

Abstract
Potentiating homologous recombination using triplex-forming peptide nucleic acids (PNAs) can be used to mediate targeted sequence editing by donor DNAs and thereby induce functional gene expression to supplant non-functional counterparts. Mutations that disrupt the normal function of the β-globin subunit cause hemoglobinopathies such as sickle cell disease and β-thalassemias. However, expression of the functional γ-globin subunit in adults, a benign condition called hereditary persistence of fetal hemoglobin (HPFH), can ameliorate the severity of these disorders, but this expression is normally silenced. Here, we harness triplex-forming PNA-induced donor DNA recombination to create HPFH mutations that increase the expression of γ-globin in adult mammalian cells, including β-yeast artificial chromosome (YAC) bone marrow and hematopoietic progenitor cells (HPCs). Transfection of human cells led to site-specific modification frequencies of 1.63% using triplex-forming PNA γ-194-3K in conjunction with donor DNAs, compared with 0.29% using donor DNAs alone. We also concurrently modified the γ-globin promoter to insert both HPFH-associated point mutations and a hypoxia-responsive element (HRE), conferring increased expression that was also regulated by oxygen tension. This work demonstrates application of oligonucleotide-based gene therapy to induce a quiescent gene promoter in mammalian cells and regulate its expression via an introduced HRE transcription factor binding site for potential therapeutic purposes.
Introduction
Elevated fetal hemoglobin (Hb F) levels occur in the benign clinical condition known as hereditary persistence of fetal hemoglobin (HPFH), in which expression of a Hb F subunit, gamma-globin (γ-globin), persists at high levels into adulthood.1 The vast majority of HPFH cases arise from point mutations or deletions in the promoter region of the γ-globin gene, which is located within the β-globin locus on human chromosome 11.2 HPFH mutations are of clinical interest because the resulting elevated Hb F levels can help ameliorate disease severity in patients with sickle cell disease or β-thalassemia, some of the most commonly inherited hemoglobinopathies. Currently, pharmacological inducers such as 5-azacytidine, hydroxyurea, and butyric acid, used in the treatment of sickle cell anemia and thalassemia, are aimed at increasing γ-globin expression,3,4 but these approaches require regular, long-term intervention and patient compliance, and the effects are limited.
There are two human γ-globin genes, Aγ-globin and Gγ-globin, and their expression is under exquisite regulation during fetal and postnatal development through a concert of actions, including stage-specific transcription factors and epigenetic chromosomal modifications. Many HPFH mutations are single base-pair changes that cluster in the 5′ promoter of these genes. It is postulated that these single base-pair mutations alter transcription factor binding and/or change promoter strength, leading to decreased silencing of γ-globin expression.
For example, the G→A HPFH mutation at the −117 position of the γ-globin promoter changes the CCAAT box of the Aγ gene,5 and results in decreased binding of GATA-1 and NF-E3,6,7,8 leading to speculation that the erythroid transcription factor GATA-1 is a negative regulator of γ-globin expression. Transgenic mice carrying the −117 Aγ HPFH mutation as a yeast artificial chromosome (YAC) show delayed γ- to β-globin switching and, consistent with human carriers of this Greek HPFH mutation, show persistent expression of the Aγ-globin chains even in adult stages.9 In humans, inheritance of the −117 G→A HPFH mutation leads to Hb F levels of up to 30% in adults, compared with <1% in the majority of the population lacking the mutation.1
Similarly, the −175 T→C HPFH mutation in the Aγ-globin promoter, which results in a reported 36–41% Hb F level in affected adults, changes the last base pair of an Oct-1 binding site and an overlapping GATA-1 binding site.1 Oct-1 affinity is dramatically decreased for the mutated site, and elimination of the GATA-1 consensus sequence at the −175 site increases γ-globin gene expression.10,11
Because pharmacological activators of γ-globin expression lose efficacy through long-term use, we sought to test an oligonucleotide-based gene therapy approach that is designed to introduce a stable and heritable HPFH mutation in the endogenous human Aγ-globin promoter. We chose to introduce known HPFH mutations at positions −117 and −175 because they have been extensively studied in various erythroid cell models as well as in transgenic mice.9 To mediate sequence-specific changes in the genome, we used a class of triplex-forming oligonucleotides known as peptide nucleic acids (PNAs). PNAs have previously been shown to stimulate recombination of donor DNAs into specific sites in the genome, leading to targeted modification of disease-related genes in human primary cells.12,13,14 PNAs show promise to be gene-targeting tools due to their resistance to nucleases and proteases, low cytotoxicity, low off-target effects, and increased stability of the PNA/DNA duplex compared with DNA/DNA. They have been used to induce site-directed mutagenesis in reporter genes,15 to correct a thalassemia-causing splicing mutation,12 and to introduce truncating mutations in the CCR5 membrane protein that serves as the HIV-1 co-receptor.16 Upon binding to duplex DNA, bifunctional PNAs, as used in the current study, form PNA/DNA/PNA triplexes and associated P-loop structures with the targeted DNA sequence. The resulting helical distortions trigger the activity of endogenous DNA repair factors, including those involved in nucleotide excision repair12,15 and homology-dependent repair.15 It is thought that these recruited DNA repair factors either create strand breaks at the target site (which by themselves stimulate recombination) or help to directly catalyze the recombination of co-introduced short donor DNAs that contain the desired DNA sequence modification.
In this study, we create HPFH mutations in the promoter region of the endogenous γ-globin gene in mammalian cells, including human hematopoietic progenitor cells (HPCs). We co-introduce the triplex-forming PNAs along with short, single-stranded donor DNA homologous to the Aγ-globin promoter except that they contain characterized HPFH mutations at positions −117 and −175, into human cells, leading to site-directed sequence changes in the endogenous Aγ-globin promoter. In addition, we demonstrate successful insertion of a novel transcription factor binding site (a hypoxia-response element; HRE) into the Aγ-globin promoter, thus permitting another regulatory mechanism of γ-globin expression. Moreover, we determine that PNA-mediated introduction of the HPFH mutations in human primary cells leads to an increase in γ-globin expression, suggesting the feasibility of an oligonucleotide-based gene therapy approach to increasing Hb F production in the treatment of inherited anemias.
Results
We identified three suitable triplex-forming PNA binding sites in the Aγ promoter, at positions −304, −287, and at −194 (numbered relative to the cap site), and designed triplex-forming PNAs to target these sites (Figure 1a). These PNAs were synthesized to contain terminal lysines to facilitate binding.17 We confirmed by electrophoretic mobility shift assays that these PNAs bind to the Aγ-globin promoter DNA sequences (Figure 1b). Note that in these mobility shift assays, the PNAs bound to DNA can produce more than one shifted band, indicating different possible binding conformations. Sometimes two PNAs can bind simultaneously the target DNA by one providing the Watson–Crick domain and the other providing the Hoogsteen domain, thus creating a complex with a higher molecular weight than one PNA binding the DNA by providing both the Watson–Crick and Hoogsteen domains, as shown in our previous work.12,13,14 Nonetheless, to further confirm high-affinity PNA/DNA/PNA triplex formation, we performed a melting temperature (Tm) analysis of PNA γ-194-6K bound to the polypurine DNA strand of the target (the predicted PNA/DNA/PNA triplex is shown in Figure 1b). We determined a Tm of 89.9 °C, consistent with high-affinity triple helix formation.
Figure 1.

Schematic of strategy to introduce naturally occurring HPFH mutations into the endogenous γ-globin gene promoter using triplex-forming PNA-stimulated recombination of donor DNAs. (a) Triplex-forming PNA-mediated recombination of a HPFH donor DNA into the human Aγ-globin promoter is expected to result in introduction of the HPFH mutation into the human genome. The HPFH mutation, in turn, should confer increased γ-globin expression in modified cells. Pictured is a schematic of HPFH mutations at the −117 and −175 sites in the Aγ-globin promoter, introduced by the donor DNAs. (b) Triplex-forming PNA sequences bound to their respective target DNA sequences, where J denotes the cytosine analog pseudoisocytidine and electrophoretic mobility shift assays of these PNAs and double-stranded, supercoiled DNAs. Melting temperature studies of the triplex-forming PNA γ-194-6K bound to the polypurine strand of the DNA target indicated a Tm of 89.9 °C. (c) Donor DNAs, with single base-pair changes from genomic DNA sequence in lowercase, and HPFH mutations underlined, used in this study. HPFH, hereditary persistence of fetal hemoglobin; PNA, peptide nucleic acid; Tm, melting temperature.
We also designed 50-nucleotide single-stranded donor DNA sequences that are homologous to the Aγ-globin promoter, except for nucleotide sequence changes that introduce the −175 T→C HPFH mutation (“−175 HPFH donor”) or the −117 G→A HPFH mutation (“−117 HPFH donor”) (Figure 1c). In addition, we designed a 100-nucleotide donor DNA that contained both the −175 and the −117 HPFH mutations (“−117/−175 HPFH donor”) to test the effect of introducing compound promoter mutations (Figure 1c).
To study the ability of these PNAs to induce recombination with donor DNA to introduce known HPFH mutations into the endogenous Aγ-globin promoter, we used a mouse bone marrow cell line derived from transgenic mice containing the multigenic human β-globin locus on a YAC (“β-YAC BMCs”).18 These cells are well suited for this application because, consistent with expression patterns in human adults, they do not express γ-globin but do express human β-globin at high levels.18 γ-Globin mRNA and protein in these cells is strongly induced by treatment with 5-azacytidine, an inhibitor of DNA methylation, consistent with the effect seen in human erythroid cells.
We first tested the donor DNAs by themselves to confirm their activity and also to validate the specificity of the allele-specific PCR assay to detect introduced mutations. β-YAC bone marrow cells (BMCs) were transfected with the donor DNAs alone, and gene modification of the Aγ-globin promoter was determined by allele-specific PCR using primers specific for the introduced mutations at the −175 or the −117 sites (Figure 2a). Donor DNAs were introduced by square wave electroporation at a final oligonucleotide concentration in the cuvette of 2 µmol/l, and the treated β-YAC BMCs were allowed to recover for 48 hours in complete medium before harvest of the genomic DNA for analysis. The primers specific for the −117 HPFH modification did not amplify genomic DNA from cells treated with the −175 donor, and vice versa. Treatment of β-YAC BMCs with the 100-nucleotide donor DNA, which harbors both the −117 and the −175 HPFH mutations, resulted in amplification using both the −117 and −175-specific PCR primers, indicating introduction of the HPFH mutations at both these sites in the Aγ-globin promoter (Figure 2a). Increasing concentrations of the −117/−175 HPFH donor DNA yielded increasing genomic amplification of the treated β-YAC BMCs (Figure 2b).
Figure 2.
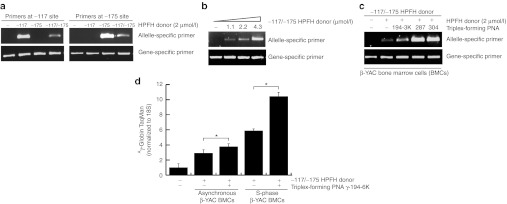
Triplex-forming PNAs can mediate recombination of the HPFH donors in β-YAC BMCs. (a) β-YAC BMCs were transfected with the −117 HPFH donor, the −175 HPFH donor, or the compound −117/−175 HPFH donor. The cells were harvested 48 hours after transfection for genomic analysis by allele-specific PCR. Specificity of the allele-specific PCR assay was demonstrated using the three different HPFH donors, and their respective allele-specific PCR primers, to detect mutation in the targeted sequence. (b) Targeted modification levels increased with increasing concentrations of HPFH donor DNA used. β-YAC BMCs were transfected with the compound −117/−175 HPFH donor and were harvested 48 hours after transfection for genomic analysis by allele-specific PCR. The HPFH mutation at the −175 site was detected by the use of allele-specific primers. (c) Cotransfection of the −117/−175 HPFH donor DNA with triplex-forming PNAs (as indicated) into β-YAC BMCs resulted in increased levels of genomic modification (lanes 3–5), relative to treatment by the donor DNA alone (lane 2). The allele-specific PCR using the −175 primer set is shown here. (d) qRT-PCR using the TaqMan assay showed increased γ-globin gene expression in β-YAC BMC samples (three independent replicates for each sample, *P < 0.05) treated with −117/−175 HPFH donor DNA and PNA γ-194-6K. Expression was even greater when β-YAC BMCs are synchronized in S-phase using double thymidine block before transfection of oligonucleotides. BMC, bone marrow cell; HPFH, hereditary persistence of fetal hemoglobin; PNA, peptide nucleic acid; qRT-PCR, quantitative reverse transcription-PCR; YAC, yeast artificial chromosome.
Next, we cotransfected the β-YAC BMCs with the triplex-forming PNAs along with the HPFH donor DNAs to test whether PNAs targeting the Aγ-globin promoter can augment donor DNA-based gene modification. β-YAC BMCs transfected with both the 100-nucleotide donor and the indicated PNAs showed higher levels of genome modification, compared with β-YAC BMCs treated with 100-nucleotide donor alone (Figure 2c). Amplification using the wild-type primer confirms similar loading of genomic DNA samples in the PCR reactions.
To quantify the degree of change in γ-globin mRNA expression conferred by the introduced HPFH mutations in treated β-YAC BMCs, we used quantitative reverse transcription-PCR (qRT-PCR), which combined RT-PCR with real-time PCR quantification. RNA samples were harvested from β-YAC BMCs 1–2 weeks following PNA and donor DNA transfection and were subjected to qRT-PCR using TaqMan probes. As shown in Figure 2d, β-YAC BMCs treated with both the 100-nucleotide donor DNA and the PNA γ-194-6K expressed relatively more γ-globin mRNA, compared with mock-transfected β-YAC BMCs or β-YAC BMCs treated with donor DNA alone. Consistent with previous work showing evidence of increased PNA targeting during S phase,12,19 β-YAC BMCs that had been synchronized in S-phase using double thymidine block and then treated with PNAs and donor DNAs showed even higher levels of γ-globin expression, compared with treatment of an asynchronous, logarithmic growth cell population (Figure 2d).
A potential and immediate application of this technology would be as a gene therapy tool for use in patients with sickle cell disease or β-thalassemia. Therefore, we next tested the ability of these triplex-forming PNAs and donor DNAs to increase γ-globin expression in HPCs, a potential target cell population for use in gene therapy. In these cells, we designed a new donor DNA that, in addition to incorporating a −117 HPFH mutation, also includes the HRE consensus sequence.20 This HRE consensus sequence is the binding site for HIF-1, a transcription factor that is upregulated by hypoxic conditions and induces expression of hypoxia-responsive genes such as VEGF. Presence of this HRE consensus sequence in the promoter region is expected to confer sensitivity of γ-globin gene expression to hypoxic conditions. For comparison, we also used a donor DNA that introduces the same HPFH mutation at the −117 site, but does not include the HRE consensus sequence and, so, is not expected to confer sensitivity to hypoxia (Figure 3a).
Figure 3.

Triplex-forming PNAs can mediate recombination of the HPFH donors in hematopoietic CD34-selected cells. (a) Human CD34+ cells were mock transfected or transfected with triplex-forming PNA γ-194-3K and one of two donors: −117 HRE/HPFH donor, which contains a HPFH mutation at −117 as well as the consensus HRE sequence, or as a control, a −117 HPFH donor, which contains a HPFH mutation at −117 but no HRE consensus sequence. (b) Allele-specific PCR of the genomic DNA from treated CD34-selected cells or K562 cells revealed the presence of the −117 HPFH mutation in cells treated with the −117 HRE/HPFH donor or the control −117 HPFH donor, in conjunction with triplex-forming PNA γ-194-3K, but not in conjunction with a non-related control triplex-forming PNA PNA-679, indicating targeted and specific gene conversion at the −117 site. Oligonucleotides were introduced into human CD34+ cells by nucleofection delivery. (c) CD34-selected cell samples (three independent replicates for each sample, *P < 0.05) treated with triplex-forming PNA γ-194-3K and either −117 HRE/HPFH donor DNA or control −117 HPFH donor, were subjected to hypoxia for 48 hours. RNA was then harvested from these hypoxia-treated cells and from a normoxia set for comparison. qRT-PCR with TaqMan probes demonstrated that γ-globin expression in triplex-forming PNA γ-194-3K and −117 HRE/HPFH donor DNA-treated cells were now responsive to hypoxia. In contrast, cells treated with triplex-forming PNA γ-194-3K and control −117 HPFH donor did not show hypoxia-based regulation of γ-globin expression. (d) CD34-selected cell samples (three independent replicates for each sample, *P < 0.05) were mock transfected or transfected with triplex-forming PNA γ-194-3K or a non-related control triplex-forming PNA PNA-679, and RNA was harvested. qRT-PCR indicated that neither triplex-forming PNA γ-194-3K nor a non-related control triplex-forming PNA (PNA-679) induced γ-globin expression by themselves. HPFH, hereditary persistence of fetal hemoglobin; HRE, hypoxia-responsive element; PNA, peptide nucleic acid; qRT-PCR, quantitative reverse transcription-PCR.
Human CD34+ cells isolated from granulocyte colony-stimulating factor-mobilized peripheral blood of normal adults were transfected with no molecules (i.e., a mock transfection), with the indicated donor DNA alone, or with the donor DNA plus the triplex-forming PNA γ-194-3K, or with the donor DNA plus the unrelated PNA-679 (that targets the CCR5 gene and not the γ-globin gene). The CD34+ cells were grown in expansion conditions (Stemcell Technologies, Vancouver, British Columbia, Canada), and harvested 2 or 4 days later for genomic DNA analysis. Allele-specific PCR revealed that site-specific gene modification was produced by the combination of the donor DNA with PNA γ-194-3K (Figure 3b). There was minimal modification produced by the donors alone, and the unrelated PNA 679 had no effect, as expected since it targets the CCR5 gene and not the γ-globin gene.
The triplex-forming PNA- and donor DNA-transfected CD34+ cells were then subjected or not to hypoxic conditions (0.1% O2) for 48 hours to determine whether γ-globin expression would be responsive to hypoxia-dependent HIF-1 regulation. qRT-PCR of cells placed in hypoxia versus normoxia using TaqMan probes specific for γ-globin mRNA revealed an increase in γ-globin expression in the CD34+ human HPCs in response to hypoxia (Figure 3c). Cells transfected with the control donor DNA, which does not contain the HRE element, showed only minimally increased γ-globin expression following hypoxia exposure. These results suggest that it is possible to specifically modify the Aγ-globin promoter in CD34+ human HPCs to incorporate an HPFH mutation along with a new transcription regulatory sequence. The combination of the −117 site mutation and an HRE introduction increased γ-globin expression additively. Importantly, this elevated γ-globin mRNA expression was detectable in the cell population even though quantification of gene modification frequencies suggests that about 1% of the cells were modified at the Aγ-globin promoter site (see below).
In addition, at the level of gene expression, qRT-PCR indicated that CD34+ cells transfected with triplex-forming PNA γ-194-3K alone (without any donor DNA to introduce the specific HPFH/HRE sequence changes) did not show elevated γ-globin expression as compared with mock-treated cells (neither did cells transfected with the unrelated control PNA-679) (Figure 3d). These results suggest that the upregulation of the γ-globin gene in these experiments is the result of the introduction into the promoter region of the new sequences carried in the donor DNA, rather than from an effect of the PNA binding by itself on the activity of the promoter. PNAs have the potential to create open DNA conformations that can act as artificial promoters, as first reported by Nielsen and colleagues.21
The frequencies of sequence modification in the PNA- and donor DNA-treated cells were estimated by establishing a standard curve for defined frequencies of a genomic DNA spiked with the plasmid containing the tested modifications within the γ-globin sequence. The curve was generated using seven pre-defined frequencies ranging from 0.0323%–25%. Using this standard curve as a benchmark, allele-specific quantitative PCR (qPCR) indicated modification frequencies of 1.63% for cells treated with both −117 donor DNA and PNA γ-194-3K and of 0.29% for cells treated with γ117 donor DNA alone (Figure 4). These results are consistent with prior work showing that triplex-forming PNAs can provoke the endogenous homologous recombination machinery to catalyze recombination of donor DNAs with their cognate sites in the genome.16
Figure 4.
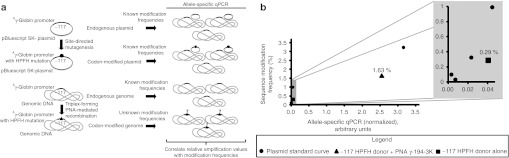
Estimation of sequence-modified frequencies. (a) Schematic representation of modification frequency standard curve assay to quantitatively determine modification frequency of human genome by triplex-forming PNA-mediated recombination. The known modification frequencies of genomes with or without endogenous or sequence-modified Aγ-globin promoters carried on plasmids can be used to estimate the unknown modification frequencies of genomes treated with donor DNA and triplex-forming PNA to introduce sequence-modified Aγ-globin promoter. (b) Allele-specific qPCR using primers for the sequence-modified and for the endogenous γ-globin region at −117 was performed on three independent replicates of genomic DNA and of calculated copies of plasmid carrying the donated mutation in genomic DNA. The resulting normalized values were plotted versus the calculated mutant allele frequency, generating a regressed standard curve that was applied to estimate mutant allele frequencies of the genomic DNA treated with both the −117 HPFH donor and triplex-forming PNA γ-194-3K and with the −117 HPFH donor alone. HPFH, hereditary persistence of fetal hemoglobin; PNA, peptide nucleic acid; qPCR, quantitative PCR.
Discussion
Increased levels of Hb F, either through co-inherited HPFH or through pharmacological activators of γ-globin expression, help reduce the clinical severity of some hemoglobinopathies. Therefore, much basic and clinical research has focused on identifying therapeutic modulators of γ-globin expression and on understanding the mechanisms of γ-globin silencing in adults.
In this study, we sought a nonviral genetic approach to increasing γ-chain synthesis. We reasoned that mimicking HPFH mutations in patients with sickle cell disease or β-thalassemia might have a clinical benefit of reducing anemia, while by itself the HPFH mutation is clinically benign. We used triplex-forming PNAs in conjunction with short, single-stranded donor DNAs to introduce known HPFH mutations into the Aγ-globin promoter. Introduction of these HPFH mutations into genomic DNA, determined by allele-specific PCR, led to an increase in γ-globin expression as verified by qRT-PCR with TaqMan probes.
That we were able to demonstrate genomic modification in human CD34-selected cells is clinically significant because these cells would be the likely targets in autologous therapy. In this study and in a previous study of a β-thalassemic gene target,13 we were able to introduce a specific genomic sequence change using cotransfection of human CD34-selected cells with PNAs and donor DNAs. In the previous study, we were able to demonstrate that the initially treated cell population was able to give rise to erythroid and neutrophil cell populations, which still carried the specific genomic sequence change 22 days following oligonucleotide delivery. Therefore treatment with the PNA and DNA oligonucleotides does not appear to change the lineage potential of this progenitor cell population.
In this study, we used a donor DNA that introduced a new regulatory element into the Aγ-globin promoter, a HRE site, in addition to a naturally occurring HPFH mutation. Following PNA and donor DNA transfection of these human HPCs, we observed that γ-globin expression of these cells was rendered responsive to hypoxia. This provides evidence for the use of PNAs to stimulate the introduction of specific sequence changes in disease-related sites in the genome, including artificial regulatory elements that, in this case, can further increase γ-globin expression in the presence of hypoxic conditions. Transfection of the triplex-forming PNA alone was not sufficient to induce γ-globin expression. The cotransfection of triplex-forming PNA and donor DNA specifically targeting and recombining, respectively, with the γ-globin locus was necessary for lasting gene conversion and γ-globin expression.
This is an initial step in the application of oligonucleotide-stimulated promoter modification as a means to increase γ-globin gene expression. This approach may ultimately be of clinical interest in the treatment of hemoglobinopathies, although improving frequencies of triplex-forming PNA-induced genome modification will be required before clinical applicability. In this regard, we have previously shown that modification in PNA design (to extend the Watson–Crick-binding domain,)16 into a next generation “tail-clamp” and in the delivery methodology (using biocompatible nanoparticles,13 or conjugated peptides,)15 can enhance the efficiency of the triplex technology. We expect that application of these approaches to target the Aγ-globin promoter in future work may help to boost the modification frequencies at this site as well. In addition, we have also found that improvements in targeting efficiency can be achieved by cell cycle synchronization by treatment of cells with chloroquine (to enhance oligonucleotide bioavailability) and by treatment of cells with a histone deacetylase inhibitor, SAHA (to modify the chromatin environment to facilitate molecular access to the genomic DNA).12,13 In addition, we expect relatively low clinical toxicity based on the similarity of composition of PNAs and other triplex-forming oligonucleotides to antisense, aptamer, and RNA interference oligonucleotides currently in clinical trials.22 We anticipate that judicious combinations of these approaches will lead to clinically meaningful frequencies of targeted gene modification using triplex-based technologies.
Materials and Methods
Cells. β-YAC mouse BMCs, from transgenic mice containing the β-globin locus YAC (“β-YAC BMCs”), were kindly provided by Dr Kenneth Peterson (University of Kansas Medical Center, Kansas City, KS). These cells, derived from transgenic mice containing the β-globin locus as a YAC, are under conditional control of a signaling molecule, chemical inducer of dimerization, which permits growth.18 Briefly, BMCs were extracted from β-YAC transgenic mice and retrovirally transduced with an artificial proliferation signal, consisting of an F36V-modified FKBP12 derivative fused to the intracellular portion of the thrombopoietin receptor Mpl. The resulting chemical inducer of dimerization-dependent β-YAC BMCs were selected and maintained in the presence of AP20187 dimerizer (100 nmol/l; Ariad Pharmaceuticals, Cambridge, MA) in Iscove's modified Dulbecco's medium containing 10% fetal bovine serum, penicillin, and streptomycin. Human CD34+ cells were isolated by magnetic sorting from granulocyte colony-stimulating factor-mobilized peripheral blood from normal, healthy adult donors, and cryopreserved in liquid nitrogen at a core facility before use (Center of Excellence in Molecular Hematology, Yale University, New Haven, CT).
Oligonucleotides. Triplex-forming PNAs, each with a three 8-amino-2,6-dioxaoctanoic acid linkers, were purchased from Bio-Synthesis (Lewisville, TX) or Applied Biosystems (Framingham, MA). All triplex-forming PNAs, purified by high performance liquid chromatography, had three terminal lysines, one at the N terminus and two at the C terminus, or six terminal lysines, three at the N terminus and three at the C terminus. Donor DNAs were synthesized at 50 or 100 nucleotides in length and with 5′- and 3′-end protected by three phosphorothioate internucleoside linkages at each end and purified by reversed phase-high performance liquid chromatography by Midland Certified Reagent (Midland, TX). The donor DNAs are homologous to the Aγ-globin promoter, except for a six-nucleotide change that is required for reliable detection of the genomic sequence modification by allele-specific PCR. The triplex-forming PNA sequences are as follows, triplex-forming PNA γ-304: Lys-JJJ TJT TJT TTT-LINKER-TTT TCT TCT CCC-Lys-Lys; triplex-forming PNA γ-287: Lys-TTT JJJ TTJ TT-LINKER- TT CTT CCC TTT-Lys-Lys; triplex-forming PNA γ-194-3K: Lys-JJJ JTT JJJ J-LINKER-C CCC TTC CCC-Lys-Lys; triplex-forming PNA γ-194-6K: Lys-Lys-Lys-JJJ JTT JJJ J-LINKER-C CCC TTC CCC-Lys-Lys-Lys; triplex-forming PNA PNA-679 (non-related control): Lys-Lys-Lys-JTJ TTJ TTJ T-LINKER-T CTT CTT CTC-Lys-Lys-Lys. The donor DNA sequences are as follows, with single base-pair changes from genomic DNA sequence in lowercase and HPFH mutations underlined. The −117 HPFH donor: 5′-AGT TTG CCT TGT CAA GGC TAa cGc gtA AGG CAA GGC TGG CCA ACC CAT GG-3′ the −175 HPFH donor: 5′-GCC AGG AAC CGT TTC AGA CAG Acg cgT Gtc gac AGA TAG TGT GGG GAA GG-3′ the −117/−175 HPFH donor: 5′-GGT CAA GTT TGC CTT GTC AAG GCT ATg aat tcc Gcg gAG GCT GGC CAA CCC ATG GGT GGA GTT TAG CCA GGA ACC GTT TCA GAC ccg ggT TTG CAT TGA G-3′ the −117 HRE/HPFH donor: 5′-GGT CAA GTT TGC CTT GTC AAG GCT ATc acg tAA GGC AAG GCT GGC CAA CCC ATG-3′.
Electrophoretic mobility shift assays. To evaluate the formation of triplex-forming PNA invasion complexes, segments of the Aγ promoter sequence containing the targeted triplex-binding sites were cloned into pBlueScript II-SK plasmid DNA. Double-stranded triplex-binding sites were generated by annealing oligonucleotides (Keck Oligonucleotide Synthesis Resource, Yale University, New Haven, CT) flanked by EcoRI and BamHI restriction sites. These duplex-binding sites were then ligated into linearized plasmid DNA which were both double-digested with EcoRI and BamHI, and extracted using Gel Extraction Kit (Qiagen, Valencia, CA). Ligation reactions with 5:1 and 3:1 insertion to vector ratios were performed with Rapid Ligation Kit (Roche Applied Science, Mannheim, Germany). Ligated products were transformed into TOP10 Chemically Competent cells (Invitrogen, Carlsbad, CA) according to manufacturer's instructions and streaked onto LB-ampicillin plates at 37 °C overnight. The next day, colonies were selected for growth in LB-ampicillin medium, purified by Miniprep Plasmid Purification Kit (Qiagen), then sequenced by the Yale Keck Sequencing facility for verification of the Aγ promoter insert.
These pBlueScript derivatives were then incubated with increasing concentrations of triplex-forming PNA in tris-EDTA buffer (pH = 7.4) with 10 mmol/l KCl and digested with restriction enzymes EcoRI and BamHI to release an about 110 bp fragment of the plasmid where the triplex-forming PNA was designed to bind. The digest was separated by molecular weight by non-denaturing 12% polyacrylamide gel electrophoresis and visualized by silver staining.
Tmstudies. To interrogate the triplex-forming properties of triplex-forming PNA to DNA, Tm studies were conducted. Triplex-forming PNA γ-194-3K with aforementioned sequence and the polypurine strand of the target DNA with sequence 5′-GGG GAA GGG G-3′ were incubated in equimolar ratios of 1 µmol/l of each strand (pH = 7.4) and annealed by heating to 95 °C and slowly cooling to 25 °C. Tm were determined by ultraviolet thermal denaturation at 260 nm at a heating rate of 1 °C/minute from 25 to 95 °C. The first derivative of the ultraviolet melting curve indicated the Tm of the triplex-forming PNA from the DNA.
Cell transfection. Cells in 100 or 250 µl complete medium were electroporated in the presence of 4 µmol/l donor DNA and 0 or 10 µmol/l triplex-forming PNA, as indicated. The β-YAC BMCs were in logarithmic growth or synchronized at S-phase using double thymidine block at the time of transfection. For double thymidine block, the cells were placed in complete medium supplemented with 2 mmol/l thymidine for 12 hours, then replaced with fresh complete medium for 12 hours and then placed in complete medium with 2 mmol/l for an additional 12 hours. Cells were released in fresh medium for 3–5 hours before electroporation with a Bio-Rad Gene Pulser electroporator (Bio-Rad Laboratories, Hercules, CA) at settings of 250 V, 960 µF. About 107 cells were thawed in StemSpan Serum-Free Expansion Media supplemented with StemSpan CC100 cytokine cocktail (STEMCELL Technologies, Vancouver, Canada) and nucleofected with oligonucleotide using the Amaxa Human CD34+ Cell Nucleofector Kit, according to manufacturer's instructions (Amaxa Biosystems, Gaithersburg, MD). At 48 hours following transfection, cells were collected for genomic DNA and RNA analyses.
Introduction of HRE and hypoxia treatment. Human CD34-selected and K562 cells were treated with triplex-forming PNAs and donor DNAs containing the HRE consensus core sequence by Amaxa nucleofection. Following treatment, the cells were maintained in cell culture in complete medium (StemSpan supplemented with CC100 cytokine cocktail, and RPMI with 10% fetal bovine serum, respectively) for an additional 4 and 14 days, respectively, whereupon they were placed for 48 hours in a hypoxia chamber with a constant 0.1% oxygen tension. A duplicate set of cells was kept in normal oxygen tension (“normoxia”) for comparison. Cells were harvested for genomic DNA and RNA analyses immediately after 48 hours incubation in hypoxia or normoxia.
RNA extraction and qRT-PCR. RNA was isolated from K562, β-YAC BMCs, or human CD34-selected cells using the Absolutely RNA Miniprep Kit (Stratagene, La Jolla, CA). The RNA was treated with DNaseI and cDNA was made using the SuperScript III RT-PCR Kit with oligo dT20 primers (Invitrogen). qRT-PCR was performed on a Mx3000P real-time PCR machine (Stratagene), with TaqMan Gene Expression Assays designed for the human Aγ-globin gene conjugated to the FAM fluorophore. Ct values were normalized to eukaryotic 18S or human actin expression as determined with specific TaqMan HEX probes (Applied Biosystems, Foster City, CA).
Allele-specific PCR and qPCR. Genomic DNA was harvested from K562 cells, β-YAC BMCs or CD34-selected cells and purified using the Wizard Genomic DNA Purification Kit (Promega, Madison WI). Equal amounts of genomic DNA were subjected to allele-specific PCR, in which the 3′ end of the forward primer corresponded to the wild-type or mutated sequence as introduced by the donor DNA. The PCR conditions are as follows, where the annealing temperature varied with primer set: 94 °C for 2 minutes; 35 cycles of 94 °C for 30 seconds, annealing for 30 seconds, and 72 °C for 1 minute; followed by 72 °C for 5 minutes. The annealing temperatures varied from 57 to 62 °C and were determined empirically for each primer pair to amplify only the appropriate DNA template.
Estimation of genome sequence modification frequencies. To estimate sequence modification frequencies of the −117 region of the human Aγ-globin promoter in genomic DNA from cells treated with −117 HPFH donor and triplex-forming PNA γ-194-3K or with −117 HPFH donor alone, known and unknown modification frequencies of samples were correlated with allele-specific qPCR relative amplification values. Known frequencies of modification of samples were formulated by mixing (or not mixing) plasmid carrying the endogenous Aγ-globin promoter sequence or codon-modified promoter sequence into human genomic DNA. These samples were subjected to allele-specific qPCR, and their relative amplification values were determined. Concurrently, samples with unknown frequencies of modification of human genomic DNA treated by our molecules were also subjected to this allele-specific qPCR, and their relative amplification values were determined. The relative amplification values were correlated with modification frequencies in order to estimate the genome sequence modification frequencies of samples of human genomic DNA treated by our molecules.
Plasmids with the endogenous Aγ-globin promoter sequence were created by PCR amplifying a 379-nucleotide fragment containing the −117 region of the endogenous human Aγ-globin promoter from genomic DNA with primers (Keck Oligo Synthesis Resource, Yale University, New Haven, CT) introducing single-occurrence restriction sites for HindIII and PstI (New England Biolabs, Ipswich, MA) which were used for subsequent digestion and overnight T4 ligation (New England Biolabs) into pBluescript SK- plasmid (Stratagene). To isolate this endogenous plasmid, DH5 α-competent cells (Invitrogen) were transformed with the plasmids ligated with amplicons, incubated for 12 hours, and then plated for overnight growth of single colonies. Single colonies were then inoculated, selected, and from them plasmid DNA was extracted (Qiagen, Valencia CA) and sequenced to verify the presence, orientation, and sequence content of the endogenous Aγ-globin promoter.
Plasmids with codon-modified Aγ-globin promoter were created from these endogenous plasmids using site-directed mutagenesis (Agilent Technologies, Santa Clara, CA) and isolated as aforementioned. Similar to the mutations introduced by the −117 HPFH donor, a total of 5 bp, including the HPFH single base pair, were mutated in order to optimize primer discriminatory sensitivity in the allele-specific qPCR assay.
For allele-specific qPCR, sequence-modified plasmid DNA concentrations were verified using OD260 spectroscopy (Thermo Scientific, West Palm Beach, FL), initial concentrations adjusted using speedvac, and serial dilutions mixed into a milieu of genomic DNA to formulate modification frequencies. Frequencies were computed based on the number of copies of the sequence-modified Aγ-globin promoter in plasmid DNA and in genomic DNA, normalized to the estimated total number of cells constituting one qPCR reaction. The qPCR reactions were performed with codon-modified allele-specific primers on a Mx3000P real-time PCR machine with SYBR Green and ROX reference dyes (Stratagene). Relative amplification values were computed based on the fold difference in threshold cycle number of each sample to that of untreated human genomic DNA with no sequence-modified plasmid, and thus having a baseline 0% modification frequency. These relative amplification values, normalized to analogous values using endogenous allele-specific primers, were plotted and regression fit to a curve with calculated modification frequencies.
Human genomic DNA from donor and triplex-forming PNA and donor alone treated cells were harvested and purified (Promega) and initial concentrations were adjusted using speedvac. This harvested genomic DNA was similarly and simultaneously subjected to the allele-specific qPCR for comparison, and, as previously described, relative amplification values were computed. These relative amplification values and the fitted curve were used to calculate estimated frequencies of sequence-modified Aγ-globin promoter in the harvested genomic DNA.
Statistical analysis. The quantitative data are shown as the means with error bars representing the SEM from multiple experiments. Statistical significance between two groups was measured using two-tailed Student's t-test. If P < 0.05, then differences were considered to be statistically significant.
Acknowledgments
This work was supported by a National Institutes of Health (NIH) grant R01HL082655 and by a Doris Duke Innovations in Clinical Research Award (to P.M.G.). A National Institute of General Medical Sciences Medical Scientist Training Program Fellowship from NIH grant T32GM07205 provided support (to J.Y.C.) and a National Institute of Diabetes and Digestive and Kidney Diseases Experimental and Human Pathobiology Postdoctoral Fellowship from NIH grant T32DK007556 provided support (to F.R.). We thank Raman Bahal for technical assistance and the Yale Center of Excellence in Molecular Hematology for providing CD34+ cells (through NIH grant P30DK072442). The authors declared no conflict of interest.
REFERENCES
- Forget BG. Molecular basis of hereditary persistence of fetal hemoglobin. Ann N Y Acad Sci. 1998;850:38–44. doi: 10.1111/j.1749-6632.1998.tb10460.x. [DOI] [PubMed] [Google Scholar]
- Levings PP., and, Bungert J. The human β-globin locus control region. Eur J Biochem. 2002;269:1589–1599. doi: 10.1046/j.1432-1327.2002.02797.x. [DOI] [PubMed] [Google Scholar]
- Swank RA., and, Stamatoyannopoulos G. Fetal gene reactivation. Curr Opin Genet Dev. 1998;8:366–370. doi: 10.1016/s0959-437x(98)80095-6. [DOI] [PubMed] [Google Scholar]
- Galanello R, Veith R, Papayannopoulou T., and, Stamatoyannopoulos G. Pharmacologic stimulation of Hb F in patients with sickle cell anemia. Prog Clin Biol Res. 1985;191:433–445. [PubMed] [Google Scholar]
- Collins FS, Metherall JE, Yamakawa M, Pan J, Weissman SM., and, Forget BG. A point mutation in the Aγ-globin gene promoter in Greek hereditary persistence of fetal haemoglobin. Nature. 1985;313:325–326. doi: 10.1038/313325a0. [DOI] [PubMed] [Google Scholar]
- Mantovani R, Superti-Furga G, Gilman J., and, Ottolenghi S. The deletion of the distal CCAAT box region of the Aγ-globin gene in black HPFH abolishes the binding of the erythroid specific protein NFE3 and of the CCAAT displacement protein. Nucleic Acids Res. 1989;17:6681–6691. doi: 10.1093/nar/17.16.6681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry M, Grosveld F., and, Dillon N. A single point mutation is the cause of the Greek form of hereditary persistence of fetal haemoglobin. Nature. 1992;358:499–502. doi: 10.1038/358499a0. [DOI] [PubMed] [Google Scholar]
- Ronchi A, Berry M, Raguz S, Imam A, Yannoutsos N, Ottolenghi S.et al. (1996Role of the duplicated CCAAT box region in γ-globin gene regulation and hereditary persistence of fetal haemoglobin EMBO J 15143–149. [PMC free article] [PubMed] [Google Scholar]
- Peterson KR, Li QL, Clegg CH, Furukawa T, Navas PA, Norton EJ.et al. (1995Use of yeast artificial chromosomes (YACs) in studies of mammalian development: production of β-globin locus YAC mice carrying human globin developmental mutants Proc Natl Acad Sci USA 925655–5659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin DI, Tsai SF., and, Orkin SH. Increased γ-globin expression in a nondeletion HPFH mediated by an erythroid-specific DNA-binding factor. Nature. 1989;338:435–438. doi: 10.1038/338435a0. [DOI] [PubMed] [Google Scholar]
- McDonagh KT, Lin HJ, Lowrey CH, Bodine DM., and, Nienhuis AW. The upstream region of the human γ-globin gene promoter. Identification and functional analysis of nuclear protein binding sites. J Biol Chem. 1991;266:11965–11974. [PubMed] [Google Scholar]
- Chin JY, Kuan JY, Lonkar PS, Krause DS, Seidman MM, Peterson KR.et al. (2008Correction of a splice-site mutation in the beta-globin gene stimulated by triplex-forming peptide nucleic acids Proc Natl Acad Sci USA 10513514–13519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNeer NA, Chin JY, Schleifman EB, Fields RJ, Glazer PM., and, Saltzman WM. Nanoparticles deliver triplex-forming PNAs for site-specific genomic recombination in CD34+ human hematopoietic progenitors. Mol Ther. 2011;19:172–180. doi: 10.1038/mt.2010.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knauert MP, Kalish JM, Hegan DC., and, Glazer PM. Triplex-stimulated intermolecular recombination at a single-copy genomic target. Mol Ther. 2006;14:392–400. doi: 10.1016/j.ymthe.2006.03.020. [DOI] [PubMed] [Google Scholar]
- Rogers FA, Vasquez KM, Egholm M., and, Glazer PM. Site-directed recombination via bifunctional PNA-DNA conjugates. Proc Natl Acad Sci USA. 2002;99:16695–16700. doi: 10.1073/pnas.262556899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schleifman EB, Bindra R, Leif J, del Campo J, Rogers FA, Uchil P.et al. (2011Targeted disruption of the CCR5 gene in human hematopoietic stem cells stimulated by peptide nucleic acids Chem Biol 181189–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peffer NJ, Hanvey JC, Bisi JE, Thomson SA, Hassman CF, Noble SA.et al. (1993Strand-invasion of duplex DNA by peptide nucleic acid oligomers Proc Natl Acad Sci USA 9010648–10652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blau CA, Barbas CF., 3rd, , Bomhoff AL, Neades R, Yan J, Navas PA.et al. (2005γ-Globin gene expression in chemical inducer of dimerization (CID)-dependent multipotential cells established from human b-globin locus yeast artificial chromosome (β-YAC) transgenic mice J Biol Chem 28036642–36647. [DOI] [PubMed] [Google Scholar]
- Majumdar A, Puri N, Cuenoud B, Natt F, Martin P, Khorlin A.et al. (2003Cell cycle modulation of gene targeting by a triple helix-forming oligonucleotide J Biol Chem 27811072–11077. [DOI] [PubMed] [Google Scholar]
- Wenger RH, Stiehl DP., and, Camenisch G. Integration of oxygen signaling at the consensus HRE. Sci STKE. 2005;2005:re12. doi: 10.1126/stke.3062005re12. [DOI] [PubMed] [Google Scholar]
- Møllegaard NE, Buchardt O, Egholm M., and, Nielsen PE. Peptide nucleic acid·DNA strand displacement loops as artificial transcription promoters. Proc Natl Acad Sci USA. 1994;91:3892–3895. doi: 10.1073/pnas.91.9.3892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rayburn ER., and, Zhang R. Antisense, RNAi, and gene silencing strategies for therapy: mission possible or impossible. Drug Discov Today. 2008;13:513–521. doi: 10.1016/j.drudis.2008.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]