay.
Figure 4.

Transduction of murine X-linked chronic granulomatous disease (X-CGD) lineage negative bone marrow (BM) cells and nitroblue tetrazolium (NBT) assay. (a) Lin− BM cells from gp91phox-deficient mice were transduced 2 days after isolation with an MOI of 15 and analyzed for gp91phox expression 5 days after transduction by flow cytometry. (b) Individually transduced cell populations (n = 4; mean + SD) were differentiated toward myeloid colony forming units (CFU) in semi-solid methylcellulose in duplicates. After 10 days functional analysis for ROS production was performed by a NBT assay upon PMA stimulation. Count of NBT positive CFUs as well as total CFUs was determined and set in relation to the total number of colonies in the plates. (c) Genomic DNA was harvested from NBT positive and negative CFUs (as indicated) and subjected to an alpharetro-provirus specific PCR (176 bp) in parallel with a murine β-actin specific reaction (138 bp). Only β-actin positive colonies are shown. PMA, phorbol myristate acetate.
AS.EFS.gp91s transduced X-CGD Lin− cells (CD45.2+) were transplanted into five lethally irradiated Bl6.SJL recipient mice (CD45.1+) to evaluate the vector's potential in an in vivo setting. Ten weeks posttransplantation donor chimerism, lineage distribution, gp91phox expression and functional reconstitution of superoxide production were determined in hematopoietic cell subsets. Donor contribution to hematopoietic reconstitution was high in peripheral blood, spleen, total bone marrow, and Lin− cells with a mean of 87.2 ± 2.2% donor-derived cells (Supplementary Figure S3b online), while hematopoietic lineage distribution was analogous to that of control animals (Supplementary Figure S3c online). Up to 27% of the peripheral donor granulocytes (CD11b+ and Gr-1+) were gp91phox positive (mean 17.6 ± 7.5%) with a median vector copy number of 0.75 (Figure 5a). In accordance, the percentage of oxidase positive donor granulocytes was 18.3 ± 11.4% with a maximum of 34% as assessed by a dihydrorhodamine (DHR) assay (Figure 5a). Therefore, the percentages of oxidase positive granulocytes and gp91phox expression in this fraction directly correlated (R2 = 0.98; Supplementary Figure S3d online). ROS production levels in myeloid cells as determined by MFI reached up to 20.2% of wild-type levels with a mean of 12.5 ± 4.2% (Figure 5b). Similarly, ROS production as determined by the NBT assay was 44.5 ± 20.5% from controls (Supplementary Figure S4a online) with a good correlation between provirus positivity and ROS production (Supplemenatry Figure S4b online). Furthermore, the fraction of rhodamine positive cells correlated with the percentage of NBT+ colonies (R2 = 0.89; Supplementary Figure S3e online).
Figure 5.

Reconstitution of gp91phox expression and NADPH oxidase activity in vivo. Alpharetrovirally transduced murine X-linked chronic granulomatous disease (X-CGD) Lin− cells were transplanted into SJL recipient mice (n = 5) and analyzed 10 weeks after transplantation for gp91phox expression by flow cytometry. Genomic DNA was isolated and vector copy numbers (VCN) was determined by qPCR. (a) Percentages of gp91phox expressing (gp91+; black) and oxidase positive (rho123+; white) granulocytes (CD11b+ Gr-1+) in CD45.2+ peripheral blood cells are plotted side by side for each individual animal. Expression data was obtained by intracellular FACS staining and oxidase activity was measured in a FACS based dihydrorhodamine assay by rho123 capture in granulocytes upon phorbol myristate acetate (PMA) stimulation. A control without PMA stimulation is shown for animal #2. Mean VCNs are indicated for each animal. (b) The mean fluorescence intensities (MFI) of rho123+ myeloid cells (CD45.2+ CD11b+) are shown for wild-type (n = 2) and alpharetrovirally treated transplanted animals (n = 5). Nontransduced cells form gp91−/− mice (X-CGD) serve as controls (n = 2). (c) gp91phox expression in peripheral blood, spleen, and bone marrow subsets for animal #7 (black line) is shown in overlay with the negative X-CGD control (gray). Only CD45.2+ cells were considered for this analysis. Granulocytes (Gran; CD11+Gr-1+), Monocytes (Mono; CD11+Gr-1-), T-cells (CD3e+), and B-cells (B220+). CD45.2+, cells indicate overall expression in donor cells; Lin−, Sca1−, cKit− (L−S−K−), committed hematopoietic cells; L−S−K+: committed myeloid progenitors; L−S+K-: include early committed lymphoid progenitors; L−S+K+: enriched for HSC and multipotent progenitors; PB, peripheral blood.
We also analyzed gp91phox expression in different hematopoietic lineages as identified by surface markers (Supplementary Figure S3a online). Noteworthy, gp91phox expression in donor cells was observed mainly in the myeloid compartment (granulocytes and monocytes) in peripheral blood, spleen, and total bone marrow (BM) with low expression levels in T- (CD3+) and B-cells (B220+) (Figure 5c and Supplementary Figure S4c online). This bias is consistent with previous in vivo data obtained with lentiviral vectors expressing eGFP driven by the EFS promoter.27 In addition, gp91phox expression was also detected at low levels in all committed progenitor populations of the BM, including the L− S+ K- subset containing lymphoid progenitors (Figure 5c and Supplementary Figure S4c online).
Rescue of X-CGD phenotype in human primary X-CGD cells
To test whether our construct could also rescue gp91phox expression and oxidase activity in primary human X-CGD cells, we transduced CD34+ cells obtained from an X-CGD patient with the AS.EFS.gp91s vector. An initial transduction efficiency of 26.2% was measured on day 3 as estimated by the number of gp91phox-expressing CD34+ cells (Figure 6a). This value correlated well with a vector copy number of 0.37 measured on genomic DNA isolated on day 10 after transduction.
Figure 6.

Phenotypic correction of human primary X-linked chronic granulomatous disease (X-CGD) cells by self-inactivating (SIN) alpharetroviral gp91phox expression. (a) Human CD34+ X-CGD cells were transduced with AS.EFS.gp91s one day after immunomagnetic enrichment. Transgene expression was analyzed in CD34+ cells 3 days after transduction by surface staining (FACS). The transduced CD34+ cells (black line) are shown in overlay with the negative CD34+ X-CGD control (grey). (b) Functional reconstitution of superoxide activity in colony forming cells derived from transduced CD34+ cells were plated on methycellulose 2 days after transduction and superoxide activity was determined by the nitroblue tetrazolium (NBT) assay. (c) Transduced CD34+ X-CGD (black line) and control cells (X-CGD = light gray; wild-type = dark gray) were cultured in the presence of human granulocyte-colony stimulating factor to induce myeloid differentiation and analyzed for extracellular gp91phox expression in CD11b+ cells. Analysis was performed 10 days after transduction. (d) After 25 days of differentiation (day 28 after transduction) functional activity was assessed by intracellular rhodamine123 (rho123) accumulation upon phorbol myristate acetate (PMA) stimulation by flow cytometry. Percentages and MFI of rho123+ cells in the CD11bhigh+ subpopulations are indicated. (e) Superoxide production (day 27 after transduction) was also measured by cytochrome C reduction upon PMA stimulation (n = 2). Values for AS.EFS.gp91s transduced cells are given relative to wild-type control (f) Transduced X-CGD CD34+ cells as well as control cells were transplanted into NSG mice 2 days after transduction. Eight weeks later CD34+ cells were isolated from bone marrow by magnetic sorting and cultivated in differentiating conditions for 3 weeks before analysis for gp91phox expression by flow cytometry. The viable CD11b+ hCD45+ subfraction is plotted for X-CGD (light gray), wild-type (dark gray) and transduced X-CGD cells (black line).
Reconstitution of superoxide activity was estimated from methylcellulose colonies plated 2 days after transduction. Accordingly, 20% of the colonies derived from transduced CD34+ cells were found to reduce NBT to its insoluble tetrazolium derivate (Figure 6b), whereas 68% of the colonies derived from CD34+ cells obtained from healthy donors were found to be NBT positive. Thus compared with controls, 1/3 of the transduced X-CGD CD34+ derived colonies showed superoxide production at 1/3 VCN relative to control cells. Upon differentiation of the transduced cells in liquid culture in the presence of human granulocyte-colony stimulating factor we observed gp91phox expression in 17.4% of the CD11b+ cells with expression levels (MFI) corresponding to 75% of that seen in wild-type cells (Figure 6c). In addition, DHR and cytochrome C assays were performed on day 24/25 of differentiation in liquid culture to evaluate the level of oxidase activity. At this time point, 7.2% rhodamine123+ cells in the CD11bhigh+ fraction gave rise to superoxide production at levels corresponding to 67% of wild-type levels (compare relative MFI of rho+ cells in Figure 6d). Noteworthy, a subpopulation of rhodamine positive cells within the transduced CD11b+ cells (rhohigh+ fraction) showed a high capacity to oxidize dihydrorhodamine123 to rhodamine123 to levels equivalent to wild-type levels (MFI 61,106 vs. 61,017 for transduced vs. wild-type cells, respectively; Figure 6d). These observations were confirmed in a quantitative cytochrome C assay. Human CD11b+ cells expressing gp91phox were found to generate superoxide at levels corresponding to 53% of wild-type levels (wt = 3.8, X-CGD td AS.EFS.gp91s = 2.0 nmol/min 106 CD11b+gp91+cells−1, Figure 6e).
In parallel, we transplanted human primary cells (wt, X-CGD and AS.EFS.gp91s transduced X-CGD cells) into NSG mice on day 2 after transduction. Human CD34+ cells were isolated from BM 8 weeks after transplantation and further subjected to granulocytic differentiation before analysis. We observed 15.3% gp91phox positive human CD45+CD11b+ cells, with expression levels corresponding to 56% of wild-type levels (compare MFI values) after a total of 11 weeks after transduction (Figure 6f). In conclusion, the AS.EFS.gp91s construct provides expression and functional rescue of X-CGD in human cells in vitro at moderate VCN and sustained transgene expression in a relevant human xenograft model.
Discussion
In this study, we evaluated the potential of the SIN alpharetroviral vector system for the gene therapy of CGD. Although several vector systems have been already described for the treatment of this disease,4,28,29,30 several features attributed to alpharetroviral vectors compare favorable to other gene delivery systems including the comparatively neutral integration site selection, the reduced risk to integrate in actively transcribed genes and the low genotoxicity potential as revealed by an in vitro immortalization assay. Further in a murine transplantation model involving secondary transplants with an eGFP expression construct (spleen focus forming virus (SFFV) promoter) already showed the absence of clonal dominance for alpharetroviral vectors. In this study, extensive analysis of integration sites in proximity to annotated cancer genes was performed before and after primary and secondary transplantation revealing only a few overlapping clones between the different integromes. This meta-analysis did not provide any evidence for dominant clones persisting between the different populations analyzed.7,8 Furthermore, the lack of canonical splice acceptor and donor sites within the leader region, including packaging signals may provide an additional argument for selecting alpharetroviral vectors as gene delivery vehicle as recent studies have shown that aberrant splicing into or out of the lentiviral vector backbone may contribute to vector genotoxicity.14,15,16,26 Our data on clonal cell populations harboring five SIN alpharetroviral vector integrations in introns together with our observation in polyclonal populations showed no evidence for aberrantly spliced transcripts. In contrast, all sense-oriented intronic lentiviral integrations led to aberrant fusion transcripts, in line with the previous reports.14,15,26 Therefore, the low genotoxicity attributed to alpharetroviral vectors can now be explained by two vector inherent characteristics, presumably acting in a synergistic fashion: the almost neutral integration profile and the low risk of aberrant splicing. However, extended high throughput analysis of the transcriptome of alpharetrovirally transduced cells are warranted to clarify if SIN alpharetroviral vectors are indeed devoid of any cryptic splice sites. Nonetheless, our data indicate that SIN alpharetroviral integrations in intronic regions are less likely to generate aberrant splice products than SIN lentiviral vectors and thus, further encourage their evaluation for gene therapy applications.
For the construction of a suitable alpharetroviral vector for X-CGD, we combined a codon-optimized gp91phox transgene maximizing titer and transgene expression with the EFS promoter as no in vitro immortalization of murine bone marrow cells has been observed using this promoter in different vector contexts.8,20,21,27 In addition, neither in the episomal nor in the integrated context the EFS promoter showed significant deregulation of surrounding genes20,22 and this promoter has already been included in gammaretroviral vectors31 for clinical trials (NCT01129544; NCT01175239) addressing X-linked severe combined immunodeficiency and has been proposed for lentiviral vectors to treat X-linked severe combined immunodeficiency and Wiskott–Aldrich syndrome.21,32 Based on these facts, we decided intentionally against the use of a myeloid specific promoter, although highly promising candidate promoters are available.28,30 The rationale of targeted expression by specific promoters is to avoid transcriptional deregulation of surrounding genes mainly in stem and progenitor cells and consequently to avoid potential side effects as these cell populations are more susceptible to insertional transformation than more mature cells.33,34 In addition, tissue specific promoters may circumvent transgene expression in cell populations, in which the transgene is potentially toxic or just not required. For CGD, there is no experimental evidence of toxicity related to ectopic gp91phox expression in this cell fraction (as reviewed elsewhere).35 Nevertheless, since gp91phox expression does not seem to be required for stem cell functions, a microRNA detargeting approach in combination with the EFS promoter may be considered.36 For example, by including the target sequence for miR-126, which is abundantly expressed in HSCs but not in myeloid cells, EFS-driven gp91phox expression could be restricted to differentiated cells to avoid a potential toxicity of the transgene in primitive progenitor cells.37
Transduction of PLB-XCGD cells with the AS.EFS.gp91s vector yields high transduction efficiencies and wild-type expression levels at low VCN. Furthermore, ~90% of integrated copies were found to be transcriptionally active and the maximum of gp91phox expression could be accomplished with a single copy as shown by the analysis of single-cell-derived clonal populations. In the murine transplantation model the mean numbers of gp91phox-expressing myeloid cells ranged between 15 and 20% in peripheral blood and bone marrow (Supplementary Figure S4c online), at a median VCN of 0.75. Likewise, transduction of primary human X-CGD cells resulted in a mean VCN of 0.37 leading to 17% detectable gp91phox expression in CD11b+ cells as assessed 10 days after transduction (Figure 6). As it has been described for other retroviral vectors including lentiviral vectors,38 the discrepancy between VCN and gp91phox expression suggests multiple integrants per cell or silent vector copies caused by integration into heterochromatin and transcriptionally silent regions rather than progressive vector silencing. The sustained expression of gp91phox in vitro and in vivo as shown here, together with previous data on secondary transplants of alpharetrovirally transduced BM cells and our own colony assay studies, in which most of the ROS-negative colony forming units-colonies lacked proviral integrations, argue against the SIN alpharetroviral vectors being more prone to silencing than other integrating vectors. This extends recent observations made with vectors expressing a marker gene (eGFP).8
At a vector copy number of 1.3 the AS.EFS.gp91s vector provided stable gp91phox expression and complete functional rescue in PLB-XCGD cells as assessed by superoxide production in gp91phox+ enriched population, even after long-term culture over 150 days. Therefore, our novel construct provides complete and stable rescue of the X-CGD phenotype of a human model cell line that had been shown previously to silence gammaretroviral vectors.25 This encouraged us to further evaluate the vector's potential in primary cells. Transplantation of AS.EFS.gp91s transduced X-CGD Lin− cells into lethally irradiated recipients showed up to 34% of oxidase positive granulocytes with a direct correlation between gp91phox expression levels and superoxide production (R2 = 0.98). Unexpectedly, gp91phox expression was high in granulocytes and monocytes but severely reduced in lymphoid cells at comparable vector copy numbers suggesting a preferential expression of the EFS promoter in myeloid cells. These observations might be either due to cell type specific regulatory mechanisms in terms of RNA or protein stability, preferential gene transfer into myeloid progenitors, differential promoter activity or a combination of these mechanisms. Our data on Lin− cells in transplanted animals revealing expression in all progenitor subsets at the same VCN as in the periphery argues against preferential vector delivery to myeloid progenitors. Moreover, expression in B-cells of transplanted animals was only slightly above the detection limits despite the fact that in wild-type B lymphocytes gp91phox is expressed.39 Therefore, the lack of gp91phox in B-cells of transplanted animals cannot be explained by effects caused by the cellular machinery conferring RNA and protein stability to gp91phox. Our results rather suggest an enhancement of EFS promoter activity in myeloid cells. In fact, the EFS promoter contains putative transcription factor binding sites typically found in myeloid promoters such as C/EBP in combination with SP1.20,28 Similar observations were reported in a recent study in which EFS-driven eGFP expression was also enhanced in murine myeloid cells as compared with other leukocytes in the peripheral blood of transplanted mice.27 In line with this, the EF1α promoter has been shown to mediate higher GFP expression levels in human CD14+ myeloid cells derived from bone marrow of transplanted NSG mice as compared with human lymphocytes.40
The expression of gp91phox was retained in myeloid cells derived from transduced X-CGD CD34+ cells in vitro and in vivo. From initially 26% gp91phox+ expressing CD34+ cells transgene expression was retained in 17% of the CD11b+ cells derived thereof after in vitro differentiation and in 15% of the CD11b+ cells derived from transplanted NSG animals. These values corresponds to 75 and 56% of the expression levels found in CD11b+ cells derived from wild-type CD34+ cells in vitro and in vivo, respectively. This was accomplished at a low VCN (≤ 0.4) and provided oxidase activity in the range of wild-type cells at the single cell level in vitro (Figure 6d) and thus within the range expected to provide a clinical benefit.4,35,41 Indeed, in a recent study comprising a total of 287 CGD patients (67.9% X-CGD patients), a strong association between long-term survival and residual superoxide production was observed.42 Accordingly, a survival benefit is expected even at levels as low as 1% of normal superoxide production. Comparing these results with our data obtained in the cytochrome C reduction assay, our absolute values are several fold higher than the minimal levels used by Kuhns et al.42 to define long-term survival. From this analysis, we anticipate a significant survival benefit for patients receiving an autologous transplant gene modified with the SIN alpharetroviral vector described here.
In summary, our work reveals that functional correction of a monogenetic disease to clinically relevant levels can be achieved at low vector copy numbers with a SIN alpharetroviral vector and highlights the potential of the alpharetroviral vector system for gene therapy of hematopoietic disorders.
MaterialS and Methods
Retroviral vectors and vector production. The alpharetroviral vector transfer plasmid pAS.EFS.gp91s.oPRE was derived from pAS.EFS.eGFP.PRE*.7,8 The transgene-wPRE cassette was substituted with the codon-optimized gp91s cDNA21 combined with the oPRE element18 by excision from pAS.fes.gp91s.oPRE, a derivate of pSER11fes91-W,48 using the restriction sites NcoI and HindIII. For the lentiviral analogue a full cassette exchange of EFS.gp91s.oPRE was performed by blunt-end ligation of the NotI and HindIII digested insert into pHR′.SEW after XhoI and EcoRI treatment and subsequent blunting. All restriction enzymes were purchased from New England Biolabs (NEB, Frankfurt, Germany).
Cell-free viral supernatants were generated by transient co-transfection on HEK293T cells as described before.7 Briefly, the alpharetroviral vector pAS.EFS.gp91s.oPRE (hereafter in proviral context referred to as AS.EFS.gp91s), the alpharetroviral codon-optimized gag/pol helper plasmid (pcDNA3.alpha.gag/pol.co) and as envelope plasmid pMD2.VSV.G (Addgene plasmid 12259: pMD2.G) encoding the glycoprotein of vesicular stomatitis virus (VSV-G) or pcoPE01 (kindly provided by Axel Rethwilm, Würzburg, Germany) encoding a modified prototype foamy virus envelope protein (PFV env) were transfected in a molar ratio of 3:1:1 either by calcium phosphate in the presence of 40 mM chloroquine or the polyethylene imine precipitation method according to standard procedures. Viral supernatants were harvested 40 hours after transfection, sterile filtered (0.22 µm pore-size filter; Millipore, Schwalbach, Germany) and concentrated by ultracentrifugation over a 20% (w/v) sucrose cushion (50000g, 2 hours, 4 °C). For resuspension, StemSpan SFEM serum-free medium (StemCell Technologies, Grenoble, France) was used without any supplements and collected supernatants were stored at −80 °C. Vector titers were determined in serial dilutions of viral supernatant by spinfection (800×g, 30 minute, 32 °C) of PLB-XCGD cells in the presence of 4 mg/ml protamine sulfate (Sigma-Aldrich, Munich, Germany) and analyzed 4–5 days after transduction by flow cytometry (BD FACSCanto II, BD FACSDiva 6.1.3 software, Becton Dickinson, Heidelberg, Germany) applying the fluorescein isothiocyanate (FITC)-conjugated murine anti-human gp91phox monoclonal 7D5 (MBL international no. D162-4).
Cell lines and culture conditions. The human X-CGD PLB-985 cell line was derived from the PLB-985 myelomonocytic leukemic cell line in which the CYBB gene encoding for gp91phox was disrupted by homologous recombination.24 Both cell lines and their derivatives were grown in RPMI 1640 (Gibco/Life Technologies, Karlsruhe, Germany) complemented with 10% heat-inactivated fetal calf serum (FCS; PAN-Biotech, Aidenbach, Germany), 4 mmol/l L-glutamine and antibiotics (100 U/ml penicillin and 100 µg/µl streptomycin; all from PAA Laboratories, Pasching, Austria). For long-term culture cell populations were kept in 12-well culture plates, passaged every 3–4 days and analyzed as indicated. HEK293T cells (ATCC) were cultured in Dulbecco's modified Eagle's medium (Gibco) containing identical supplements as stated above. For granulocytic differentiation PLB-985, PLB-XCGD and derivatives were individually cultured (n ≥ 3) at a density of 2.0–2.5 × 105 cells/ml in RPMI 1640 supplemented with 2.5 % heat-inactivated FCS, 4 mmol/l L-glutamine, 100 U/ml penicillin, 100 µg/µl streptomycin and 1.25% dimethyl sulfoxide (Sigma-Aldrich) for at least 7 days. Differentiation status was verified by expression of the marker gene CD11b (flow cytometry).
Immunomagnetic selection of gp91phox-expressing cells. For enrichment of gp91phox -positive cells transduced populations were incubated with the gp91phox-specific 7D5 antibody from hybridoma supernatant43 and incubated for 20 minutes at room temperature. After washing with phosphate-buffered saline (PBS) supplemented with 0.5% bovine serum albumin, 2.5 mmol/l EDTA cells were resuspended in the same buffer containing GAM-IgG1 FITC (Jackson ImmunoResearch, Suffolk, England) and incubated for 15 minutes, room temperature. Following several washing steps, cells were incubated with Anti-FITC MicroBeads and applied to the separation column according to manufacturer's instructions (Miltenyi Biotec GmbH, Bergisch-Gladbach, Germany). Purity was assessed by flow cytometry.
Cytochrome C reduction assay. Individually differentiated cell populations were subjected to analysis in duplicates or triplicates. Cytochrome C assays were carried out as described before44 using a Spectra MAX 340 reader (Molecular Devices, Sunnyvale, CA) and the SOFTmax Version 2.02 PRO software. The mean absorbance values of duplicates at 550 nm were converted based on the extinction coefficient of cytochrome C: ΔE550 = 21 × 103 mol/l/cm. All chemicals were purchased from Sigma-Aldrich.
Determination of viral vector copy number by real-time quantitative PCR. Genomic DNA from different sources (cell lines, primary cells, peripheral blood cells) was extracted using the DNeasy Blood & Tissue Kit (Qiagen GmbH, Hilden, Germany). Real-time quantitative PCR to determine VCN was performed using a Roche LightCycler 480 (Roche, Mannheim, Germany) applying advanced quantification (LightCycler 480 Software 1.5.0, Roche) with a reference sample known to harbor one single vector integrant in serial dilution (either human PLB-XCGD clone or murine BAF3 clone). The gDNA templates of 20–50 ng per sample were mixed with Roche LC480 Probes Master Mix (Roche). All samples were run in triplicates. The primer sequences for viral copy determination (oPRE) were as follows: forward, 5′-ATTGCCACCACCTGTCAACT-3′ reverse, 5′GCAA CCTAGCCCCTGTCC-3′. The FAM-labeled Universal Probe #95 (Roche) was applied for detection. Either murine β-actin or human erythropoietin receptor were used as internal reference as described previously.45
LM-PCR for vector integration site-mapping. Genomic DNA isolated from clonal populations established by limiting dilution of transduced PLB-XCGD cells was subjected to ligation mediated PCR (LM-PCR).46 Briefly, a MseI-adaptor (5́-GTAATACGACTCACTATAGGGCTCCGCTTAAGGGAC-3́ hybridized to 3́-GAGGCGAATT CCCTGAT-5́/phosphorylated) was ligated to 100–500 ng of MseI (NEB) digested gDNA by T4-Ligase (NEB). Following enzymatic clean up (MinElute Reaction Clean up Kit, Qiagen) first and nested PCR (annealing at 55 °C, 25 cycles) was performed applying the following primers: avian sarcoma and leukosis virus LTR1: 5́-TACGTAGTGCCTAGCTCGATAC-3́ avian sarcoma and leukosis virus LTR2-nested: 5́-ACGCCATTTGACCATTCACCAC-3́ HIV LTR1: 5́-AGTGCTTCAAGT AGTGTGTGCC-3́ HIV LTR2-nested: 5́-GTCTGTTGTGTGACTCTGGTAAC-3́ Adaptor primer 1 (AP1): 5́-GTAATACGACTCACTATAGGGC-3́ Adaptor primer 2 (AP2): 5́-AG GGCTCCGCTTAAGGGAC-3́. PCR products were subcloned applying the TOPO TA Cloning Kit (Life Technologies) followed by Sanger sequencing. BLAT (UCSC Genome Browser; GRCh37/hg19) was used to map genomic loci of the integrations sites, which were verified by integration specific genomic PCR (primers: Supplementary Table S1 online).
Reverse transcription, RT-PCR and 5′RACE. Total RNA was extracted from PLB-XCGD cells and derivatives using the RNeasy Mini Kit (Qiagen) with on column DNaseI digestion and 1.8 µg of RNA was subsequently subjected to reverse transcription with random primers applying the RETROscript Kit (Ambion, Darmstadt, Germany) according to the manufacturer's instructions. For subsequent gene expression analysis by PCR on integration site flanking exons and for primary PCR to detect cellular exon–vector fusion transcripts 0.5 µl of cDNA was used as template according to standard protocols (55 °C annealing temperature; 30 cycles). A 1:50 dilution of the latter primary PCR products was subjected to semi-nested PCR applying the same conditions as before. Primers were applied as indicated in Supplementary Table S1 online. PCR products were visualized by gel electrophoresis (1% agarose gels) and either directly or after gel purification (QIAquick Gel Extraction Kit, Qiagen) sent for Sanger sequencing (GATC Biotech, Konstanz, Germany). Retrieved sequences were analyzed using SeqMan Pro Software (DNASTAR, Madison, WI), the LALIGN Server (http://www.ch.embnet.org/ software/LALIGN_form.html) and BLAT (UCSC Genome Browser; GRCh37/hg19). Primers for RT-PCR positive controls were as follows: HGR 5-2: 5́-TACAGTCCTCACCCTGATGACCTTGA-3́ HGR 3-2: 5́-AAAGTATGCAGATCGCCTGG GAG-3́ GAPDH-fw: 5́-AG CCACATCGCTCAGACACCATGG-3́ GAPDH-rev: 5́-GCCC AATACG ACCAAATCCGTTG-3́.
For 5'RACE 0.8 µg RNA (HEK293T: 0.4 µg) was reverse transcribed by appling the SMARTer RACE cDNA Amplification Kit (Clontech, Mountain View, CA) according to manufacturer's instructions with the exception that the 5′RACE CDS Primer A was substituted by biotinylated vector leader sequence specific primers (aDWSAf1 or DWLVSA1, 20 µmol/l). After clean up (MinElute Reaction Clean up Kit, Qiagen) vector specific cDNA was purified by applying Dynabeads M-280 Streptavidin (Life Technologies) and resuspended in 10 µl H2O (100 µl for 293T derived cDNA) following standard protocols. Double strand cDNA synthesis and amplification was performed by 20 cycles of PCR with the provided Universal Primer A Mix (Clontech) and the adequate nested leader specific oligonucleotide (aDWSAf2; DWcrypSA2). Following additional clean up as before, 1 µl PCR product was reamplified (35 cycles) by the Nested Universal Primer A (NUP; Clontech) together with either aDWSAf2 or DWcrypSA2. Following another clean up procedure to remove primers PCR products were shotgun cloned into TOPO TA vector (Life Technologies) and positive clones according to blue/white selection were sequenced by the Sanger method (GATC Biotech). As positive control subconfluent 293T cells (100 mm dish) were transfected with 5 µg of either transfer vector plasmid (pAS.EFS.gp91s, pHR′.EFS.gp91s) by standard polyethylene imine method and cells were harvested 30 hours after transfection for RNA isolation.
Isolation, culture and ex vivo transduction of primary hematopoietic progenitor cells. Murine bone marrow cells were harvested from femurs and tibias of gp91phox knockout mice (X-CGD) or wild-type C57/Bl6 mice as control at ~8 weeks of age. Lin− progenitors were isolated using lineage-specific antibodies (Lineage cell depletion kit; Miltenyi, Bergisch-Gladbach, Germany). Lin– cells were prestimulated at a density of 0.5–1.0 × 106 cells per ml for 2 days in StemSpan medium (Stem Cell Technologies, Grenoble, France) supplemented with 1% penicillin/streptomycin and the following cytokines: 10 ng/ml hFGF-acidic, 10 ng/ml mSCF, 20 ng/ml mTPO (all from Peprotech, Hamburg, Germany), 20 ng/ml mIGF (R&D Sytems, Minneapolis, MN) and 1:1000 heparin (Ratiopharm, Ulm, Germany; added 1 day after transduction).47 Transduction of murine gp91−/− Lin- cells (X-CGD) was performed 48 hours after isolation at an multiplicity of infection of 15 in the presence of 4 µg/ml protamine sulfate.
Human CD34-positive cells from cryopreserved G-CSF mobilized peripheral blood of healthy or X-CGD donors were thawed and purified using the human CD34 Microbead Kit (Miltenyi Biotec GmbH) according to manufacturer's instructions. For expansion cells were cultured in X-VIVO-10 (Lonza, Basel, Switzerland), 1% penicillin-streptomycin (PAA), 300 ng/ml hSCF 300 ng/ml hFLT3, 100 ng/ml hTPO, 20 ng/ml interleukin-3. Following overnight prestimulation cells were transduced by a 20-fold dilution of the PFV env pseudotyped SIN alpharetroviral vector. Therefore, cells were plated at a density of 1 × 106 cells per ml in a 96-well-plate in the presence of 4 µg/ml protamine sulfate and transduction was supported by spinoculation (600×g, 30 minutes, 32 °C). At day 2 after transduction, cells were washed, resuspended in fresh medium and further cultivated or transplanted. For granulocytic differentiation in liquid culture, human CD34+ derived cells were cultured in Iscove's modified Dulbecco's media (Lonza) complemented with 20% heat-inactivated FCS, 4 mmol/l L-glutamine and antibiotics (1% penicillin-streptomycin) in the presence of 50 ng/ml hG-CSF. All cytokines were purchased from Peprotech (Hamburg, Germany). The local Ethics Review Board of the University of Frankfurt Medical School approved the use of these cells and informed consent was obtained in accordance with the Declaration of Helsinki.
NBT assay. Primary cells were cultured in semi-solid methylcellulose media (M3434 or H4433, Stem cell Technologies) supplemented with 10% Iscove's modified Dulbecco's media (Gibco) and 1% penicillin/streptomycin at a cell number of 1,000 (murine) or 2,000 (human) per 35 mm dish (duplicates) either at day 2–6 after transduction or directly after isolation in case derived from transplanted animals. After 10–11 days (murine) or ≥15 days (human) thereof derived colonies were overlaid with 1 mg/ml NBT (Sigma-Aldrich) in PBS supplemented with 2 µmol/l phorbol myristate acetate (Sigma-Aldrich). After 25–40 minutes at 37 °C total colony count and number of NBT-positive colonies were determined. Colonies were picked and DNA was isolated according to standard proteinase K lysis. PCR was performed applying the following primers specific for SIN alpharetro-provirus and murine β-actin: alpha-dU3for: 5́-CTCTTGTACGTAGTGCCTAGCTC-3́ alpha-PBSrev: 5́GG CCGACCACTATTCCCTAAC3́ mβactin-fw: 5́-AGAGGGAAATCGTG CGTGAC-3́ mβactin-rev: 5́CAATAGTGATGACCTGGCCGT-3́.
Flow cytometry. For surface staining of the different hematopoietic lineages of peripheral blood, total bone marrow, and spleen, erythrocytes were lysed by incubating cells in BD Pharm Lyse Buffer (BD Pharmingen, San Diego, CA) for 10 minutes at room temperature. Cells were washed and resuspended in PBS and unspecific Fc-binding was blocked by the addition of FcR blocking reagent (Miltenyi Biotech). The added antibodies for surface staining are described elsewhere45 except hCD45-PE (Miltenyi Biotech). Dead cell exclusion was facilitated by the fixable viability dye eFluor 780 (eBioscience, Frankfurt, Germany). After 20–30 minutes incubation at room temperature in the dark, cells were washed, resuspended in PBS and either analyzed or further subjected to intracellular staining for gp91phox detection (7D5-FITC; MBL international no. D162-4) applying BD Cytofix/Cytoperm (BD Biosciences) according to manufacturer's instructions. Data acquisition was performed on a BD FACSCanto II flow cytometer and analyzed using BD FACSDiva 6.1.3 software (Becton Dickinson) or FlowJo 9.4.10 software (Tree Star, Ashland, OR).
DHR assay. The DHR assays were performed as described previously.45 Briefly, washed samples (lysed blood or cultivated cells) were subjected to surface marker staining in HBSS (Gibco) supplemented with 7.5 mmol/l D-Glucose, 0.5% bovine serum albumin, 2000 U/ml catalase (C3155) and 5 µg/ml DHR123 at 37 °C. Following incubation at 37 °C for 10 minutes phorbol myristate acetate was added to a final concentration of 0.2 µmol/l as indicated. Samples were further incubated for 15–20 minutes at 37 °C, the reaction was stopped on ice and the fluorescence signal was recorded on a flow cytometer (BD FACS Canto II), immediately. All chemicals were purchased from Sigma-Aldrich.
Animals and transplantation. The gp91−/− deficient mice (X-CGD; B6.129S6-Cybbtm1Din/J; CD45.2),48 congenic B6.SJL-PtprcaPepcb/BoyJ mice (Bl6/SJL; CD45.1), wt C57/Bl6/N (Bl6; CD45.2), and NSG (NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ) were obtained from Jackson Laboratories (Bar Harbor, ME). Animal experiments were approved by the regional council (Regierungspräsidium, Darmstadt, Germany) and conducted in compliance with the local animal experimentation guidelines. One day after transduction, transduced murine BM Lin− cells and wild-type controls (CD45.2+) were washed twice, resuspended in PBS and 106 cells were injected intravenously into lethally irradiated (9.5 Gy) recipient Bl6/SJL mice (CD45.1+). Peripheral blood was drawn and mice were sacrificed at week 10 after transplantation. Human CD34+ cells (0.5–1 × 106) were washed twice and resuspended in X-VIVO-10 medium (Lonza, Basel, Switzerland) before intravenous injection into sublethally irradiated (2.5 Gy) NSG mice on day 2 after transduction. Mice were killed 8 weeks later, BM was isolated, subjected to human CD34+ selection via magnetic sorting (CD34 Microbead Kit; Miltenyi Biotec GmbH) and purified cells were cultured in granulocytic differentiating conditions. Transplanted mice were kept in individually ventilated cages and drinking water was supplemented with 1.6 g/l neomycin for 2 weeks.
Statistical analysis. Experimental data is plotted as means ± SD. Student's two-tailed t-test was performed for comparison of differences between groups as indicated. A P value of <0.05 was considered significant.
SUPPLEMENTARY MATERIAL Figure S1. Vector performance assessed by transduction efficiency and VCN in PLB-XCGD cells. Figure S2. Gene expression analysis of genes harboring intronic vector integrations and detection of aberrant splice events in monoclonal and polyclonal PLB-XCGD cells by RT-PCR and 5′RACE. Figure S3. In vivo evaluation in X-CGD transplantation model. Figure S4. NBT assay on colonies derived from transplanted animals and gp91phox expression data. Table S1. Primers applied for integration specific PCR and RT-PCR. Table S2. Summary of 5′RACE sequencing results. Table S3. Aberrantly spliced cellular transcripts as detected by 5′RACE. Table S4. 5′RACE sequencing results.
Acknowledgments
This work was supported by a grant from the European Union (FP7 integrated project CELL-PID HEALTH-2010-261387), the Bundesministerium für Bildung und Forschung (E-RARE 01GM1012), the LOEWE Center for Cell and Gene Therapy Frankfurt funded by the Hessische Ministerium für Wissenschaft und Kunst (HMWK; funding reference number: III L 4- 518/17.004 (2010)) and by the Deutsche Forschungsgemeinschaft (DFG) Graduate Program GK1172-Biologicals (KBK) and by the Excellence Cluster REBIRTH. The Georg-Speyer-Haus is supported by the Bundesministerium für Gesundheit and the Hessisches Ministerium für Wissenschaft und Kunst. We thank Axel Rethwilm, Würzburg for providing us with the plasmid pcoPE01. A.S., J.D.S. and C.B. are inventors on a patent application describing alpharetroviral SIN vectors. The other authors declared no conflict of interest.
Supplementary Material
Vector performance assessed by transduction efficiency and VCN in PLB-XCGD cells.
Gene expression analysis of genes harboring intronic vector integrations and detection of aberrant splice events in monoclonal and polyclonal PLB-XCGD cells by RT-PCR and 5′RACE.
In vivo evaluation in X-CGD transplantation model.
NBT assay on colonies derived from transplanted animals and gp91phox expression data.
Primers applied for integration specific PCR and RT-PCR.
Summary of 5′RACE sequencing results.
Aberrantly spliced cellular transcripts as detected by 5′RACE.
5′RACE sequencing results.
REFERENCES
- Segal BH, Veys P, Malech H., and, Cowan MJ. Chronic granulomatous disease: lessons from a rare disorder. Biol Blood Marrow Transplant. 2011;17 1 Suppl:S123–S131. doi: 10.1016/j.bbmt.2010.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seger RA. Modern management of chronic granulomatous disease. Br J Haematol. 2008;140:255–266. doi: 10.1111/j.1365-2141.2007.06880.x. [DOI] [PubMed] [Google Scholar]
- Johnston RB., Jr Clinical aspects of chronic granulomatous disease. Curr Opin Hematol. 2001;8:17–22. doi: 10.1097/00062752-200101000-00004. [DOI] [PubMed] [Google Scholar]
- Ott MG, Schmidt M, Schwarzwaelder K, Stein S, Siler U, Koehl U.et al. (2006Correction of X-linked chronic granulomatous disease by gene therapy, augmented by insertional activation of MDS1-EVI1, PRDM16 or SETBP1 Nat Med 12401–409. [DOI] [PubMed] [Google Scholar]
- Stein S, Ott MG, Schultze-Strasser S, Jauch A, Burwinkel B, Kinner A.et al. (2010Genomic instability and myelodysplasia with monosomy 7 consequent to EVI1 activation after gene therapy for chronic granulomatous disease Nat Med 16198–204. [DOI] [PubMed] [Google Scholar]
- Rivat C, Santilli G, Gaspar HB., and, Thrasher AJ. Gene therapy for primary immunodeficiencies. Hum Gene Ther. 2012;23:668–675. doi: 10.1089/hum.2012.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suerth JD, Maetzig T, Galla M, Baum C., and, Schambach A. Self-inactivating alpharetroviral vectors with a split-packaging design. J Virol. 2010;84:6626–6635. doi: 10.1128/JVI.00182-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suerth JD, Maetzig T, Brugman MH, Heinz N, Appelt JU, Kaufmann KB.et al. (2012Alpharetroviral self-inactivating vectors: long-term transgene expression in murine hematopoietic cells and low genotoxicity Mol Ther 201022–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell RS, Beitzel BF, Schroder AR, Shinn P, Chen H, Berry CC.et al. (2004Retroviral DNA integration: ASLV, HIV, and MLV show distinct target site preferences PLoS Biol 2E234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narezkina A, Taganov KD, Litwin S, Stoyanova R, Hayashi J, Seeger C.et al. (2004Genome-wide analyses of avian sarcoma virus integration sites J Virol 7811656–11663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derse D, Crise B, Li Y, Princler G, Lum N, Stewart C.et al. (2007Human T-cell leukemia virus type 1 integration target sites in the human genome: comparison with those of other retroviruses J Virol 816731–6741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barsov EV., and, Hughes SH. Gene transfer into mammalian cells by a Rous sarcoma virus-based retroviral vector with the host range of the amphotropic murine leukemia virus. J Virol. 1996;70:3922–3929. doi: 10.1128/jvi.70.6.3922-3929.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greger JG, Katz RA, Taganov K, Rall GF., and, Skalka AM. Transduction of terminally differentiated neurons by avian sarcoma virus. J Virol. 2004;78:4902–4906. doi: 10.1128/JVI.78.9.4902-4906.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moiani A, Paleari Y, Sartori D, Mezzadra R, Miccio A, Cattoglio C.et al. (2012Lentiviral vector integration in the human genome induces alternative splicing and generates aberrant transcripts J Clin Invest 1221653–1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cesana D, Sgualdino J, Rudilosso L, Merella S, Naldini L., and, Montini E. Whole transcriptome characterization of aberrant splicing events induced by lentiviral vector integrations. J Clin Invest. 2012;122:1667–1676. doi: 10.1172/JCI62189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavazzana-Calvo M, Payen E, Negre O, Wang G, Hehir K, Fusil F.et al. (2010Transfusion independence and HMGA2 activation after gene therapy of human ß-thalassaemia Nature 467318–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno-Carranza B, Gentsch M, Stein S, Schambach A, Santilli G, Rudolf E.et al. (2009Transgene optimization significantly improves SIN vector titers, gp91phox expression and reconstitution of superoxide production in X-CGD cells Gene Ther 16111–118. [DOI] [PubMed] [Google Scholar]
- Schambach A, Bohne J, Baum C, Hermann FG, Egerer L, von Laer D.et al. (2006Woodchuck hepatitis virus post-transcriptional regulatory element deleted from X protein and promoter sequences enhances retroviral vector titer and expression Gene Ther 13641–645. [DOI] [PubMed] [Google Scholar]
- Schambach A, Bohne J, Chandra S, Will E, Margison GP, Williams DA.et al. (2006Equal potency of gammaretroviral and lentiviral SIN vectors for expression of O6-methylguanine-DNA methyltransferase in hematopoietic cells Mol Ther 13391–400. [DOI] [PubMed] [Google Scholar]
- Zychlinski D, Schambach A, Modlich U, Maetzig T, Meyer J, Grassman E.et al. (2008Physiological promoters reduce the genotoxic risk of integrating gene vectors Mol Ther 16718–725. [DOI] [PubMed] [Google Scholar]
- Zhou S, Mody D, DeRavin SS, Hauer J, Lu T, Ma Z.et al. (2010A self-inactivating lentiviral vector for SCID-X1 gene therapy that does not activate LMO2 expression in human T cells Blood 116900–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombardo A, Cesana D, Genovese P, Di Stefano B, Provasi E, Colombo DF.et al. (2011Site-specific integration and tailoring of cassette design for sustainable gene transfer Nat Methods 8861–869. [DOI] [PubMed] [Google Scholar]
- Zhang F, Frost AR, Blundell MP, Bales O, Antoniou MN., and, Thrasher AJ. A ubiquitous chromatin opening element (UCOE) confers resistance to DNA methylation-mediated silencing of lentiviral vectors. Mol Ther. 2010;18:1640–1649. doi: 10.1038/mt.2010.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhen L, King AA, Xiao Y, Chanock SJ, Orkin SH., and, Dinauer MC. Gene targeting of X chromosome-linked chronic granulomatous disease locus in a human myeloid leukemia cell line and rescue by expression of recombinant gp91phox. Proc Natl Acad Sci USA. 1993;90:9832–9836. doi: 10.1073/pnas.90.21.9832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zentilin L, Qin G, Tafuro S, Dinauer MC, Baum C., and, Giacca M. Variegation of retroviral vector gene expression in myeloid cells. Gene Ther. 2000;7:153–166. doi: 10.1038/sj.gt.3301057. [DOI] [PubMed] [Google Scholar]
- Heckl D, Schwarzer A, Haemmerle R, Steinemann D, Rudolph C, Skawran B.et al. (2012Lentiviral vector induced insertional haploinsufficiency of Ebf1 causes murine leukemia Mol Ther 201187–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montiel-Equihua CA, Zhang L, Knight S, Saadeh H, Scholz S, Carmo M.et al. (2012The ß-globin locus control region in combination with the EF1a short promoter allows enhanced lentiviral vector-mediated erythroid gene expression with conserved multilineage activity Mol Ther 201400–1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santilli G, Almarza E, Brendel C, Choi U, Beilin C, Blundell MP.et al. (2011Biochemical correction of X-CGD by a novel chimeric promoter regulating high levels of transgene expression in myeloid cells Mol Ther 19122–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatziandreou I, Siapati EK., and, Vassilopoulos G. Genetic correction of X-linked chronic granulomatous disease with novel foamy virus vectors. Exp Hematol. 2011;39:643–652. doi: 10.1016/j.exphem.2011.03.003. [DOI] [PubMed] [Google Scholar]
- Barde I, Laurenti E, Verp S, Wiznerowicz M, Offner S, Viornery A.et al. (2011Lineage- and stage-restricted lentiviral vectors for the gene therapy of chronic granulomatous disease Gene Ther 181087–1097. [DOI] [PubMed] [Google Scholar]
- Thornhill SI, Schambach A, Howe SJ, Ulaganathan M, Grassman E, Williams D.et al. (2008Self-inactivating gammaretroviral vectors for gene therapy of X-linked severe combined immunodeficiency Mol Ther 16590–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avedillo Díez I, Zychlinski D, Coci EG, Galla M, Modlich U, Dewey RA.et al. (2011Development of novel efficient SIN vectors with improved safety features for Wiskott-Aldrich syndrome stem cell based gene therapy Mol Pharm 81525–1537. [DOI] [PubMed] [Google Scholar]
- Newrzela S, Cornils K, Li Z, Baum C, Brugman MH, Hartmann M.et al. (2008Resistance of mature T cells to oncogene transformation Blood 1122278–2286. [DOI] [PubMed] [Google Scholar]
- Kustikova OS, Schiedlmeier B, Brugman MH, Stahlhut M, Bartels S, Li Z.et al. (2009Cell-intrinsic and vector-related properties cooperate to determine the incidence and consequences of insertional mutagenesis Mol Ther 171537–1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grez M, Reichenbach J, Schwäble J, Seger R, Dinauer MC., and, Thrasher AJ. Gene therapy of chronic granulomatous disease: the engraftment dilemma. Mol Ther. 2011;19:28–35. doi: 10.1038/mt.2010.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown BD., and, Naldini L. Exploiting and antagonizing microRNA regulation for therapeutic and experimental applications. Nat Rev Genet. 2009;10:578–585. doi: 10.1038/nrg2628. [DOI] [PubMed] [Google Scholar]
- Gentner B, Visigalli I, Hiramatsu H, Lechman E, Ungari S, Giustacchini A.et al. (2010Identification of hematopoietic stem cell-specific miRNAs enables gene therapy of globoid cell leukodystrophy Sci Transl Med 258ra84. [DOI] [PubMed] [Google Scholar]
- Ellis J. Silencing and variegation of gammaretrovirus and lentivirus vectors. Hum Gene Ther. 2005;16:1241–1246. doi: 10.1089/hum.2005.16.1241. [DOI] [PubMed] [Google Scholar]
- Kobayashi S, Imajoh-Ohmi S, Nakamura M., and, Kanegasaki S. Occurrence of cytochrome b558 in B-cell lineage of human lymphocytes. Blood. 1990;75:458–461. [PubMed] [Google Scholar]
- Sumiyoshi T, Holt NG, Hollis RP, Ge S, Cannon PM, Crooks GM.et al. (2009Stable transgene expression in primitive human CD34+ hematopoietic stem/progenitor cells, using the Sleeping Beauty transposon system Hum Gene Ther 201607–1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malech HL. Progress in gene therapy for chronic granulomatous disease. J Infect Dis. 1999;179 Suppl 2:S318–S325. doi: 10.1086/513852. [DOI] [PubMed] [Google Scholar]
- Kuhns DB, Alvord WG, Heller T, Feld JJ, Pike KM, Marciano BE.et al. (2010Residual NADPH oxidase and survival in chronic granulomatous disease N Engl J Med 3632600–2610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura M, Murakami M, Koga T, Tanaka Y., and, Minakami S. Monoclonal antibody 7D5 raised to cytochrome b558 of human neutrophils: immunocytochemical detection of the antigen in peripheral phagocytes of normal subjects, patients with chronic granulomatous disease, and their carrier mothers. Blood. 1987;69:1404–1408. [PubMed] [Google Scholar]
- Mayo LA., and, Curnutte JT. Kinetic microplate assay for superoxide production by neutrophils and other phagocytic cells. Meth Enzymol. 1990;186:567–575. doi: 10.1016/0076-6879(90)86151-k. [DOI] [PubMed] [Google Scholar]
- Brendel C, Müller-Kuller U, Schultze-Strasser S, Stein S, Chen-Wichmann L, Krattenmacher A.et al. (2012Physiological regulation of transgene expression by a lentiviral vector containing the A2UCOE linked to a myeloid promoter Gene Ther 191018–1029. [DOI] [PubMed] [Google Scholar]
- Wu X, Li Y, Crise B., and, Burgess SM. Transcription start regions in the human genome are favored targets for MLV integration. Science. 2003;300:1749–1751. doi: 10.1126/science.1083413. [DOI] [PubMed] [Google Scholar]
- Zhang CC., and, Lodish HF. Murine hematopoietic stem cells change their surface phenotype during ex vivo expansion. Blood. 2005;105:4314–4320. doi: 10.1182/blood-2004-11-4418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollock JD, Williams DA, Gifford MA, Li LL, Du X, Fisherman J.et al. (1995Mouse model of X-linked chronic granulomatous disease, an inherited defect in phagocyte superoxide production Nat Genet 9202–209. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Vector performance assessed by transduction efficiency and VCN in PLB-XCGD cells.
Gene expression analysis of genes harboring intronic vector integrations and detection of aberrant splice events in monoclonal and polyclonal PLB-XCGD cells by RT-PCR and 5′RACE.
In vivo evaluation in X-CGD transplantation model.
NBT assay on colonies derived from transplanted animals and gp91phox expression data.
Primers applied for integration specific PCR and RT-PCR.
Summary of 5′RACE sequencing results.
Aberrantly spliced cellular transcripts as detected by 5′RACE.
5′RACE sequencing results.