

Abstract
The potency of immunotherapies targeting endogenous tumor antigens is hindered by immune tolerance. We created a therapeutic agent comprised of a tumor-homing module fused to a functional domain capable of selectively rendering tumor cells sensitive to foreign antigen-specific CD8+ T cell-mediated immune attack, and thereby, circumventing concerns for immune tolerance. The tumor-homing module is comprised of a single-chain variable fragment (scFv) that specifically binds to mesothelin (Meso), which is commonly overexpressed in human cancers, including ovarian tumors. The functional domain is comprised of the Fc portion of IgG2a protein and foreign immunogenic CD8+ T cell epitope flanked by furin cleavage sites (R), which can be recognized and cleaved by furin that is highly expressed in the tumor microenvironment. We show that our therapeutic protein specifically loaded antigenic epitope onto the surface of mesothelin-expressing tumor cells, rendering tumors susceptible to antigen-specific cytotoxic CD8+ T lymphocytes (CTL)-mediated killing in vitro and in vivo. Our findings have important implications for bypassing immune tolerance to enhance cancer immunotherapy.
Introduction
Antigen-specific immunotherapy is important for its ability to harness the immune system to specifically target tumors without the toxicity associated with traditional chemoradiation. Cytotoxic CD8+ T lymphocytes (CTLs) can selectively kill tumor cells at multiple sites throughout the body. Furthermore, antigen-specific immunotherapy is unlikely to generate nonspecific autoimmunity (for review, see ref. 1). Despite this, antigen-specific immunotherapy targeting tumor-associated endogenous antigen faces the major obstacle of immune tolerance.
We hypothesize that by selectively coating tumor cells with foreign immunogenic CD8+ T cell peptide(s) recognized by preexisting immunity, immune tolerance can be bypassed and preexisting immunity could control the tumor. We designed a therapeutic agent containing a tumor-homing module fused to a functional cargo domain using a murine ovarian cancer model. We chose ovarian cancer because it is a highly lethal gynecological malignancy. The tumor-homing portion of our therapeutic chimeric protein is comprised of an antimesothelin single-chain variable fragment (meso-scFv), which has high binding affinity to mesothelin. Mesothelin is overexpressed by most ovarian cancers,2,3 and therefore is an ideal target molecule for the therapeutic protein carrying foreign immunogenic CTL epitope. The cargo domain of the therapeutic chimeric protein is comprised of Fc portion of the IgG2a protein and major histocompatibility complex (MHC) class I-restricted foreign immunogenic CTL epitope flanked by furin cleavage sites. The antigenic CTL epitope is loaded onto tumor cells because furin selectively cleaves the amino acid sequence, RVKR. Many tumors, including ovarian tumors (for review, see ref. 4), highly express furin.5,6,7,8,9,10 The results of this study indicate that meso-scFv can preferentially shuttle functional cargo to mesothelin-expressing ovarian cancer cells, where cleavage by furin releases the foreign immunogenic CTL epitope to be loaded on MHC class I molecules of tumor cells and render tumor cells susceptible to antigen-specific CTL-mediated killing, both in vitro and in vivo. This system may serve as a foundation leading to the development of future clinical therapies.
Results
Meso-scFv-ROR-Fc binds mesothelin-expressing tumor cells in vitro and leads to MHC class I presentation of OVA peptide to OVA-specific CD8+ T cells
We created a chimeric antihuman mesothelin scFv (Meso-scFv) conjugated with Fc (IgG2a) protein containing ovalbumin (OVA) peptide flanked by furin cleavage sites (Meso-scFv-ROR-Fc). Figure 1a shows the schematic diagram of chimeric Meso-scFv-ROR-Fc construct, control Meso-scFv-Fc protein without OVA peptide, and control Meso-scFv-O-Fc protein without furin cleavage sites. As shown in Figure 1b, only the furin-expressing baby hamster kidney 21 cells transfected with Meso-scFv-ROR-Fc generated a 30-kDa band that is consistent with the size of Fc fragment, suggesting cleavage of the chimeric Meso-scFv-ROR-Fc. In comparison, baby hamster kidney 21 cells transfected with Meso-scFv-O had a 60-kDa band that is consistent with the size of uncleaved full-length protein. In addition, furin-deficient FD11 cells were transfected with various Meso-scFv-Fc chimeric proteins. As shown in Figure 1c, the FD11 cells transfected with Meso-scFv-ROR-Fc showed an ~60-kDa band consistent with the size of the uncleaved, full-length protein. These results indicate furin expressed by cancer cells can act on the furin cleavage sites of chimeric Meso-scFv-ROR-Fc.
Figure 1.

Generation and characterization of therapeutic chimeric proteins. (a) Schematic diagram of different chimeric proteins (Meso-scFv-Fc) containing antihuman mesothelin scFv (Meso-scFv), ovalbumin peptide (SIINFEKL), furin cleavage sites (RVKR), and Fc protein (IgG2a). (b,c) Gel electrophoresis of isolated proteins from (b) furin-positive BHK21 cells or (c) furin-deficient FD11 cells26 transfected with the different variants of Meso-scFv-Fc. Protein was isolated from supernatant of cultured cells and then characterized by sodium dodecyl sulfate-PAGE and Coomassie Brilliant Blue staining. (d) Representative flow cytometry analysis of chimeric protein bound to ID8-meso with ID8 cells28,29 as the control. ID8-meso was generated through transfection of ID8 cells with DNA encoding human mesothelin as previously described.24 Purified proteins were incubated with either ID8-meso or ID8 cells and then stained with phycoerythrin (PE)-labeled antimouse Fc antibody. (e) Immunofluorescence detection of Meso-scFv-ROR-Fc binding to ID8-meso cells and cleavage of furin recognition sites over time. The controls were meso-scFv-Fc and ID8 cells. Tumor cells were treated with different chimeric proteins and then stained with PE-labeled antimouse Fc antibody. BHK, baby hamster kidney; scFv, single-chain variable fragment.
To determine whether various chimeric Meso-scFv-Fc proteins can selectively bind to human mesothelin-expressing murine ovarian tumor cell line, ID8-meso, we performed flow cytometry analysis. As shown in Figure 1d, human mesothelin-expressing ID8-meso cells incubated with various Meso-scFv-Fc proteins displayed a shift consistent with increased cell binding as compared with non mesothelin-expressing ID8 cells. This suggests meso-scFv-Fc chimeric proteins binds specifically to human mesothelin-expressing ID8-meso tumor cells.
We then determined if the binding of Meso-scFv-ROR-Fc to human mesothelin-expressing ID8-meso cells could facilitate the cleavage of furin recognition sites in the chimeric protein. ID8-meso or control ID8 tumor cells were incubated with chimeric Meso-scFv-Fc or Meso-scFv-ROR-Fc and then stained with phycoerythrin (PE)-labeled antimouse Fc antibody for visualization by fluorescence microscopy. As shown in Figure 1e, ID8-meso incubated with Meso-scFv-ROR-Fc or Meso-scFv-Fc had similar levels of red fluorescent activity at 0 minute. However, the red fluorescent activity of ID8-meso incubated with Meso-scFv-ROR-Fc was greatly reduced at 60 minutes. The reduced fluorescence indicates that furin-mediated proteolysis of the chimeric Meso-scFv-ROR-Fc protein cleavage sites resulted in loss of the Fc fragment.
We further determined if the binding of Meso-scFv-ROR-Fc to ID8-meso could enable MHC class I presentation of OVA peptide and activate OVA-specific CD8+ T cells. As shown in Figure 2a,b, ID8-meso incubated with Meso-scFv-ROR-Fc had the greatest OVA-specific CD8+ T-cell activation (>20-fold). Furthermore, the activation of OVA-specific CD8+ T cells was positively correlated with the amount of Meso-scFv-ROR-Fc incubated with ID8-meso in a concentration-dependent manner. As ID8 incubated with Meso-scFv-ROR-Fc activated some OVA-specific CD8+ T cells, furin alone could lead to low-level cleavage of the Meso-scFv-ROR-Fc protein, resulting in peptide coating of tumor cells. However, the importance of mesothelin-binding in mediating furin cleavage of chimeric protein is exemplified by the >10-fold difference in the activation of OVA-specific CTL between ID8-meso and ID8 incubated with Meso-scFv-ROR-Fc.
Figure 2.

Major histocompatibility complex class I presentation of ovalbumin (OVA) peptide to OVA-specific CD8+T cells by ID8-meso cells treated with Meso-scFv-ROR-Fc. (a) Flow cytometry characterization of OVA-specific CD8+ T-cell activation by ID8-meso cells treated with different chimeric proteins. ID8-meso or control ID8 tumor cells were incubated with different protein concentrations followed by incubation with 2 × 105 OVA-specific cytotoxic CD8+ T lymphocytes (CTLs). OVA-specific CD8+ T-cell activation was determined by CD8 and intracellular interferon γ (IFN-γ) staining. (b) Representative bar graph depicting the % of IFN-γ-secreting OVA-specific CD8+ T cells out of total OVA-specific T cells (mean ± SD). (c) Representative luminescence imaging of in vitro OVA-specific CTL killing of luciferase-expressing ID8-meso cells treated with different concentrations of Meso-scFv-ROR-Fc. Luciferase-expressing ID8-meso or control ID8 tumor cells treated with chimeric proteins were later incubated with 2 × 105 OVA-specific CD8+ T cells. CTL-mediated tumor cell death was determined by decreasing luminescence activity. (d) Bar graph depiction of tumor cell viability after treatment with protein and/or OVA-specific cytotoxic T cells (mean ± SD) (data representative of two experiments). Meso-scFv, mesothelin single-chain variable fragment.
We then determined if ID8-meso cells bound by Meso-scFv-ROR-Fc were susceptible to OVA-specific CD8+ T-cell killing. As shown in Figure 2c,d, ID8-meso incubated with Meso-scFv-ROR-Fc had the greatest amount of OVA-specific CTL-mediated tumor cell death, as seen by the greatest reduction in luminescence activity. In addition, the amount of Meso-scFv-ROR-Fc incubated with ID8-meso was positively correlated with OVA-specific CTL killing of tumor cells in a dose-dependent manner. This dose-dependent killing was observed in both ID8-meso and ID8 tumor cells, though Meso-scFv-ROR-Fc binding to tumor cells increased OVA-specific CTL killing of ID8-meso (as compared with ID8) eightfold. Taken together, these results indicate Meso-scFv-ROR-Fc specifically binds ID8-meso and facilitates MHC class I presentation of OVA peptide to activate OVA-specific CD8+ T cells. Furthermore, the binding of Meso-scFv-ROR-Fc to tumor cells renders bound tumor cells susceptible to OVA-specific CTL killing.
Meso-scFv-ROR-Fc binds mesothelin-expressing tumor cells in vivo and leads to MHC class I presentation of OVA peptide to OVA-specific CD8+ T cells
To determine if the chimeric proteins could target human mesothelin-expressing ID8-meso in vivo, we isolated and characterized tumor cells from tumor-bearing mice intraperitoneally (i.p.) injected with various Meso-scFv-Fc chimeric proteins. As shown in Figure 3a, ID8-meso cells isolated from tumor-bearing mice injected with various chimeric proteins showed significant positive staining, indicating the chimeric protein had bound to tumor cells. ID8-meso from mice injected with Meso-scFv-ROR-Fc had less staining whereas ID8-meso from mice injected with Meso-scFv-Fc and Meso-scFv-O-Fc had similarly higher levels of staining. This suggests the ID8-meso from mice injected with Meso-scFv-ROR-Fc had fewer Fc-positive molecules due to furin-mediated loss of the Fc fragment. In addition, reduced binding of Meso-scFv-ROR-Fc to ID8-meso (Figure 3a) was not observed under in vitro conditions (Figure 1d) as the cell binding assay was performed at 4 °C, which inhibits furin function. We also observed that no binding occurred between chimeric Meso-scFv-Fc proteins and ID8 tumor cells. This suggests that the Meso-scFv portion of chimeric proteins can specifically bind human mesothelin-expressing tumor cells, and that the furin cleavage sites flanking the OVA peptide enable the proteolytic release of the Fc fragment from the chimeric proteins.
Figure 3.

Major histocompatibility complex class I presentation of ovalbumin (OVA) peptide to OVA-specific CD8+ T cells by ID8-meso cells treated with Meso-scFv-ROR-Fc in vivo. (a) Flow cytometry characterization of Meso-scFv-ROR-Fc binding to ID8-meso tumor cells. PBS was used as a negative control. Mice were i.p. injected with GFP-expressing ID8-meso or control ID8 tumor cells followed by i.p injection of different Meso-scFv-Fc chimeric proteins. Tumor cells isolated from peritoneal wash of treated tumor-bearing mice were stained with phycoerythrin (PE)-labeled antimouse Fc antibody and analyzed by flow cytometry analysis. GFP-positive cells were gated for analysis. (b) Flow cytometry characterization of OVA-specific CD8+ T-cell activation by tumor cells treated with different chimeric proteins. Tumor cells from peritoneal wash of tumor-bearing mice treated with different chimeric proteins were incubated with OVA-specific CD8+ T cells. OVA-specific CD8+ T-cell activation was determined by CD8 and intracellular interferon γ (IFN-γ) staining. (c) Representative bar graph depicting % of IFN-γ-secreting OVA-specific CD8+ T cells out of total OVA-specific T cells (mean ± SD). (d) Flow cytometry analysis to characterize the proliferation of OVA-specific CD8+ T cells in ID8-meso tumor-bearing mice treated with Meso-scFv-ROR-Fc. C57BL/six mice (five mice/group) were injected with GFP-expressing ID8-meso tumor cells (1 × 106 cells/mouse). One day later, 50 µg of different chimeric proteins and 2.5 × 106 carboxyfluorescein succinimidyl ester (CFSE)-labeled OVA-specific CD8+ T cells were i.p. injected. Three days later, washed intraperitoneal cells were stained with PE-conjugated anti-CD8 antibody then analyzed by flow cytometry to determine the extent of CFSE dilution in CFSE-labeled CD8+ T cells. Meso-scFv, mesothelin single-chain variable fragment; PBS, phosphate buffered saline.
We then analyzed whether the binding of the chimeric proteins to human mesothelin-expressing ID8-meso tumor cells could load OVA peptide onto MHC class I molecules and activate OVA-specific CD8+ T cells in vivo. Tumor cells isolated from the peritoneal wash of tumor-bearing mice i.p. injected with various Meso-scFv-Fc chimeric proteins were incubated with OVA-specific CD8+ T cells. CD8+ T-cell activation was characterized by intracellular cytokine staining for interferon γ (IFN-γ) and CD8, followed by flow cytometry analysis.
As shown in Figure 3b,c, tumor cells derived from the peritoneal wash of ID8-meso tumor-bearing mice i.p. injected with Meso-scFv-ROR-Fc generated the highest level of activation of OVA-specific CD8+ T cells. In contrast, tumor cells derived from peritoneal wash of mesothelin-negative ID8 tumor-bearing mice injected with the Meso-scFv-ROR-Fc chimeric proteins did not lead to significant activation of OVA-specific CD8+ T cells. Our data suggest that cleavage of Meso-scFv-ROR-Fc by furin in ID8-meso tumor cells can free OVA peptide to be loaded onto MHC class I molecules of ID8-meso cells and activate OVA-specific CD8+ T cells.
The therapeutic potential of this strategy and candidacy for clinical translation hinges on whether or not antigen-specific CD8+ T cells can infiltrate the tumor site and proliferate in vivo. To demonstrate whether the OVA-specific CD8+ T cells could proliferate in the mesothelin expressing tumor-bearing mice treated with the chimeric Meso-scFv-ROR-Fc, we labeled the OVA-specific CD8+ T cells with carboxyfluorescein succinimidyl ester (CFSE) and injected the i.p. labeled T cells. As shown in Figure 3d, treatment of tumor-bearing mice only with Meso-scFv-ROR-Fc, but not Meso-scFv-Fc or Meso-scFv-O-Fc, lead to significant proliferation of CSFE-labeled OVA-specific CD8+ T cells. These results indicate that treatment of tumor-bearing mice with Meso-scFv-ROR-Fc can render OVA-specific CD8+ T cells capable of proliferation in the abdominal cavity of the tumor-bearing mice. Such results serve as an important step toward developing an effective form of immunotherapy.
Meso-scFv-ROR-Fc combined with adoptive transfer of OVA-specific CD8+ T cells produces a potent antitumor effect
We then determined if i.p. administration of Meso-scFv-ROR-Fc could render human mesothelin-expressing ID8-meso tumors susceptible to OVA-specific CTL killing in vivo. Figure 4a displays the schematic diagram of the treatment regimen. As shown in Figure 4b,c, significant tumor growth reduction, as evidenced by decreased luminescence, was seen only in tumor-bearing mice treated with Meso-scFv-ROR-Fc in conjunction with adoptive transfer of OVA-specific CD8+ T cells. Administration of Meso-scFv-ROR-Fc without adoptive transfer elicited no therapeutic effects, suggesting antitumor effects are mediated by OVA-specific CD8+ T cells. Furthermore, tumor-bearing mice treated with Meso-scFv-ROR-Fc in conjunction with OVA-specific CD8+ T cells had the longest survival (Figure 4d). This suggests that ID8-meso treated with Meso-scFv-ROR-Fc presents OVA peptide-loaded MHC class I on the cell surface, resulting in OVA-specific CTL-mediated therapeutic antitumor effects in vivo.
Figure 4.
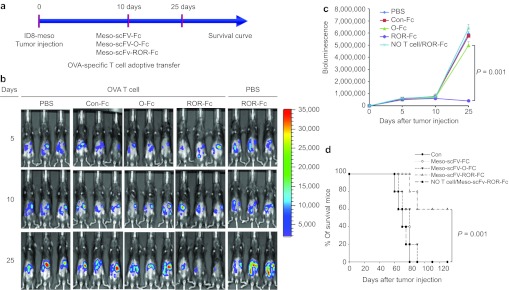
In vivo therapeutic antitumor effects by various chimeric proteins combined with adoptive transfer of ovalbumin (OVA)-specific CD8+ T cells. (a) Schematic diagram of treatment regimen. Mice were i.p. injected with luciferase-expressing ID8-meso cells on day 0. On day 10, tumor-bearing mice were i.p injected with one type of chimeric protein in conjunction with adoptive transfer of OVA-specific CD8+ T cells. Red bars represent time of imaging. (b) Representative luminescence image of fluorescence intensity (tumor load) in tumor-bearing mice treated with different chimeric proteins with or without adoptive transfer of OVA-specific CD8+ T cells. (c) Line graph depicting the fluorescence intensity (tumor load) in tumor-bearing mice treated with different regimens. (d) Kaplan–Meier survival analysis of tumor-bearing mice treated with different regimens.
Meso-scFv-ROR-Fc facilitated activation of OVA-specific CD8+ T cells is not specific to ID8-meso cells
We tested our approach in another human mesothelin-expressing murine tumor cell line, TC-1/Meso. TC-1/Meso incubated with the various Meso-scFv-Fc proteins stained positive for cell binding, unlike mesothelin-negative TC-1 cells (Figure 5a). In addition, TC-1/Meso incubated with Meso-scFv-ROR-Fc had the highest activation of OVA-specific CD8+ T cells (Figure 5b,c). However, mesothelin-negative TC-1 incubated with Meso-scFv-ROR-Fc activated OVA-specific CD8+ T cells to a small degree. This suggests that some of the Meso-scFv-ROR-Fc may have been cleaved by the furin expressed in TC-1, leading to the loading of OVA peptides on the MHC class I molecules and activation of OVA-specific CD8+ T cells. Similar to the ID8-meso tumor system, the OVA-specific CD8+ T cell activation by mesothelin-negative TC-1 is (~sevenfold) lower than that of mesothelin-positive TC-1/Meso. As shown in Figure 5d,e, Meso-scFv-ROR-Fc binding to TC-1/Meso led to the greatest OVA-specific CTL-mediated tumor cell death. We also observed low level tumor killing in TC-1 tumor cells pulsed with Meso-scFv-ROR-Fc as compared with TC-1/Meso (~15-fold difference). Our data demonstrate that our system works in another mesothelin-expressing tumor model, TC-1/Meso.
Figure 5.
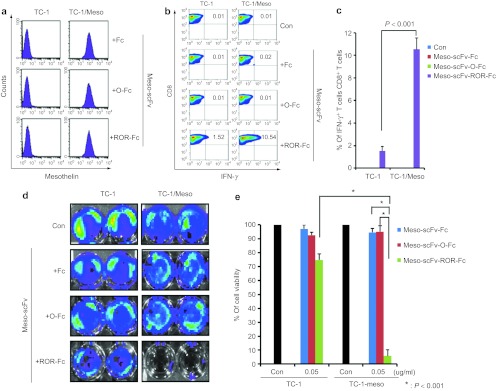
Major histocompatibility complex class I presentation of ovalbumin (OVA) peptide to OVA-specific CD8+ T cells by TC-1/Meso tumor cells treated with Meso-scFv-ROR-Fc. (a) Representative flow analysis of Meso-scFv-ROR-Fc protein binding to TC-1/Meso. The purified chimeric proteins were incubated with TC-1/Meso or control TC-1 tumor cells followed by staining with phycoerythrin-labeled antimouse Fc antibody. (b) Flow cytometry characterization of OVA-specific CD8+ T-cell activation by TC-1/Meso cells treated with different chimeric proteins. OVA-specific CD8+ T-cell activation was determined by CD8 and intracellular IFN-γ staining. (c) Representative bar graph depicting the % of IFN-γ-secreting OVA-specific CD8+ T cell out of total OVA-specific CD8+ T cells (mean ± SD). (d) Representative luminescence imaging of in vitro OVA-specific cytotoxic CD8+ T lymphocytes (CTL) killing of luciferase-expressing TC-1/Meso cells treated with Meso-scFv-ROR-Fc. Degree of CTL-mediated tumor cell death is indicated by decrease of luminescence activity. (e) Bar graph depicting viability of tumor cells treated with protein and/or OVA-specific cytotoxic T cells (mean ± SD) (representative data of two experiments). Meso-scFv, mesothelin single-chain variable fragment.
We were interested in determining whether the release of OVA peptide by furin cleavage of Meso-scFv-ROR-Fc in bound tumor cells could also make mesothelin-negative nonbound tumor cells to become targets of OVA-specific CTL-mediated killing. The supernatants from ID8-meso or TC-1/Meso tumor cells treated with different Meso-scFv-Fc chimeric proteins were collected and incubated with mesothelin-negative ID8 and TC-1 tumor cells. OVA-specific CD8+ T cells were then added and activation was analyzed by intracellular cytokine staining for IFN-γ and CD8 followed by flow cytometry analysis. As shown in Supplementary Figure S1a,b, only the supernatant from ID8-meso or TC-1/Meso cells treated with Meso-scFv-ROR-Fc could activate OVA-specific CD8+ T cells on incubation with mesothelin-negative ID8 or TC-1 cells. Furthermore, as shown in Supplementary Figure S1c,d, luciferase-expressing ID8 or TC-1 tumor cells incubated with supernatant from ID8-meso or TC-1/Meso tumor cells treated with Meso-scFv-ROR-Fc had the greatest tumor cell death (reduced luminescence) upon addition of OVA-specific CD8+ T cells. In addition, the furin inhibitor experiment suggests the importance of furin in releasing OVA peptide for loading of MHC class I molecules (see Supplementary Figure S2). These findings suggest that the specific binding of Meso-scFv-ROR-Fc to human mesothelin-expressing tumors can lead to the release of OVA peptide and sensitize nonbound tumor cells to OVA-specific CTL-mediated killing.
OVA peptide located at the carboxyl end of Meso-scFv-Fc chimeric protein can lead to MHC class I presentation of OVA peptide in different human mesothelin-expressing tumor cells, ID8-meso and TC-1/Meso
The Meso-scFv-ROR-Fc chimeric protein design leads to the loss of Fc fragment upon cleavage by furin on the bound tumor cell, which abolishes the sensitivity of tumor cells to antibody-dependent cellular cytotoxicity and complement-dependent cytotoxicity. Thus, we generated antihuman mesothelin scFv conjugated with Fc (IgG2a) protein containing a C-terminal OVA peptide preceded by a furin cleavage site (Meso-scFv-Fc-RO) (Figure 6a) and tested whether the chimeric protein was still capable of MHC class I presentation of the OVA peptide in human mesothelin-expressing tumor cells. As shown in Figure 6b,c, when luciferase-expressing ID8-meso and TC-1/Meso were incubated with Meso-scFv-Fc-RO, these human mesothelin-expressing tumor cell lines were more susceptible to OVA-specific CD8+ T-cell killing, as demonstrated by the dramatically reduced luciferase activity as compared to incubation with Meso-scFv-Fc or Meso-scFv-Fc-O. Our results indicate that the binding of Meso-scFv-Fc-RO to human mesothelin-expressing tumor cells can lead to MHC class I presentation of OVA peptide, rendering the human mesothelin-expressing tumor cells susceptible to OVA-specific CD8+ T-cell killing. Furthermore, this data in conjunction with the data in Figure 2, suggest that the efficacy of the chimeric protein is retained regardless of the location of the OVA peptide, as long as the OVA peptide is flanked by furin recognition sequence(s). Meso-scFv-Fc-RO can lead to antigen-specific CD8+ T cell-mediated killing and allow the Fc fragment to remain and maintain tumor cell sensitivity to antibody-dependent cellular cytotoxicity and complement-dependent cytotoxicity.
Figure 6.

In vitro cytotoxicity assay to demonstrate the major histocompatibility complex class I presentation of ovalbumin (OVA) peptide in ID8/meso or TC-1/Meso tumor cells treated with Meso-scFv-Fc-RO. (a) Schematic diagram depicting antihuman mesothelin scFv fragment conjugated with Fc protein (Meso-scFv-Fc) containing OVA peptide SIINKFEL at the carboxy end with (Meso-scFv-Fc-RO) or without (Meso-scFv-Fc-O) flanking by the furin recognition sequence RVKR. (b) Representative luminescence imaging to demonstrate in vitro cytotoxicity by OVA-specific CD8+ T cells of ID8/meso or TC-1/Meso tumor cells treated with Meso-scFv-Fc-RO. Luciferase expressing ID8-meso and TC-1-meso cells were treated with 0.5 µg of purified chimeric proteins (PBS, Meso-scFv-Fc, Meso-scFv-Fc-O, or Meso-scFv-Fc-RO). After 18 hours, treated tumor cells were incubated with 2 × 105 OVA-specific CD8+ T cells. The degree of cytotoxic CD8+ T lymphocytes-mediated killing of the tumor cells was indicated by the decrease of luminescence activity using the IVIS luminescence imaging system series 2000. Bioluminescence signals were acquired for 1 minute. (c) Bar graph depicting viability of tumor cells treated with various chimeric protein and/or OVA-specific cytotoxic T cells (mean ± SE). Data shown are representative of two experiments performed. scFv, single-chain variable fragments; PBS, phosphate buffered saline.
The Meso-scFv-Fc chimeric protein can be extended to HPV-16 E7 CTL epitope to result in the loading of E7 peptide on MHC class I molecules of mesothelin-expressing tumors
We generated antihuman mesothelin scFv conjugated with Fc (IgG2a) protein containing a C-terminal HPV-16 E7 peptide preceded by a furin cleavage site (Meso-scFv-Fc-RE7) (Figure 7a). This was used to test whether the chimeric protein with an alternate antigenic peptide, the HPV E7 peptide, could elicit the desired antitumor response against human mesothelin-expressing tumor cells. As expected, ID8-meso cells that were treated with Meso-scFv-Fc-RE7 were able to activate E7-specific CD8+ T cells (Figure 7b,c). When luciferase-expressing ID8-meso tumor cells were treated with Meso-scFv-Fc- RE7, E7-specific CTL-mediated killing of ID8-meso occurred as demonstrated by the reduction in luminescence (Figure 7d,e). This demonstrates the versatility of the chimeric protein system and suggests that other CTL epitopes can potentially be incorporated into the chimeric protein to be targeted to mesothelin-expressing tumors, resulting in loading of the CTL epitope on MHC class I molecules.
Figure 7.

Characterization of the major histocompatibility complex class I presentation of E7 peptide to E7-specific CD8+ T cells by ID8-meso tumor cells treated with Meso-scFv-Fc-RE7. (a) Schematic diagram depicting antihuman mesothelin scFv fragment (Meso-scFv) conjugated to Fc protein-containing E7 peptide, RAHYNIVTE, at the carboxyl end proceeded by furin recognition sequence, RVKR. (b) Flow cytometry analysis to characterize the activation of E7-specific CD8+ T cells by ID8-meso tumor cells treated with the various Meso-scFv-Fc chimeric proteins. ID8-meso tumor cells (1 × 105 cells/well) were added to 48-well plates and incubated with 0.5 µg/ml of proteins. After 18 hours, treated tumor cells were incubated with 2 × 105 E7-specific cytotoxic CD8+ T lymphocytes (CTLs). Activated interferon γ (IFN-γ)-secreting E7-specific CD8+ T cells were determined by staining for surface CD8+ and intracellular IFN-γ before flow cytometry analysis. (c) Representative bar graph depicting the % of IFN-γ-secreting E7-specific CD8+ T cells out of total E7-specific T cells (mean ± SD). (d) Representative luminescence imaging to demonstrate in vitro cytotoxicity by E7-specific CD8+ T cells of ID8-meso tumor cells treated with Meso-scFv-Fc- RE7. Luciferase-expressing ID8-meso tumor cells were plated in a 24-well plate (1 × 105 cells/well) and incubated with 0.5 µg/ml of Meso-scFv-Fc chimeric proteins. After 18 hours, treated tumor cells were incubated with 2 × 105 ovalbumin (OVA)-specific CD8+ T cells. Luciferase-expressing ID8 tumor cells not incubated with chimeric protein and treated with 2 × 105 OVA-specific CTL alone were the control. The degree of CTL-mediated killing of tumor cells was seen as a reduction in luminescence activity and measured by IVIS luminescence imaging system series 2000. Bioluminescence signals were acquired for 1 minute. (e) Bar graph depicting viability of tumor cells treated with E7-specific cytotoxic T cells and/or protein (mean ± SD). Data shown are representative of two experiments performed. IFN, interferon.
Human tumors expressing human mesothelin can also be targeted by the chimeric protein resulting in loading of CTL epitopes on MHC class I molecules
In our study, we also characterized several human tumors for the expression of human mesothelin. As shown in Supplementary Figure S3, human ovarian cancer cell lines expressed human mesothelin at different levels. In addition, HeLa cells were found to express human mesothelin. To ensure the chimeric protein therapy is efficacious against human cancer cells and is also capable of coating cancer cells with foreign peptides of different MHC class I restrictions, human mesothelin-expressing HeLa cells were transfected with MHC class I alleles H2-Kb or H2-Db and then treated with Meso-scFv-Fc-RO or Meso-scFv-Fc-RE7 (Figure 8a). Flow cytometry analysis shows that H2-Kb transfected HeLa cells treated with the chimeric proteins containing H2-Kb-restricted OVA peptide was able to activate the largest percentage of OVA-specific CD8+ T cells. Similarly, H2-Db-transfected HeLa cells treated with chimeric proteins containing H2-Db-restricted E7 peptide were able to promote the greatest activation of E7-specific CD8+ T cells. Taken together, our data suggest that the Meso-scFv-Fc chimeric protein can also be targeted to mesothelin-expressing human tumors resulting in loading of CTL epitopes on MHC class I molecules of the tumor cells.
Figure 8.

Activation of ovalbumin (OVA)-specific or E7-specific CD8+ T cells by HeLa cells transfected with mouse major histocompatibility complex class I Kb or Db and treated with Meso-scFv-Fc-RO or Meso-scFv-Fc-RE7. (a) T-cell activation of human cancer cells. HeLa cells, a human cervical cancer cell line, were added to 48-well plates (5 × 104 per well). HeLa cells were transfected with 0.5 µg of pcDNA Kb or Db plasmid using Lipofectamine 2000 transfection reagent (Invitrogen). One day after, the cells were incubated with 0.5 µg of Meso-scFv-Fc-RE7 or Meso-scFv-Fc-RO proteins. After 18 hours, treated tumor cells were incubated with 2 × 105 OVA-specific or E7-specific CD8+ T cells for 1 day. CD8+ T-cell activation was determined by intracellular cytokine staining for IFN-γ and analyzed by flow cytometry. (b) Representative bar graph depicting the % of IFN-γ positive cells out of total CD8+ T cells (mean ± SD). Meso-scFv, mesothelin single-chain variable fragment.
Discussion
Our study represents an innovative approach to use preexisting immunity against various common viruses to generate antitumor effects by targeting the viral antigen-specific CTLs against tumor cells that have been coated with foreign antigens. Most individuals have T cell-mediated immunity against a host of common viruses, such as Epstein-Barr virus, human cytomegalovirus, and influenza. Conveniently, many of these viral MHC class I-restricted epitopes have already been identified.11 To generate a chimeric protein that is suitable for different individuals, who have different human leukocyte antigen alleles, it will be important to come up with a chimeric protein containing many CTL epitopes from different viruses which are restricted by different human leukocyte antigen alleles. In our study, we are able to generate a chimeric protein with the CTL epitopes flanked by furin cleavage sites at the 3′ end of Fc fragment (see Figure 6). It is conceivable that multiple CTL epitopes restricted by different human leukocyte antigen alleles can be generated, flanked by furin cleavage sites, and placed at the 3′ end of the Fc fragment to be suitable for individuals with different human leukocyte antigen alleles. When such chimeric proteins bind with the target tumor cell, furin will be able to cleave the protein at furin cleavage sites and release the CTL epitopes to coat the tumor cell rendering the tumor cell susceptible to CTL-mediated killing. Furthermore, the use of multiple CTL epitopes flanked by furin-sensitive linkers has already been demonstrated by Lu et al.12 to be both feasible and capable of presentation through different MHC class I molecules.
The Meso-scFv fragment has been examined in a multitude of studies that focused on finding a way to target mesothelin-expressing tumor cells for direct killing or indirect killing through immune-mediated mechanisms. For example, Meso-scFv has been used to target a cytotoxic recombinant immunotoxin, Pseudomonas exotoxin A,13 to mesothelin-expressing tumor cells. Meso-scFv has also been used linked to MHC-peptide complexes to introduce MHC-peptide complexes to tumor cells for T cell-mediated killing.14 The success of our mesothelin-targeting approach suggests that other tumor-specific target molecules could also be used. For example, epidermal growth factor receptor is widely expressed in cancers such as cervical,15 head and neck,16 esophageal,17 ovarian,18 and bladder19 cancers (for review, see ref. 20,21). Other targets of interest include Her2/neu22 and other surface molecules highly expressed by tumors.
We chose furin as the proteolytic enzyme to release the foreign immunogenic peptide from the chimeric protein because furin is highly expressed by tumor cells.5,6,7,8,9,10 Our strategy can also be extended to other tumor microenvironment enzymes, such as matrix metalloproteinase-2 and matrix metalloproteinase-9 and both of them have unique cleavage sites. However, furin can also be highly expressed at sites of inflammation.10 Thus, it will be important to consider the potential side effects as clinical translation of related treatments advances. Nevertheless, the specificity of targeting mesothelin may alleviate these concerns.
For clinical translation of this therapy, it will be important to consider safety as well as efficacy. Some possible concerns are that the chimeric protein may be allergenic and potentially rapidly eliminated due to immune responses against the chimeric protein. However, we believe this issue can be potentially minimized in humans through the use of humanized antibodies, whose similarity to human antibodies should make the generation of a potent immune response against the chimeric protein less likely, thereby alleviating concern for significant allergic reaction or rapid elimination. Furthermore, immunogenic and allergenic molecules are typically larger than the small CTL epitope used in our approach. On the basis of knowledge of the common characteristics of allergens (molecular weight, hydrophobicity, post-translational modification, etc.), we may be able to choose viral epitopes that are less likely to promote an allergic response. Furthermore, an added benefit of marking cancer cells with foreign viral peptides is that unlike many other forms of cancer immunotherapy, the risk of autoimmunity is minimal. Thus, these concerns can potentially be mitigated.
In this study, we observed that the tumor cells isolated from tumor-bearing mice injected with Meso-scFv-ROR-Fc led to activation of a significant number of OVA-specific CD8+ T cells, but not all of the OVA-specific CD8+ T cells (Figure 3b,c). This phenomenon may be explained by several factors. First, the number of the OVA-specific CD8+ T cells is significantly higher than the number of tumor cells. Thus, not all the OVA-specific CD8+ T cells have contact with tumor cells. Second, some normal cells, such as macrophages, may contaminate the isolated tumor cells. This would also contribute to the observed phenomenon. Although the cross-presentation of the chimeric protein by macrophages is possible and may contribute to the activation of OVA-specific CD8+ T cells, it is not the major factor given that the use of Meso-scFv-O-Fc, which also contains OVA peptide, did not generate a similar activation of OVA-specific T cells (Figure 3b,c).
This study has successfully demonstrated the use of chimeric proteins encoding tumor antigens to overcome the issue of immune tolerance to generate potent antitumor effects. However additional issues with this innovative therapeutic method remain to be addressed including effector cell trafficking, survival, and function at the tumor site through their interactions with multiple regulatory checkpoints in the tumor microenvironment. Further research on these issues will facilitate the control of tumors through the innovative approach described in our study.
Our novel strategy has high translational value because the therapeutic chimeric protein has the advantages of targeted delivery, immunogenic CTL epitope release at the tumor site, and potent tumor-targeted killing through existing CTL immunity. This strategy may significantly advance the field of cancer immunotherapy as it can bypass immune tolerance. This system may serve as a platform technology for the development of cancer immunotherapy targeting other types of cancers.
Materials and Methods
Mice. Female C57BL/six mice (6–8 weeks) were purchased from National Cancer Institute (Frederick, MD). All animal procedures were performed according to approved protocols and in accordance with recommendations for the proper use and care of laboratory animals.
Constructs. Construction of human mesothelin-specific single-chain variable region (Meso-scFv):Meso-scFv was synthesized by Genescript (Piscataway, NJ) in the sequence reported by Chowdhury et al.13 and cloned into EcoRI/Bgl II sites of pfuse-migg2aFC2 (San Diego, CA).
Meso-scfv-O-Fc was constructed by PCR amplification of Meso-scfv-O using the primers: TTTGAATTCGatgcaggtacaactgcagca and aaactcgagcagtttttcaaagttgattatactttttatttcc aactttgtccc, then cloned into EcoRI/Bgl II sites of pfuse-migg2aFC2.
Meso-scfv-ROR-Fc was constructed by PCR amplifying Meso-scfv-O with the primers: TTTGAATTCGatgcaggtacaactgcagca and aaactcgagCCTTTTTACCCTcagtttttcaaagttgattata ctCCTTTTCACCCTttttatttccaactttgtccc, then cloned into EcoRI/Bgl II sites of pfuse-migg2aFC2.
Meso-scfv-Fc-O was created by cloning the oligos CTAGAagtataa tcaactttgaaaaactgTAAc and tcgagTTAcagtttttcaaa gttgattatactT into XabI/Xho sites of Meso-scfv-Fc.
Meso-scfv-Fc-RO was constructed by cloning the oligos CTAGAC GGGTGAAGCGGagtataatcaactttgaaaaactgTAAc and tc gagTTAcagtttttcaaagttgattatactCCGCTTCACCCGT into XabI/Xho sites of Meso-scfv-Fc.
Meso-scfv-Fc-R7 was constructed by cloning the oligos: CTAGAAGGG TGAAGAGACGCGCTCACTACAACATCGTGACCTTTTAAC and TCG AGTTAAAAGGTCACGATGT TGTAGTGAGCGCGTCT CTTCACCCTT into XabI/Xho sites of Meso-scfv-Fc.
Cell lines. Luciferase-expressing syngeneic mouse ovarian epithelial cancer cell lines, ID8 and ID8-meso, and luciferase-expressing TC-1 and TC-1/meso were generated in the same manner as previously described.23
The generation of pMSCV(n)-Hmeso and methods used to generate the human mesothelin expressing murine tumor cells have been previously described.24 Briefly, pMSCV(n)-Hmeso was transfected into Phoenix packaging cell line. After 48 hours, virion-containing supernatant was collected, clarified with 0.45-mm cellulose acetate syringe filter (Nalgene, Rochester, NY), and then used to infect TC-1 or ID8 cells in the presence of 8 µg/ml Polybrene (Sigma, St. Louis, MO). TC-1/meso or ID8-meso cells were isolated by preparative flow cytometry through staining with human mesothelin antibody (Signet laboratories, Dedham, MA). Baby hamster kidney 21 cells were also obtained from the ATCC (ATCC, Manassas, VA).
The generation of OVA-specific T cell line has been described previously.25 EG.7 cell line, a murine EL4 lymphoma cell line purchased from ATCC, was transfected with OVA-expressing vector. EG.7 cells were cultured in complete RPMI-1640 medium containing 10% heat-inactivated fetal bovine serum. OVA-specific CD8+ T cells were produced via the stimulation of splenocytes obtained from OT-1 transgenic mice with irradiated EG.7 cells in the presence of interleukin-2 (20 IU/ml, Pepro-Tech, Rock Hill, NJ). FD11 (a furin-deficient Chinese hamster ovary cell line) has been well characterized26 and was kindly provided by Dr Stephen H. Leppla at National Institutes of Health.
The generation of the E7-specific T cell line has been previously described.27 C57/BL6 (H-2b) mice were immunized using intraperitoneal injection of 107 p.f.u. of Sig/E7/LAMP-1 vaccinia virus. Splenocytes were harvested at day 8 after immunization. Autologous, irradiated splenocytes pulsed with 1 µmol/l E7 peptide (amino acid 49–57) were used as stimulators. Recombinant human interleukin-2 was added to the culture at a final concentration of 30 U/ml.
Transfection and protein purification. For purification of the various antihuman mesothelin scFv fragments, 50 µg of plasmid was transfected into 1 × l07 furin-deficient FD11 cells in T-150 flask using Lipofectamine 2000 (Invitrogen, Carlsbad, CA). After 3 days, cell cultured media was accumulated, filtered using 0.22 µm syringe filter (Millipore, Billerica, MA), and concentrated with Amicon cut-off 50 kDa Ultra-15 (Millipore, Billerica, MA). Concentrated recombinant protein containing media was applied to a HiTrap Protein G HP column (GE Healthcare, Pittsburg, PA) and followed vendor's protocol. Protein concentrations were determined by the Coomassie Plus protein assay (Pierce, Rockford, IL) and purity was estimated by sodium dodecyl sulfate polyacrylamide gel electrophoresis.
In vivo T-cell proliferation assay.1 × 105 luciferase-expressing ID8-meso cells were i.p. injected into C57BL/six mice (three per group). One day later, mice received each scFv-protein (20 µg) with 2.5 × 106 of OVA T cells labeled with 5 µmol/l of CFSE by i.p. injection. After 3 days, washed intraperitoneal cells were stained with PE conjugated anti-CD8 antibody and flow cytometry was performed for the detection of CFSE dilution in CD8+ T cells.
OVA and E7-specific T-cell activation and in vitro cytotoxicity assay. T-cell activation experiment was performed by adding tumor cells to 48-well plates (1 × 105/well) before incubation with each type of chimeric proteins at different concentrations. After 18 hours, treated tumor cells were incubated with 2 × 105 OVA-CTLs. One day after activation, IFN-γ-secreting OVA or E7-specific CD8+ T cells were identified by intracellular cytokine staining for IFN-γ and then analyzed by flow cytometry.
In vitro cytotoxicity experiment was performed by incubating luciferase-expressing tumor cells (1 × 105) with different concentrations of each type of proteins in a 24-well plate for 18 hours before the addition of 2 × 105 of OVA- or E7-specific CD8+ T cells. Degree of CTL-mediated tumor cell killing was measured by IVIS luminescence imaging system series 2000 (Xenogen, Alameda, CA).
T-cell activation experiment with human cervical cancer cell line, HeLa, was performed by first transfecting HeLa cells with pcDNA encoding MHC class I alleles H2-Kb or H2-Db using Lipofectamine 2000 transfection reagent (Invitrogen). A day later, transfected HeLa cells were incubated with Meso-scFv-Fc-RE7 or Meso-scFv-Fc-RO proteins. After 18 hours, treated tumor cells were incubated with 2 × 105 OVA-specific or E7-specific CTLs. CD8+ T-cell activation was identified by intracellular cytokine staining for IFN-γ and flow cytometry analysis as previously described.
Cell staining and flow cytometry analysis. For flow cytometry analysis, tumor cells were stained with 0.5 µg of purified various antihuman mesothelin scFv fragments and PE-conjugated antimouse antibody was used as a detection antibody (BD Bioscience, San Jose, CA). The percentage of OVA-specific IFN-γ-secreting CD8+ T cells was determined using intracellular cytokine staining and FACScan analysis with CELLQuest software (Becton Dickinson Immunocytometry System, Mountain View, CA). The positive control indicates maximum CD8+ T-cell activation was ~90%. For immunofluorescence staining, cells were stained with each protein, followed by PE conjugated antimouse Fc antibody after incubation for 0 minute or 60 minutes at 37 °C. Stained cells were examined using fluorescence microscopy (Carl Zeiss, Oberkochen, Germany).
OVA-specific T-cell activation and in vitro cytotoxicity assay. For T-cell activation, tumor cells were added to 48-well plates at a dose of 1 × 105 cells/well and incubated with different concentrations of proteins. After 18 hours, treated tumor cells were incubated with 2 × 105 OVA-specific CTLs. One day after activation, IFN-γ-secreting OVA specific CD8+ T cells were identified by intracellular cytokine staining and analyzed by flow cytometry analysis. For in vitro cytotoxicity experiment, 1 × 105 of luciferase-expressing tumor cells were stained with each chimeric protein on 24-well plate for 18 hours and treated with 2 × 105 OVA-specific cytotoxic T cells. The degree of CTL-mediated killing of the tumor cells was measured by the IVIS luminescence imaging system series 2000. Bioluminescence signals were acquired for 1 minute.
In vivo cell staining of scFv-proteins and T-cell activation by stained cells. For cell-specific binding experiment, 1 × 106 GFP-expressing ID8 or ID8-meso cells were injected into C57BL/six mice (three per group) using intraperitoneal injection. After 24 hours, mice were injected intraperitoneally with 10 µg of each scFv protein followed by i.p. wash after 18 hours. Intraperitoneally washed cells were stained with PE-conjugated antimouse Fc antibody and analyzed by flow cytometry. For T-cell activation, 1 × 106 intraperitoneally washed cells from each group were incubated with 1 × 105 OVA-specific T cells and activated IFN-γ-secreting OVA-specific CD8+ T cells were analyzed by flow cytometry analysis.
In vivo tumor treatment experiments. For in vivo tumor treatment experiments, 1 × 105 luciferase-expressing ID8-meso cells were injected into C57BL/six mice (five per group) using intraperitoneal injection. After 10 days, 20 µg of each scFv protein was injected with or without 5 × 106 OVA T cell using intraperitoneal injection. Fluorescence intensity in tumor-bearing mice treated with each scFv protein was measured by the IVIS luminescence imaging system series 2000.
Statistical analysis. The data presented in this study are representative of at least two experiments performed, and are expressed as means ± SD. The number of samples in each group for any given experiment was more than 3. Results for intracellular cytokine staining with flow cytometry analysis and tumor treatment experiments were evaluated by one-way analysis of variance and the Tukey-Kramer multiple comparison test. Comparisons between individual data points were performed using Student's t-test. The event time distributions for different mice were compared using the Kaplan–Meier method and the log-rank statistic. All P values <0.05 were considered significant.
SUPPLEMENTARY MATERIAL Figure S1. MHC class I presentation of OVA peptide by mesothelin- negative tumor cells following incubation with supernatant from ID8-meso or TC-1/meso treated with Meso-scFv-ROR-Fc. Figure S2. Furin facilitates the coating of tumor cells with antigenic peptides. Figure S3. Characterization of human mesothelin expression in human cancer cells.
Acknowledgments
We thank R.B. Roden for helpful discussions and critical review of the manuscript. This work was supported by the American Cancer Society grant RSG-07-199-01-LIB, the National Institutes of Health/National Cancer Institute (NIH/NCI) 2 P50 CA098252-06, NIH/NCI 2 P50 CA96784-06, and NIH/NCI 1 RO1 CA114425-06.
Supplementary Material
MHC class I presentation of OVA peptide by mesothelin- negative tumor cells following incubation with supernatant from ID8-meso or TC-1/meso treated with Meso-scFv-ROR-Fc.
Furin facilitates the coating of tumor cells with antigenic peptides.
Characterization of human mesothelin expression in human cancer cells.
REFERENCES
- Sznol M., and, Holmlund J. Antigen-specific agents in development. Semin Oncol. 1997;24:173–186. [PubMed] [Google Scholar]
- Scholler N, Fu N, Yang Y, Ye Z, Goodman GE, Hellström KE.et al. (1999Soluble member(s) of the mesothelin/megakaryocyte potentiating factor family are detectable in sera from patients with ovarian carcinoma Proc Natl Acad Sci USA 9611531–11536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassan R, Bera T., and, Pastan I. Mesothelin: a new target for immunotherapy. Clin Cancer Res. 2004;10 12 Pt 1:3937–3942. doi: 10.1158/1078-0432.CCR-03-0801. [DOI] [PubMed] [Google Scholar]
- Coppola JM, Bhojani MS, Ross BD., and, Rehemtulla A. A small-molecule furin inhibitor inhibits cancer cell motility and invasiveness. Neoplasia. 2008;10:363–370. doi: 10.1593/neo.08166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page RE, Klein-Szanto AJ, Litwin S, Nicolas E, Al-Jumaily R, Alexander P.et al. (2007Increased expression of the pro-protein convertase furin predicts decreased survival in ovarian cancer Cell Oncol 29289–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercapide J, Lopez De Cicco R, Bassi DE, Castresana JS, Thomas G., and, Klein-Szanto AJ. Inhibition of furin-mediated processing results in suppression of astrocytoma cell growth and invasiveness. Clin Cancer Res. 2002;8:1740–1746. [PubMed] [Google Scholar]
- Bassi DE, Mahloogi H, Al-Saleem L, Lopez De Cicco R, Ridge JA., and, Klein-Szanto AJ. Elevated furin expression in aggressive human head and neck tumors and tumor cell lines. Mol Carcinog. 2001;31:224–232. doi: 10.1002/mc.1057. [DOI] [PubMed] [Google Scholar]
- Cheng M, Watson PH, Paterson JA, Seidah N, Chrétien M., and, Shiu RP. Pro-protein convertase gene expression in human breast cancer. Int J Cancer. 1997;71:966–971. doi: 10.1002/(sici)1097-0215(19970611)71:6<966::aid-ijc10>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- Schalken JA, Roebroek AJ, Oomen PP, Wagenaar SS, Debruyne FM, Bloemers HP.et al. (1987Fur gene expression as a discriminating marker for small cell and nonsmall cell lung carcinomas J Clin Invest 801545–1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas G. Furin at the cutting edge: from protein traffic to embryogenesis and disease. Nat Rev Mol Cell Biol. 2002;3:753–766. doi: 10.1038/nrm934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Currier JR, Kuta EG, Turk E, Earhart LB, Loomis-Price L, Janetzki S.et al. (2002A panel of MHC class I restricted viral peptides for use as a quality control for vaccine trial ELISPOT assays J Immunol Methods 260157–172. [DOI] [PubMed] [Google Scholar]
- Lu J, Higashimoto Y, Appella E., and, Celis E. Multiepitope Trojan antigen peptide vaccines for the induction of antitumor CTL and Th immune responses. J Immunol. 2004;172:4575–4582. doi: 10.4049/jimmunol.172.7.4575. [DOI] [PubMed] [Google Scholar]
- Chowdhury PS, Viner JL, Beers R., and, Pastan I. Isolation of a high-affinity stable single-chain Fv specific for mesothelin from DNA-immunized mice by phage display and construction of a recombinant immunotoxin with anti-tumor activity. Proc Natl Acad Sci USA. 1998;95:669–674. doi: 10.1073/pnas.95.2.669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lev A, Noy R, Oved K, Novak H, Segal D, Walden P.et al. (2004Tumor-specific Ab-mediated targeting of MHC-peptide complexes induces regression of human tumor xenografts in vivo Proc Natl Acad Sci USA 1019051–9056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kersemaekers AM, Fleuren GJ, Kenter GG, Van den Broek LJ, Uljee SM, Hermans J.et al. (1999Oncogene alterations in carcinomas of the uterine cervix: overexpression of the epidermal growth factor receptor is associated with poor prognosis Clin Cancer Res 5577–586. [PubMed] [Google Scholar]
- Maurizi M, Almadori G, Ferrandina G, Distefano M, Romanini ME, Cadoni G.et al. (1996Prognostic significance of epidermal growth factor receptor in laryngeal squamous cell carcinoma Br J Cancer 741253–1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inada S, Koto T, Futami K, Arima S., and, Iwashita A. Evaluation of malignancy and the prognosis of esophageal cancer based on an immunohistochemical study (p53, E-cadherin, epidermal growth factor receptor) Surg Today. 1999;29:493–503. doi: 10.1007/BF02482343. [DOI] [PubMed] [Google Scholar]
- Fischer-Colbrie J, Witt A, Heinzl H, Speiser P, Czerwenka K, Sevelda P.et al. (1997EGFR and steroid receptors in ovarian carcinoma: comparison with prognostic parameters and outcome of patients Anticancer Res 171B613–619. [PubMed] [Google Scholar]
- Mellon K, Wright C, Kelly P, Horne CH., and, Neal DE. Long-term outcome related to epidermal growth factor receptor status in bladder cancer. J Urol. 1995;153 3 Pt 2:919–925. [PubMed] [Google Scholar]
- Normanno N, De Luca A, Bianco C, Strizzi L, Mancino M, Maiello MR.et al. (2006Epidermal growth factor receptor (EGFR) signaling in cancer Gene 3662–16. [DOI] [PubMed] [Google Scholar]
- Nicholson RI, Gee JM., and, Harper ME. EGFR and cancer prognosis. Eur J Cancer. 2001;37 Suppl 4:S9–15. doi: 10.1016/s0959-8049(01)00231-3. [DOI] [PubMed] [Google Scholar]
- Ross JS., and, Fletcher JA. The HER-2/neu oncogene in breast cancer: prognostic factor, predictive factor, and target for therapy. Stem Cells. 1998;16:413–428. doi: 10.1002/stem.160413. [DOI] [PubMed] [Google Scholar]
- Chang CL, Wu TC., and, Hung CF. Control of human mesothelin-expressing tumors by DNA vaccines. Gene Ther. 2007;14:1189–1198. doi: 10.1038/sj.gt.3302974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung CF, Calizo R, Tsai YC, He L., and, Wu TC. A DNA vaccine encoding a single-chain trimer of HLA-A2 linked to human mesothelin peptide generates anti-tumor effects against human mesothelin-expressing tumors. Vaccine. 2007;25:127–135. doi: 10.1016/j.vaccine.2006.06.087. [DOI] [PubMed] [Google Scholar]
- Peng S, Monie A, Kang TH, Hung CF, Roden R., and, Wu TC. Efficient delivery of DNA vaccines using human papillomavirus pseudovirions. Gene Ther. 2010;17:1453–1464. doi: 10.1038/gt.2010.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu M, Rappaport J., and, Leppla SH. Furin is important but not essential for the proteolytic maturation of gp160 of HIV-1. FEBS Lett. 1995;365:95–97. doi: 10.1016/0014-5793(95)00447-h. [DOI] [PubMed] [Google Scholar]
- Wang TL, Ling M, Shih IM, Pham T, Pai SI, Lu Z.et al. (2000Intramuscular administration of E7-transfected dendritic cells generates the most potent E7-specific anti-tumor immunity Gene Ther 7726–733. [DOI] [PubMed] [Google Scholar]
- Conejo-Garcia JR, Benencia F, Courreges MC, Kang E, Mohamed-Hadley A, Buckanovich RJ.et al. (2004Tumor-infiltrating dendritic cell precursors recruited by a beta-defensin contribute to vasculogenesis under the influence of Vegf-A Nat Med 10950–958. [DOI] [PubMed] [Google Scholar]
- Roby KF, Taylor CC, Sweetwood JP, Cheng Y, Pace JL, Tawfik O.et al. (2000Development of a syngeneic mouse model for events related to ovarian cancer Carcinogenesis 21585–591. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
MHC class I presentation of OVA peptide by mesothelin- negative tumor cells following incubation with supernatant from ID8-meso or TC-1/meso treated with Meso-scFv-ROR-Fc.
Furin facilitates the coating of tumor cells with antigenic peptides.
Characterization of human mesothelin expression in human cancer cells.