

Abstract
Even though other γδ T-cell subsets exhibit antitumor activity, adoptive transfer of γδ Tcells is currently limited to one subset (expressing Vγ9Vδ2 T-cell receptor (TCR)) due to dependence on aminobisphosphonates as the only clinically appealing reagent for propagating γδ T cells. Therefore, we developed an approach to propagate polyclonal γδ T cells and rendered them bispecific through expression of a CD19-specific chimeric antigen receptor (CAR). Peripheral blood mononuclear cells (PBMC) were electroporated with Sleeping Beauty (SB) transposon and transposase to enforce expression of CAR in multiple γδ T-cell subsets. CAR+γδ T cells were expanded on CD19+ artificial antigen-presenting cells (aAPC), which resulted in >109 CAR+γδ T cells from <106 total cells. Digital multiplex assay detected TCR mRNA coding for Vδ1, Vδ2, and Vδ3 with Vγ2, Vγ7, Vγ8, Vγ9, and Vγ10 alleles. Polyclonal CAR+γδ T cells were functional when TCRγδ and CAR were stimulated and displayed enhanced killing of CD19+ tumor cell lines compared with CARnegγδ T cells. CD19+ leukemia xenografts in mice were reduced with CAR+γδ T cells compared with control mice. Since CAR, SB, and aAPC have been adapted for human application, clinical trials can now focus on the therapeutic potential of polyclonal γδ T cells.
Introduction
Chimeric antigen receptors (CARs) re-direct T-cell specificity to tumor-associated antigens (TAAs), such as CD19, independent of major histocompatibility complex.1,2,3,4,5 This genetic modification of T cells has clinical applications as adoptive transfer of CAR+ T cells with specificity for CD19 can lead to antitumor responses in patients with refractory B-cell malignancies.6,7,8,9 Current trials administer CAR+ T cells coexpressing αβ T-cell receptor (TCRαβ) derived from a population that represents 95% of the peripheral T-cell pool. However, the remaining 1–5% of circulating T cells expressing TCRγδ (γδ T cells) have clinical appeal based on their endogenous cytotoxicity toward tumor cells as well as their ability to present TAA and elicit an antitumor response.10,11,12 This population of T cells directly recognizes TAA, e.g., heat shock proteins, major histocompatibility complex class I chain-related gene A/B, F1-ATPase, and intermediates in cholesterol metabolism (phosphoantigens), in humans.13 Therefore, broad recognition of tumor cells and antitumor activity is achieved by these T cells expressing a diverse TCRγδ repertoire (combination of Vδ1, Vδ2, or Vδ3 with one of fourteen Vγ chains).14
T cells expressing Vδ1 and Vδ2 have been associated with antitumor immunity, but current adoptive immunotherapy approaches are limited to the Vδ2 subpopulation due to limited expansion methods of Vδ1 to clinically sufficient numbers of cells for human applications. For the most part, γδ T cells have been numerically expanded in vivo and ex vivo using Zoledronic acid (Zol),15 an aminobisphosphonate that results in selective proliferation of T cells expressing Vγ9Vδ2 TCR.10,12,16 This treatment modality has resulted in objective clinical responses against both solid and hematologic tumors, but has not been curative as a monotherapy. Vδ1 γδ T cells have not yet been infused, but their presence has correlated with complete responses observed in patients with B-cell acute lymphoblastic leukemia after undergoing αβ T cell-depleted allogeneic hematopoietic stem cell transplantation.17,18,19,20 As both of these subpopulations of γδ T cells are associated with antitumor activity, but have not been combined for cell therapy, we sought a clinically appealing approach to propagate T cells that maintain a polyclonal TCRγδ repertoire.
Recognizing that a CD19-specific CAR can sustain the proliferation of αβ T cells on artificial antigen-presenting cells (aAPC) independent of TCRαβ usage,21 we hypothesized that γδ T cells would expand on aAPC independent of TCRγδ. Our approach was further stimulated by the observation that K562, the cell line from which the aAPC were derived, is a natural target for γδ T cells.18 We report that CAR+γδ T cells can be propagated to clinically relevant numbers on designer aAPC while maintaining a polyclonal population of TCRγδ as assessed by our “direct TCR expression assay ” (DTEA), a novel digital multiplexed gene expression analysis that we adapted to interrogate all TCRγδ isotypes.22 These CAR+γδ T cells displayed enhanced killing of CD19+ tumor cell lines in vitro compared with polyclonal γδ T cells not expressing CAR. Leukemia xenografts in immunocompromised mice were significantly reduced when treated with CAR+γδ T cells compared with control mice. This study highlights the ability of aAPC to numerically expand bispecific T cells that exhibit introduced specificity for CD19 and retain endogenous polyclonal TCRγδ repertoire.
Results
CAR+γδ T cells numerically expand on aAPC
To date, it has been problematic to synchronously manipulate and expand multiple γδ T-cell subpopulations for application in humans. Viral-mediated gene transfer typically requires cell division to achieve stable gene transfer and CARs have been introduced into transduced T cells expressing just Vδ2 TCR following the use of aminobisphosphonates to drive proliferation.23 In contrast, nonviral gene transfer with Sleeping Beauty (SB) transposition can be achieved in quiescent peripheral blood mononuclear cells (PBMC) with the full complement of peripheral γδ T cells initially present. Thus, stable expression of CAR can be achieved without prior T-cell propagation, enabling us to investigate whether a population of T cells expressing polyclonal TCRγδ chains could then be numerically expanded in a CAR-dependent manner on designer aAPC. PBMC were electroporated (day 0) with SB transposon/transposase system to enforce expression of a second generation CD19-specific CAR (CD19RCD28)5 that signals through chimeric CD28 and CD3-ζ. Electroporated cells were sorted using paramagnetic beads to separate the 4.0 ± 1.5% (mean ± SD; n = 4) CAR+γδ T cells from the majority of CAR+αβ T cells. The CAR+γδ T cells were selectively propagated by the recursive additions of γ-irradiated K562-derived aAPC (clone #4, genetically modified to coexpress CD19, CD64, CD86, CD137L, and membrane bound interleukin (IL)-15)5 with soluble IL-2 and IL-21. IL-21 is included in the manufacture of our CAR+αβ T cells so it was used to propagate CAR+γδ T cells.5 Prior experiments predicted that IL-2 and IL-15 enhance the proliferative potential of γδ T cells, and synergy between IL-2 and IL-21 has led to improved antitumor activity compared with γδ T cells grown with either IL-2 or IL-21 alone.24,25,26,27,28 Sham electroporations were undertaken to provide staining control T cells that were propagated by cross-linking CD3 using aAPC loaded with OKT3 to numerically expand CARnegαβ T cells.29 As expected, CAR was expressed on the day following electroporation (day 1) in most of the T cells, including γδ T cells, which comprised up to 10% of the mononuclear cells (Figure 1a, left). After 36 days of co-culture on aAPC, the majority of cells coexpressed CD3 and TCRγδ with 30.7 ± 23.3% (n = 4) CAR expression (Figure 1a, right). The absolute CAR proportions at day 36 varied in frequency depending on the donor, but increased compared with the initial populations of CAR+ γδ T cells at day 1(Figure 1b). As we have demonstrated, our aAPC co-culture system enforces CAR expression in αβ T cells (>90% CAR+ T cells by 28 days of co-culture),5 but the apparent lack of the same degree of selective pressure when combined with γδ T cells was attributed to an inherent ability of CARnegγδ T cells to sustain proliferation on aAPC derived from K562. Continuous proliferation of both CARneg and CAR+γδ T cells was observed over the tissue culture period. Even so, we could generate up to 1.5 × 109 ± 1.2 × 109 (n = 3) CAR+γδ T cells from the 2.8 × 105 ± 1.5 × 105 (n = 3) CAR+γδ T cells at the start of the culture (Figure 1c). Most of the propagated cells coexpressed CD3 and TCRγδ, but did not express TCRαβ (Figure 1d). These data demonstrate that aAPC could be used to sustain proliferation of CAR+ T cells coexpressing TCRγδ.
Figure 1.
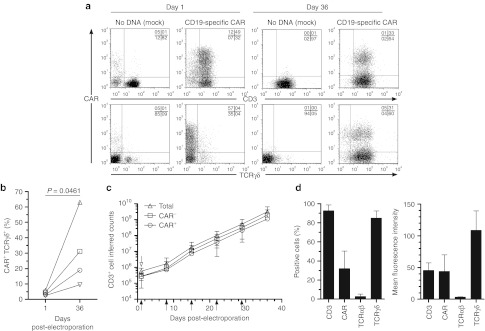
CAR+γδ T cells propagate on designer aAPC. (a) Transient (day 1) and stable (day 36) expression of CAR in T cells (top) and γδ T cells (bottom) in mock electroporated (“no DNA”) or CD19-specific CAR-electroporated cells (CD19RCD28). (b) Percentage of CAR+γδ T cells in the culture as transient (day 1) and stable (day 36) expression, where each shape represents an individual donor. (c) Rate of expansion of total γδ T cells (open triangles), CARnegγδ T cells (open squares), and CAR+γδ T cells (open circles) over tissue culture period following paramagnetic bead sorting (open arrow) and recursive stimulation (closed arrows) with aAPC and exogenous IL-2 and IL-21 administration. (d) Percentage-positive cells and mean fluorescence intensity of CD3, CAR, TCRαβ, and TCRγδ at day 36. Data are mean ± SD (n = 4) and quadrant percentages of flow plots are in upper right corner. aAPC, artificial antigen-presenting cell; ***P < 0.001. CAR, chimeric antigen receptor; IL, interleukin; TCR, T-cell receptor.
Immunophenotype of numerically expanded CAR+γδ T cells
Multiparameter flow cytometry was used to gate on CAR+ T cells and analyze their expression of cell surface markers (Figure 2). TCRγδ was expressed at high and low densities (Figure 2a, top). CD56, a marker of major histocompatibility complex-unrestricted lytic ability,30 was also expressed on T cells, but the culture contained <1% CD3negCD56+ natural killer cells and <1% CD3+Vα25TCR+NK T cells (data not shown). In contrast to αβ T cells, no CAR+γδ T cells expressed CD4, some were CD8+, but most were CD4negCD8neg, which is consistent with what is known for γδ T cells.31 The relative frequencies for each donor are shown in Figure 2b. Markers associated with memory, e.g., CD27, CD28, CD62L, and CCR7, were expressed by CAR+γδ T cells (Figure 2a, bottom). Both naive (CD45RA) and antigen-experienced (CD45RO) cells were present after propagation on aAPC, and the T cells were not exhausted as measured by low expression of CD57 (Figure 2b). In aggregate, cultures contained a heterogonous mixture of naive (CD45RA+CD27+CD28+CCR7+; 26.5 ± 6.2%), central memory (CD45RAnegCD27+CD28+CCR7+; 7.8 ± 3.6%), effector memory (CD45RAnegCD27+CD28negCCR7neg; 10.1 ± 5.4%), and effector memory RA (CD45RA+CD27negCD28negCCR7neg; 7.6 ± 3.4%) T-cell phenotypes.32,33 Costimulation by enforced expression of CD86 and CD137L (4-1BBL) on aAPC may be important for CAR+γδ T-cell numeric expansion due to expression of their receptors CD28 and CD137 (4-1BB), respectively. Molecules associated with homing to bone marrow (cutaneous lymphocyte antigen and CXCR4) and lymph nodes (CD62L and CCR7) were present on CAR+γδ T cells suggesting that they could migrate to sites known to harbor leukemia. In sum, propagated CAR+γδ T cells expressed T cell-associated surface markers that indicate desired potential for memory and homing.
Figure 2.
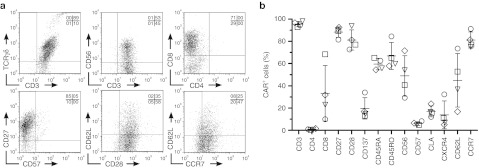
Immunophenotype of electroporated, separated, and propagated CAR+γδ T cells. (a) Expression by flow cytometry of cell surface markers associated with T cells and memory as gated on CD3+CAR+ cells. (b) Percentages of CAR+ T cells expressing T-cell markers, where each shape represents a different donor. Data are mean ± SD (n = 4). Quadrant percentages of flow plots are in upper right corner. CAR, chimeric antigen receptor; TCR, T-cell receptor.
Direct TCR expression assay to reveal γ and δTCR usage in CAR+γδ T cells
We sought to determine that aAPC-propagated CAR+ T cells were indeed bispecific as defined by the presence of a polyclonal population of TCRγδ alleles. Up to now, it has been difficult to determine the pattern of expression of the γ and δTCR chains. Therefore, we adapted our DTEA to assess the complete TCRγδ transcriptome. This approach takes advantage of the nCounter assay system to measure multiple bar-coded genes in a single reaction with high sensitivity and linearity across a broad range of expression.34 A multiplexed CodeSet was designed with two sequence-specific probes for each allele to evaluate TCRγδ isotypes. The DTEA was initially validated using Zol to preferentially propagate Vγ9Vδ2 cells from PBMC and, as expected, the resultant TCR usage was dominated by both Vδ2 and Vγ9 at protein and mRNA levels (Supplementary Figure S1). A second validation employed antibodies directed against γδ T-cell subsets (Vδ1 and Vδ2) to measure their mRNA expression. Vδ1negVδ2neg, Vδ1+Vδ2neg, and Vδ1negVδ2+cells were sorted from CARneg T cells (to maximize the number of Vδ2 cells recovered by fluorescence-activated cell sorting (FACS)) and subjected to DTEA (Figure 3a). As expected, Vδ1+Vδ2neg, Vδ1negVδ2+, and Vδ1negVδ2neg expressed Vδ1*01, Vδ2*02, and Vδ3*01 mRNA species, respectively (Figure 3b). These two strategies supported the validity of the DTEA panel enabling the identity of TCRγδ to be determined in CAR+ T cells. Therefore, we measured the mRNA levels for all three Vδ alleles as present in electroporated, separated, and propagated CAR+γδ T cells which correlated with multiparameter flow cytometry on gated CAR+ T cells to reveal the frequencies of Vδ subsets based on protein expression. The three Vδ populations were present in ascending frequency (Vδ1>Vδ3>>>Vδ2) in the electroporated and propagated T cells (Figure 3c). CARnegγδ T cells displayed similar frequencies of VδTCR usage as CAR+γδ T cells. DTEA array also assessed Vγ usage, which is of particular utility because only one antibody against Vγ9 is commercially available, thus limiting the tools with which to detect Vγ usage. Of note, Vγ2, Vγ7, Vγ8 (both alleles), Vγ9, and Vγ10 were present in CAR+ T-cell cultures (Figure 3d). A lack of commercially available antibodies prevented assessment of pairing between individual Vδ and Vγ chains on the T cells. The TCR usage described for γδ T cells was that which was present at the time of functional assays. Our ability to digitally quantify the presence of mRNA species enabled us to determine that the propagated CAR+ T cells expressed a polyclonal population of TCRγδ chains.
Figure 3.
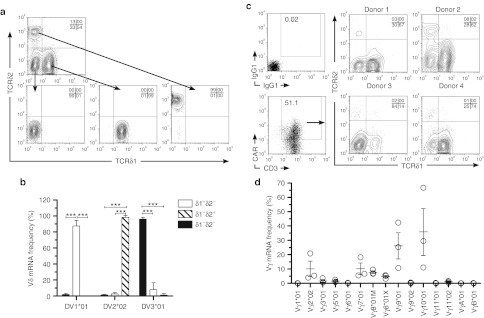
Distribution of Vδ and Vγ in CAR+γδ T cells. (a) Representative FACS of Vδ populations (top) into Vδ1negVδ2neg (left), Vδ1+Vδ2neg (middle), and Vδ1negVδ2+ (right) populations and (b) Vδ allele mRNA expression in sorted T cells. (c) Vδ1negVδ2neg, Vδ1+Vδ2neg, and Vδ1negVδ2+ frequencies in gated CAR+γδ T-cell populations from four donors. (d) Vγ allele mRNA expression in CAR+γδ T cells. Data are mean ± SD (n = 3). Quadrant percentages of flow plots are in upper right corner. ***P <0.001. CAR, chimeric antigen receptor; FACS, fluorescence-activated cell sorting; TCR, T-cell receptor.
T cells produced proinflammatory cytokines in response to stimulation through endogenous TCRγδ and introduced CAR
The functional activity of the CAR+ T cells was assessed by activation with leukocyte activation cocktail, which was comprised of phorbol 12-myristate 13-acetate and ionomycin. Leukocyte activation cocktail mimics activation through TCR by simulating protein kinase C and increasing intracellular Ca2+ to activate phospholipase C. Measurement of secreted and intracellular cytokines (in the presence of the inhibitor GolgiPlug, which contains brefeldin A) were performed on genetically modified T cells with and without leukocyte activation cocktail (Figure 4a,b). A broad range of cytokines were produced by γδ T cells, with the highest expression of interferon-γ (IFNγ), tumor necrosis factor-α, and chemokines macrophage inflammatory protein (MIP)-1α, MIP-1β, and regulated and normal T cell expressed and secreted (Figure 4b). IL-17 has been shown to be important for antitumor efficacy of γδ T cells and this cytokine was secreted by CAR+γδ T cells. These results suggest that TCRγδ can be activated to produce cytokines that could promote inflammation within the tumor. Next, CAR-specific cytokine production was assessed by activation using the murine T-cell lymphoma line EL4 and a genetically modified derivate to enforce expression of human CD19. Both tumor necrosis factor-α and IFNγ were produced by CAR+γδ T cells in response to CD19 (Figure 4c). A less diverse repertoire of cytokines was secreted following CAR stimulation when compared with stimulation of TCRγδ, but IFNγ, tumor necrosis factor-α, MIP-1α, MIP-1β, and regulated and normal T cell expressed and secreted were all increased in response to activation through CAR (Figure 4d). In aggregate, proinflammatory cytokines were upregulated by bispecific CAR+γδ T cells through their TCR and CAR.
Figure 4.
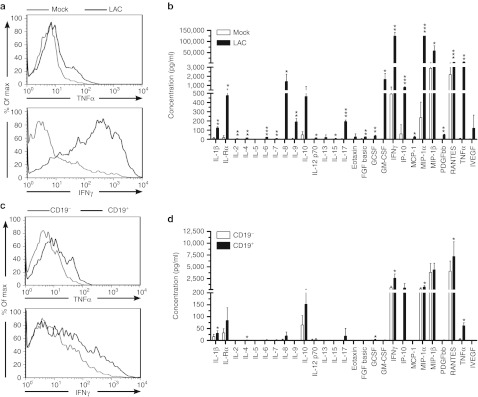
Bispecific γδ T cells produce proinflammatory cytokines when endogenous TCR and introduced CAR are stimulated. (a) CAR+γδ T cells at day 35 of co-culture on aAPC were stimulated for 4 hours with a mock cocktail (media alone) or leukocyte activation cocktail (LAC, PMA/ionomycin) to induce TCR stimulation and then analyzed by flow cytometry. CAR+ T cells were gated and tumor necrosis factor-α (TNF-α, top) and interferon-γ (IFN-γ, bottom) production is shown. (b) Luminex array (27-Plex) of cytokines secreted by CAR+γδ T cells in conditions described in a. (c) Similar to a except that EL4-CD19neg and EL4-CD19+ were used instead of mock/LAC. (d) Same as b but with EL4-CD19neg and EL4-CD19+ targets. Student's t-test for statistical analysis between mock and LAC (in b) and EL4-CD19neg and EL4-CD19+ (in d) where *P < 0.05, **P < 0.01, and ***P < 0.001. Data are representative of four donors for a and c and mean ± SD (n = 3) for b and d. aAPC, artificial antigen-presenting cell; CAR, chimeric antigen receptor; FGF, fibroblast growth factor; GM-CSF, granulocyte-macrophage colony-stimulating factor; IL, interleukin; MCP, monocyte chemoattractant protein; MIP, macrophage inflammatory protein; PDGFβ, platelet-derived growth factor-β PMA, phorbol 12-myristate 13-acetate; RANTES, regulated and normal T cell expressed and secreted; TCR, T-cell receptor; VEGF, vascular endothelial growth factor.
CAR+γδ T cells exhibit enhanced antitumor effects against CD19+ targets in vitro
It was anticipated that γδ T cells would display endogenous cytotoxicity to leukemia cells. Therefore, γδ T cells without CAR were numerically expanded on aAPC in order to test their antileukemia activity. Human CD19+ B-cell acute lymphoblastic leukemia cell lines (REH, Kasumi-2, and Daudi genetically modified to express β2M) were lysed by CARnegγδ T cells while primary, healthy CD19+ B cells were not killed by the same effectors (Figure 5a). However, not all B-cell acute lymphoblastic leukemia cell lines were susceptible to efficient lysis by CARnegγδ T cells. In particular, EL4 and NALM-6 cells were largely resistant to cytolysis by γδ T cells. Thus, the ability of the CD19-specific CAR to amplify the inherent antitumor activity of γδ T cells was investigated. Enforced expression of CD19 on the surface of EL4 cells improved targeting and killing of this cell line by CAR+γδ T cells at significantly higher (P = 0.0001) levels compared with the parental CD19neg EL4 cell line (Figure 5b). Similarly, CAR+γδ T cells exhibited improved ability (P = 0.001) to kill CD19+ NALM-6 cells compared with CARnegγδ T cells (Figure 5c). In summary, the introduced CAR enhanced the specific killing capability of genetically modified γδ T cells.
Figure 5.
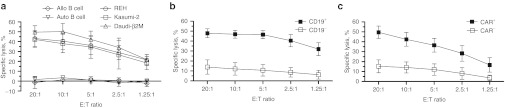
Specific lysis of CD19+ tumor cell lines by CAR+γδ T cells. (a) Standard 4-hour CRA of (a) CARnegγδ T cells against CD19+ B-ALL cell lines (REH, Kasumi-2, and Daudi-β2M) or primary CD19+ B cells from autologous (Auto) or allogeneic (Allo) donors, (b) CAR+γδ T cells against EL4-CD19neg (open squares) and EL4-CD19+ (closed squares) tumor cells, and (c) CARnegγδ T cells (open squares) and CAR+γδ T cells (closed squares) against CD19+ NALM-6 tumor cells. Data are mean ± SD from four healthy donors (average of triplicate measurements for each donor) that were pooled from two independent experiments. B-ALL, B-cell acute lymphoblastic leukemia; CAR, chimeric antigen receptor; CRA, chromium release assay; E:T, effector to target ratio.
CAR+γδ T cells can target CD19+ tumor in vivo
The ability of electroporated and propagated γδ T cells to target CD19+ tumor was then investigated in vivo. NALM-6 is an aggressive CD19+ B-cell leukemia model and immunocompromised mice engrafted with 105 NALM-6 are moribund in 20–25 days when untreated. Control of disseminated NALM-6 tumor in vivo is dependent on the infused T cells homing to tumor and activating cytolytic machinery in the tumor microenvironment. After adoptive immunotherapy, the burden of tumor was significantly decreased in mice receiving CAR+γδ T cells (donor no. 4 from Figure 3c) compared with untreated mice (Figure 6). Mice in treatment group receiving CAR+ T cells displayed fewer characteristics of the untreated and thus unwell mice, which included lethargy, ruffled coat, temporary hind limb paralysis, and difficulty entering and exiting anesthesia at late stages of the experiment. A uniform date for euthanasia was chosen to measure the antitumor effect based on flow cytometry for NALM-6 in lymphoid tissue. There was significant antitumor activity by the CAR+γδ T cell as measured by bioluminescent imaging of NALM-6-eGFP-ffLuc (Figure 6b) as exemplified at 22 days after injection of tumor (Figure 6c). Noninvasive imaging was corroborated by analysis of presence of tumor cells at necroscopy. Mice that received CAR+γδ T cells exhibited significant reductions in tumor burden (CD19+eGFP+) in the bone marrow, spleen, and peripheral blood (Figure 6d,e). These data reveal that polyclonal CAR+γδ T cells exhibit therapeutic activity in vivo.
Figure 6.
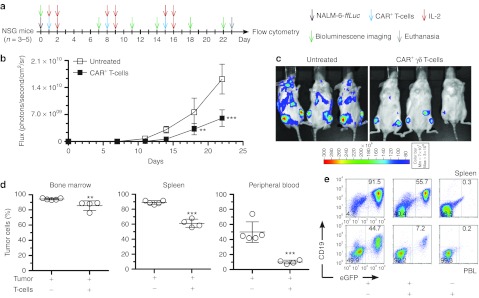
In vivo antitumor activity of CAR+γδ T cells. (a) Schematic of experiment. (b) BLI derived from eGFP+ffLuc+CD19+ NALM-6 tumor and (c) representative images of mice at day 22. (d) Postmortem analysis of tissues and blood where tumor cells (CD19+eGFP+) were detected by flow cytometry. (e) Representative flow plots from d. Data are mean ± SD (n = 3–5 mice per group, representative of two independent experiments) and gating frequencies in e are displayed. The percentage of tumor cells is derived from detecting CD19+eGFP+NALM-6 by flow cytometry from postmortem samples. Statistics performed with (in b) two-way ANOVA with Bonferroni's post-tests and (in d) Student's t-test between treated and untreated mice. **P < 0.01 and ***P < 0.001. ANOVA, analysis of variance; BLI, bioluminescent imaging; CAR, chimeric antigen receptor; eGFP, enhanced green fluorescent protein; IL, interleukin; PBL, peripheral blood leukocyte.
Discussion
We established that introduction of a second generation CAR could (i) drive the numeric expansion of T cells independent of usage of TCRγδ chains and (ii) augment the lytic potential of CD19+ tumors by γδ T cells. Propagating bispecific CAR+ T cells with a broad diversity of TCRγδ chains are desirable based on their therapeutic potential. Indeed, γδ T cells other than those expressing Vγ9Vδ2 have been generated from PBMC using TCRγδ-specific and CD3-specific monoclonal antibodies.35,36,37 These prior approaches did not comprehensively measure TCRγδ isotype expression nor did they yield Vδ1 and Vδ3 at frequencies as high as seen in this study. The Vγ2 TCR chain was detected on our T cells, which has been described to pair with Vδ2, and these T cells can have antigen presentation capabilities.38 Our CAR+γδ T cells expressed molecules consistent with antigen presentation, e.g., CD86, CD137L, and human leukocyte antigen-DR (data not shown), and Vγ9Vδ2 cells have served as aAPC for αβ T cells.11 Future experiments will investigate whether our polyclonal CAR+γδ T cells also have an ability to serve as aAPC. Also present were T-cell subpopulations expressing Vγ7, Vγ8, and Vγ10, where the first two chains have been associated with intestinal intraepithelial lymphocytes39,40 and the latter chain's functional significance is not yet apparent. In all, our approach is the first to report expansion of CAR+ T cells that maintained a polyclonal TCRγδ expression.
The repertoire of TCRγδ chains employed by CAR+ T cells was similar to the initial pool of γδ T cells in PBMC with two exceptions. We noted an increase in Vδ3 usage, but this may be advantageous as it is associated with specificity for viruses that could offer enhanced immune responses to viral infections in immunocompromised patients receiving therapy.41 A decrease in Vγ9Vδ2 usage was also observed compared with the starting frequency of this TCR in PBMC, but this could potentially be increased by priming aAPC with Zol to increase Vγ9Vδ2 ligand expression in the co-culture. Whether this loss of Vγ9Vδ2 TCR expression was due to preferential activation induced cell death or selective out-growth of T cells expressing Vδ1 and Vδ3TCR is not known. Nonetheless, Vγ9Vδ2 chains were still present in the final T-cell cultures indicating that aminobisphosphonate therapy could drive expansion of this subset of T cells after administration.
Recombinant retroviruses have been previously employed to achieve stable expression of CARs in γδ T cells, but this required using an aminobisphosphonate to achieve numeric expansion of T cells before transduction.16,42 We now demonstrate propagation of T cells after, rather than before, gene transfer using SB-mediated transposition results in a polyclonal population of bispecific γδ T cells capable of CAR-mediated (i) production and secretion of proinflammatory cytokines in response to CD19, (ii) enhanced lysis of CD19+ tumor targets, and (iii) in vivo antitumor activity against a CD19+ tumor. The ability of these T cells to exhibit effector functions was not correlated to a particular Vδ or Vγ usage as cells with different VδTCR frequencies (Figure 3c) produced the same cytokines (Figure 4) and displayed similar cytolysis of CD19+ targets (Figure 5b). We noted that frequency of CAR expression was more variable on γδ T cells compared with αβ T cells. This was likely due to an endogenous ability of K562 cells to sustain proliferation of γδ T cells independent of CAR. Nevertheless, adoptive transfer of γδ T cells of which 60% expressed CAR could still yield the same in vitro lytic ability as 98% CAR+γδ T cells (Supplementary Figure S2). This indicated that (i) CAR+γδ T cells are potent tumor killers and (ii) >90% CAR expression may not be a critically limiting parameter for predicting therapeutic efficacy. Nonetheless, we are undertaking improvements to increase the expression of CAR on propagated γδ T cells. Furthermore, the chimeric signaling molecules in the CAR endodomain could be specifically designed to enhance triggering of γδ T cells. For example, γδ T cells can be activated through FcγRIIIA (CD16) in the TCR complex,43 which raises the possibility that signaling through chimeric FcRγ (as compared with CD3-ζ in our current design) in a CAR endodomain may improve activation. However, CD16 was not detected on CAR+γδ T cells in this study (data not shown). Since clinical responses against CD19+ chronic lymphocytic leukemia have been achieved with T cells expressing a CAR that signaled through 4-1BB (CD137) endodomain,7,8 another option is to swap CD28 for CD137 for activation of γδ T cells.
In addition to improving CAR expression on γδ T cells, the type of γδ T cell arising after electroporation with SB system and propagation on aAPC could be manipulated to further improve antitumor activity. For instance, some γδ T cells were observed to secrete IL-17, a proinflammatory cytokine that has potent, yet context-dependent, antitumor effects.44,45,46,47,48 IL-17 producing lineages of T cells can be mutually exclusive from those that secrete IFNγ.49 Inducible costimulator of T cells (ICOS) leads to IL-17 polarization in CD4+ T cells and CD28 costimulation overcame this effect to dictate that CD4+ T cells now produce IFNγ.50 CD86 is one of the costimulatory molecules on our aAPC and the majority of CAR+γδ T cells secrete IFNγ in response to CD19 with diminished production of IL-17. Furthermore, the CAR contains a chimeric CD28 endodomain which may contribute to IFNγ polarization in genetically modified T cells. Substitution of chimeric CD28 for ICOS in the CAR and replacement of CD86 on the aAPC with ICOS-ligand could potentially reverse the polarization to IL-17. Given that we can propagate CAR+γδ T cells on aAPC, we are prepared to design aAPC to evaluate whether we can skew the cytokine profile to reflect the propagation of desired T-cell subsets.
The human application of CAR+γδ T cells is appealing given their inherent potential for antitumor effects and their apparent lack of alloreactivity.19 The CAR, SB system, and aAPC are all already in use in our clinical trials. Therefore, we plan to modify our manufacturing scheme in compliance with current good manufacturing practice to generate bispecific CAR+γδ T cells. Our data provides a clinically appealing approach to numerically expand and manipulate CAR+ T cells with multiple Vγ and Vδ pairings enabling clinical trials to evaluate their therapeutic potential.
Materials and Methods
Plasmids and cell lines. Codon-optimized DNA plasmids for SB transposase (SB11) and a second generation CD19-specific CAR (designated CD19RCD28) transposon are described elsewhere.5 NALM-6 and EL4 cell lines were acquired from Deutsche Sammlung von Mikroorganismen und Zellkulturen (DSMZ, Braunschweig, Germany) and American Type Culture Collection (Manassas, VA), respectively. Daudi expressing β2 microglobulin and NALM-6 expressing firefly luciferase and enhanced green fluorescent protein (NALM-6-ffLuc-eGFP) were generated as previously described.5 Kasumi-2 and REH cell lines were provided by Dr Jeff Tyner (Oregon Health and Science University). A transposon (Supplementary Figure S3) containing neomycin phosphotransferase linked via F2A self-cleavable peptide sequences to human CD19 (truncated following its transmembrane domain) was used to express this TAA on EL4 cells following electroporation with SB11 transposase and Mouse T Cell Nucleofector Kit (cat. no. VPA-1006; Lonza, Basel, Switzerland) followed by subsequent selection under 0.8 mg/ml G418 (InvivoGen, San Diego, CA). K562-derived aAPC (clone #4) were used as previously described.5,51 Cell lines were maintained in complete media (RPMI, 10% heat-inactivated fetal bovine serum (Hyclone, Logan, UT), and 1% Glutamax-100 (Gibco, Grand Island, NY)), in humidified conditions with 5% CO2 at 37 °C. All cell line identities were confirmed by STR DNA Fingerprinting at the MDACC's Cancer Center Support Grant (CCGS) core facility.
T-cell propagation. PBMC were obtained after informed consent from healthy volunteers and isolated by Ficoll-Paque (GE Healthcare, Milwaukee, WI).1 108 thawed PBMC were electroporated using program U-014 (on day 0 of co-culture) with 75 µg supercoiled DNA plasmid coding for CD19RCD28 transposon and 25 µg supercoiled DNA plasmid coding for SB11 transposase in cuvettes (2 × 107 cells per cuvette) using Nucleofector II and Human T cell Nucleofector Kit (Lonza).5 The following day (day 1), paramagnetic separation was performed with TCRγ/δ+ T-cell isolation kit (cat. no. 130-092-892; Miltenyi Biotec, Auburn, CA) and LS columns (cat. no. 130-042-401; Miltenyi Biotec), which separated untouched γδ T cells in the negative fraction from αβ T cells attached to magnet. Also on day 1, CAR+γδ T cells were stimulated at a ratio of one CAR+ T cell to two γ-irradiated (100 Gy) aAPC (clone #4) in presence of exogenous IL-2 (50 U/ml, added three times per week; Novartis, Basel, Switzerland) and IL-21 (30 ng/ml, added three times per week; eBioscience, San Diego, CA). Cells were serially re-stimulated with addition of aAPC as on day 1 of co-culture every 7 days for 5 weeks. Six donors were tested in three independent experiments. Validation of coexpression of CD19, CD64, CD86, CD137L, and IL-15 (eGFP) on aAPC were performed before addition to T-cell cultures as described.5 When CD3negCD56+ populations exceeded 10% of the culture, these natural killer cells were depleted using CD56 microbeads (Miltenyi Biotec) and LS columns.5 As negative control for CAR and TCRγδ expression, αβ T cells from sham-electroporated PBMC (no DNA electrotransferred) were propagated in parallel with OKT3 (CD3-specific monoclonal antibody; Orthoclone)-loaded γ-irradiated aAPC added every 7 days with thrice weekly administration of IL-2, as above.29 CARnegγδ T cells were also sorted and expanded on aAPC with IL-2 and IL-21 as undertaken with CAR+γδ T cells, except that they were not electroporated, and these cells were used for cytotoxicity experiments. 107 PBMC were cultured with a single dose of Zol (1 µg/ml; Novartis) with thrice weekly additions of IL-2 and IL-21, as above, to expand CARneg Vγ9Vδ2 T cells.
Flow cytometry. Cultures were phenotyped using antibodies detailed in Supplementary Table S1. Appropriate isotype controls were used to validate gating. Staining was performed in FACS buffer (phosphate-buffered saline, 2% fetal bovine serum, 0.1% sodium azide) for 20–30 minutes at 4 °C, and two washes with FACS buffer were performed before staining and between stains. Intracellular staining was done following fixation and permeabilization for 20 minutes at 4 °C with BD Cytofix/Cytoperm (BD Biosciences, San Diego, CA). Intracellular staining was performed in Perm/Wash buffer, 10% human AB serum for 30 minutes at 4 °C. FITC, PE, PerCP/Cy5.5, and APC antibodies were used at 1:20, 1:40, 1:33, and 1:40 dilutions, respectively. All samples were acquired on FACSCalibur (BD Biosciences) and analyzed with FlowJo software (version 7.6.3; TreeStar, Ashland, OR).
Direct imaging of mRNA molecules by DTEA. At days 0 and 36 of co-culture on aAPC, at least 105 T cells were lysed at a ratio of 5 µl RLT Buffer (Qiagen, Valencia, CA) per 3 × 104 cells and frozen at -80 °C in replicate vials for one time use. RNA lysates were thawed and immediately analyzed using nCounter Analysis System (NanoString Technologies, Seattle, WA) following a minimum of 12 hours hybridization at 65 °C using multiplexed target-specific color-coded reporter and biotinylated capture probes to detect mRNAs of interest. Two CodeSets were generated from RefSeq accession numbers for selected mRNA transcripts and were used to generate the specific reporter and capture probe pairs for the designer TCR expression array (DTEA, Supplementary Table S2). Reporter-capture nCounter probe pairs were identified that (i) minimized off-target effects due to cross-hybridization of reporter-capture probe pairs to non-target molecules, (ii) target most, if not all, of the transcript variants for a particular gene, and (iii) efficiently hybridize. Five reference genes that span the dynamic range of RNA expression in lymphocytes (ACTB, OAZ1, POLR1B, POLR2A, and RPL27) were included to normalize transcript levels between different samples and to account for differences in the amount of total RNA present in the samples. A normalization factor for each sample was derived from the formula (σtotal/σsample) and then applied to each sample that had been background subtracted. Percentage of Vδ and Vγ were then calculated based on normalized values.
Cytokine production and secretion. T cells harvested at day 35 of co-culture on γ-irradiated aAPC were examined for cytokine secretion by multiplex analysis and expression by intracellular staining. The former was set up with triplicate overnight co-cultures of 105 genetically modified T cells and (i) mock (complete media), (ii) leukocyte activation cocktail (5 ng/ml PMA (Sigma, St Louis, MO), 500 ng/ml ionomycin (Sigma)), (iii) 105 CD19neg EL4 cells, or (iv) 105 CD19+ EL4 cells. Supernatants were harvested the following day and like wells were pooled and frozen at -80 °C until time of analysis. Samples were then thawed on ice, diluted 1:8 in complete media, and interrogated on Bio-Plex Pro Human Cytokine 27-plex Assay (Bio-Rad, Hercules, CA) according to manufacturer's instructions using Luminex100 (xMap Technologies, Austin, TX). Intracellular cytokine production was established from similar conditions as above, except (i) 5 × 105 EL4 cells were used, (ii) the incubation period was 6 hours, and (iii) the secretory pathway inhibitor GolgiPlug (BD Biosciences) containing brefeldin A was added at 1:1,000 dilution.
Chromium release assay. In vitro cytolytic capability was assessed using standard 4-hour chromium release assay as previously described.5
Mouse experiments. In vivo antitumor efficacy was assessed in NSG mice (NOD.Cg-Prkdcscid Il2rγtm1Wjl/SzJ; Jackson Laboratories, Bar Harbor, ME). The day after intravenous injection of 105 NALM6-ffLuc-eGFP, experimental groups (n = 5) received (i) no treatment or (ii) 107 CAR+γδ T cells. As controls for potential graft-versus-host-disease, three NSG mice received CAR+γδ T cells without tumor. T cells were administered every 7 days for three doses along with 6 × 104 IU/injection recombinant human IL-2 at the time of infusion and twice on the day after (Figure 6a). Noninvasive bioluminescent imaging to measure tumor burden of NALM-6-ffLuc-eGFP was performed during the course of the experiments following subcutaneous D-Luciferin (Caliper, Hopkinton, MA) administration on IVIS-100 Imager (Caliper). Bioluminescent imaging was analyzed using Living Image software (version 2.50, Xenogen; Caliper). Tumor burden in peripheral blood leukocytes, spleens, and bone marrow was evaluated by flow cytometry postmortem.
SUPPLEMENTARY MATERIAL Figure S1. Distribution of Vδ and Vγ in γδ T cells expanded on aminobisphosphonate. Figure S2. Specific lysis of CD19+ tumor cell lines by CAR+, CAR++, and CAR+++ γδ T cells. Figure S3. Sleeping Beauty DNA transposon (designated δCD19-F2A-Neo) to coexpress truncated human CD19 and neomycin phosphotransferase for in vitro selection. Table S1. Antibodies used in study. Table S2. Direct TCR expression assay.
Acknowledgments
D.C.D. is a Teal Pre-doctoral Scholar (DOD Ovarian Cancer Research Program), American Legion Auxiliary Fellow in Cancer Research (UT-GSBS at Houston), and Andrew Sowell-Wade Huggins Scholar in Cancer Research (UT-GSBS at Houston and Cancer Answers Foundation). The Immune Monitoring Core Lab (IMCL, MD Anderson Cancer Center) performed Luminex analysis. We thank Perry Hackett from University of Minnesota for help with Sleeping Beauty system and Carl June from University of Pennsylvania for assistance in generating the K562-derived artificial antigen-presenting cell (clone #4). This work was supported by funding from: Cancer Center Core grant (CA16672); RO1 (CA124782, CA120956, CA141303; CA163587); R33 (CA116127); P01 (CA148600); SPORE (CA136411, CA100632); S10RR026916; AdeeHeebe; Albert J. Ward Foundation; Ahuja family; Burroughs Wellcome Fund; Cancer Prevention and Research Institute of Texas; Caryn Papantonakis; CLL Global Research Foundation; Department of Defense; Estate of Noelan L. Bibler; Gillson Longenbaugh Foundation; Harry T Mangurian, Jr., Fund for Leukemia Immunotherapy; Fund for Leukemia Immunotherapy; Institute of Personalized Cancer Therapy; Leukemia and Lymphoma Society; Lymphoma Research Foundation; Miller Foundation; Mr and Mrs Rick Calhoon; Mr Herb Simons; Mr and Mrs Joe H Scales; Mr Thomas Scott; National Foundation for Cancer Research; Paula Gavrel Asher Foundation; Pediatric Cancer Research Foundation; Production Assistance for Cellular Therapies; Robert J. Kleberg, Jr. and Helen C. Kleberg Foundation; Team Connor; Thomas Scott; William Lawrence and Blanche Hughes Children's Foundation. This work was performed in Houston, TX, USA. The authors declared no conflict of interest.
Supplementary Material
References
- Singh H, Manuri PR, Olivares S, Dara N, Dawson MJ, Huls H.et al. (2008Redirecting specificity of T-cell populations for CD19 using the Sleeping Beauty system Cancer Res 682961–2971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowolik CM, Topp MS, Gonzalez S, Pfeiffer T, Olivares S, Gonzalez N.et al. (2006CD28 costimulation provided through a CD19-specific chimeric antigen receptor enhances in vivo persistence and antitumor efficacy of adoptively transferred T cells Cancer Res 6610995–11004. [DOI] [PubMed] [Google Scholar]
- Jena B, Dotti G., and, Cooper LJ. Redirecting T-cell specificity by introducing a tumor-specific chimeric antigen receptor. Blood. 2010;116:1035–1044. doi: 10.1182/blood-2010-01-043737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper LJ, Topp MS, Serrano LM, Gonzalez S, Chang WC, Naranjo A.et al. (2003T-cell clones can be rendered specific for CD19: toward the selective augmentation of the graft-versus-B-lineage leukemia effect Blood 1011637–1644. [DOI] [PubMed] [Google Scholar]
- Singh H, Figliola MJ, Dawson MJ, Huls H, Olivares S, Switzer K.et al. (2011Reprogramming CD19-specific T cells with IL-21 signaling can improve adoptive immunotherapy of B-lineage malignancies Cancer Res 713516–3527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brentjens RJ, Rivière I, Park JH, Davila ML, Wang X, Stefanski J.et al. (2011Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias Blood 1184817–4828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter DL, Levine BL, Kalos M, Bagg A., and, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med. 2011;365:725–733. doi: 10.1056/NEJMoa1103849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalos M, Levine BL, Porter DL, Katz S, Grupp SA, Bagg A.et al. (2011T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia Sci Transl Med 395ra73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochenderfer JN, Wilson WH, Janik JE, Dudley ME, Stetler-Stevenson M, Feldman SA.et al. (2010Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19 Blood 1164099–4102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castella B, Vitale C, Coscia M., and, Massaia M. Vγ9Vδ2 T cell-based immunotherapy in hematological malignancies: from bench to bedside. Cell Mol Life Sci. 2011;68:2419–2432. doi: 10.1007/s00018-011-0704-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandes M, Willimann K, Bioley G, Lévy N, Eberl M, Luo M.et al. (2009Cross-presenting human gammadelta T cells induce robust CD8+ alphabeta T cell responses Proc Natl Acad Sci USA 1062307–2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes AQ, Martins DS., and, Silva-Santos B. Targeting γδ T lymphocytes for cancer immunotherapy: from novel mechanistic insight to clinical application. Cancer Res. 2010;70:10024–10027. doi: 10.1158/0008-5472.CAN-10-3236. [DOI] [PubMed] [Google Scholar]
- Scotet E, Martinez LO, Grant E, Barbaras R, Jenö P, Guiraud M.et al. (2005Tumor recognition following Vgamma9Vdelta2 T cell receptor interactions with a surface F1-ATPase-related structure and apolipoprotein A-I Immunity 2271–80. [DOI] [PubMed] [Google Scholar]
- Hayday AC. [gamma][delta] cells: a right time and a right place for a conserved third way of protection. Annu Rev Immunol. 2000;18:975–1026. doi: 10.1146/annurev.immunol.18.1.975. [DOI] [PubMed] [Google Scholar]
- Kondo M, Sakuta K, Noguchi A, Ariyoshi N, Sato K, Sato S.et al. (2008Zoledronate facilitates large-scale ex vivo expansion of functional gammadelta T cells from cancer patients for use in adoptive immunotherapy Cytotherapy 10842–856. [DOI] [PubMed] [Google Scholar]
- Chiplunkar S, Dhar S, Wesch D., and, Kabelitz D. gammadelta T cells in cancer immunotherapy: current status and future prospects. Immunotherapy. 2009;1:663–678. doi: 10.2217/imt.09.27. [DOI] [PubMed] [Google Scholar]
- Lamb LS, Jr, Henslee-Downey PJ, Parrish RS, Godder K, Thompson J, Lee C.et al. (1996Increased frequency of TCR gamma delta + T cells in disease-free survivors following T cell-depleted, partially mismatched, related donor bone marrow transplantation for leukemia J Hematother 5503–509. [DOI] [PubMed] [Google Scholar]
- Lamb LS, Jr, Gee AP, Hazlett LJ, Musk P, Parrish RS, O'Hanlon TP.et al. (1999Influence of T cell depletion method on circulating gammadelta T cell reconstitution and potential role in the graft-versus-leukemia effect Cytotherapy 17–19. [DOI] [PubMed] [Google Scholar]
- Lamb LS, Jr, Musk P, Ye Z, van Rhee F, Geier SS, Tong JJ.et al. (2001Human gammadelta(+) T lymphocytes have in vitro graft vs leukemia activity in the absence of an allogeneic response Bone Marrow Transplant 27601–606. [DOI] [PubMed] [Google Scholar]
- Godder KT, Henslee-Downey PJ, Mehta J, Park BS, Chiang KY, Abhyankar S.et al. (2007Long term disease-free survival in acute leukemia patients recovering with increased gammadelta T cells after partially mismatched related donor bone marrow transplantation Bone Marrow Transplant 39751–757. [DOI] [PubMed] [Google Scholar]
- Numbenjapon T, Serrano LM, Singh H, Kowolik CM, Olivares S, Gonzalez N.et al. (2006Characterization of an artificial antigen-presenting cell to propagate cytolytic CD19-specific T cells Leukemia 201889–1892. [DOI] [PubMed] [Google Scholar]
- Zhang M, Maiti S, Bernatchez C, Huls H, Rabinovich B, Champlin RE.et al. (2012A new approach to simultaneously quantify both Tcr α- and β-chain diversity after adoptive immunotherapy Clin Cancer Res: Off J Am Assoc Cancer Res 18(17)4733–4742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rischer M, Pscherer S, Duwe S, Vormoor J, Jürgens H., and, Rossig C. Human gammadelta T cells as mediators of chimaeric-receptor redirected anti-tumour immunity. Br J Haematol. 2004;126:583–592. doi: 10.1111/j.1365-2141.2004.05077.x. [DOI] [PubMed] [Google Scholar]
- Hanrahan CF, Kimpton WG, Howard CJ, Parsons KR, Brandon MR, Andrews AE.et al. (1997Cellular requirements for the activation and proliferation of ruminant gammadelta T cells J Immunol 1594287–4294. [PubMed] [Google Scholar]
- Nagamine I, Yamaguchi Y, Ohara M, Ikeda T., and, Okada M. Induction of gamma delta T cells using zoledronate plus interleukin-2 in patients with metastatic cancer. Hiroshima J Med Sci. 2009;58:37–44. [PubMed] [Google Scholar]
- García VE, Jullien D, Song M, Uyemura K, Shuai K, Morita CT.et al. (1998IL-15 enhances the response of human gamma delta T cells to nonpeptide [correction of nonpetide] microbial antigens J Immunol 1604322–4329. [PubMed] [Google Scholar]
- Do JS., and, Min B. IL-15 produced and trans-presented by DCs underlies homeostatic competition between CD8 and {gamma}{delta} T cells in vivo. Blood. 2009;113:6361–6371. doi: 10.1182/blood-2008-12-192997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thedrez A, Harly C, Morice A, Salot S, Bonneville M., and, Scotet E. IL-21-mediated potentiation of antitumor cytolytic and proinflammatory responses of human V gamma 9V delta 2 T cells for adoptive immunotherapy. J Immunol. 2009;182:3423–3431. doi: 10.4049/jimmunol.0803068. [DOI] [PubMed] [Google Scholar]
- O'Connor CM, Sheppard S, Hartline CA, Huls H, Johnson M, Palla SL.et al. (2012Adoptive T-cell therapy improves treatment of canine non-Hodgkin lymphoma post chemotherapy Sci Rep 2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly-Rogers J, Madrigal-Estebas L, O'Connor T., and, Doherty DG. Activation-induced expression of CD56 by T cells is associated with a reprogramming of cytolytic activity and cytokine secretion profile in vitro. Hum Immunol. 2006;67:863–873. doi: 10.1016/j.humimm.2006.08.292. [DOI] [PubMed] [Google Scholar]
- Smetak M, Kimmel B, Birkmann J, Schaefer-Eckart K, Einsele H, Wilhelm M.et al. (2008Clinical-scale single-step CD4(+) and CD8(+) cell depletion for donor innate lymphocyte infusion (DILI) Bone Marrow Transplant 41643–650. [DOI] [PubMed] [Google Scholar]
- Klebanoff CA, Gattinoni L., and, Restifo NP. CD8+ T-cell memory in tumor immunology and immunotherapy. Immunol Rev. 2006;211:214–224. doi: 10.1111/j.0105-2896.2006.00391.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Mitri D, Azevedo RI, Henson SM, Libri V, Riddell NE, Macaulay R.et al. (2011Reversible senescence in human CD4+CD45RA+CD27- memory T cells J Immunol 1872093–2100. [DOI] [PubMed] [Google Scholar]
- Kulkarni MM. Digital multiplexed gene expression analysis using the NanoString nCounter system. Curr Protoc Mol Biol. 2011;Chapter 25:Unit25B.10. doi: 10.1002/0471142727.mb25b10s94. [DOI] [PubMed] [Google Scholar]
- Kang N, Zhou J, Zhang T, Wang L, Lu F, Cui Y.et al. (2009Adoptive immunotherapy of lung cancer with immobilized anti-TCRgammadelta antibody-expanded human gammadelta T-cells in peripheral blood Cancer Biol Ther 81540–1549. [DOI] [PubMed] [Google Scholar]
- Dokouhaki P, Han M, Joe B, Li M, Johnston MR, Tsao MS.et al. (2010Adoptive immunotherapy of cancer using ex vivo expanded human gammadelta T cells: A new approach Cancer Lett 297126–136. [DOI] [PubMed] [Google Scholar]
- Lopez RD, Xu S, Guo B, Negrin RS., and, Waller EK. CD2-mediated IL-12-dependent signals render human gamma delta-T cells resistant to mitogen-induced apoptosis, permitting the large-scale ex vivo expansion of functionally distinct lymphocytes: implications for the development of adoptive immunotherapy strategies. Blood. 2000;96:3827–3837. [PubMed] [Google Scholar]
- Brandes M, Willimann K., and, Moser B. Professional antigen-presentation function by human gammadelta T Cells. Science. 2005;309:264–268. doi: 10.1126/science.1110267. [DOI] [PubMed] [Google Scholar]
- Newton DJ, Andrew EM, Dalton JE, Mears R., and, Carding SR. Identification of novel gammadelta T-cell subsets following bacterial infection in the absence of Vgamma1+ T cells: homeostatic control of gammadelta T-cell responses to pathogen infection by Vgamma1+ T cells. Infect Immun. 2006;74:1097–1105. doi: 10.1128/IAI.74.2.1097-1105.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olofsson K, Hellström S., and, Hammarström ML. The surface epithelium of recurrent infected palatine tonsils is rich in gammadelta T cells. Clin Exp Immunol. 1998;111:36–47. doi: 10.1046/j.1365-2249.1998.00446.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight A, Madrigal AJ, Grace S, Sivakumaran J, Kottaridis P, Mackinnon S.et al. (2010The role of Vδ2-negative γδ T cells during cytomegalovirus reactivation in recipients of allogeneic stem cell transplantation Blood 1162164–2172. [DOI] [PubMed] [Google Scholar]
- Stresing V, Daubiné F, Benzaid I, Mönkkönen H., and, Clézardin P. Bisphosphonates in cancer therapy. Cancer Lett. 2007;257:16–35. doi: 10.1016/j.canlet.2007.07.007. [DOI] [PubMed] [Google Scholar]
- Angelini DF, Borsellino G, Poupot M, Diamantini A, Poupot R, Bernardi G.et al. (2004FcgammaRIII discriminates between 2 subsets of Vgamma9Vdelta2 effector cells with different responses and activation pathways Blood 1041801–1807. [DOI] [PubMed] [Google Scholar]
- Haas JD, Nistala K, Petermann F, Saran N, Chennupati V, Schmitz S.et al. (2011Expression of miRNAs miR-133b and miR-206 in the Il17a/f locus is co-regulated with IL-17 production in αβ and γδ T cells PLoS ONE 6e20171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kryczek I, Wei S, Zou L, Altuwaijri S, Szeliga W, Kolls J.et al. (2007Cutting edge: Th17 and regulatory T cell dynamics and the regulation by IL-2 in the tumor microenvironment J Immunol 1786730–6733. [DOI] [PubMed] [Google Scholar]
- Lai D, Wang F, Chen Y, Wang C, Liu S, Lu B.et al. (2012Human ovarian cancer stem-like cells can be efficiently killed by γδ T lymphocytes Cancer Immunol Immunother 61979–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Middleton GW, Annels NE., and, Pandha HS. Are we ready to start studies of Th17 cell manipulation as a therapy for cancer. Cancer Immunol Immunother. 2012;61:1–7. doi: 10.1007/s00262-011-1151-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cua DJ., and, Tato CM. Innate IL-17-producing cells: the sentinels of the immune system. Nat Rev Immunol. 2010;10:479–489. doi: 10.1038/nri2800. [DOI] [PubMed] [Google Scholar]
- Haas JD, González FH, Schmitz S, Chennupati V, Föhse L, Kremmer E.et al. (2009CCR6 and NK1.1 distinguish between IL-17A and IFN-gamma-producing gammadelta effector T cells Eur J Immunol 393488–3497. [DOI] [PubMed] [Google Scholar]
- Paulos CM, Carpenito C, Plesa G, Suhoski MM, Varela-Rohena A, Golovina TN.et al. (2010The inducible costimulator (ICOS) is critical for the development of human T(H)17 cells Sci Transl Med 255ra78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manuri PV, Wilson MH, Maiti SN, Mi T, Singh H, Olivares S.et al. (2010piggyBac transposon/transposase system to generate CD19-specific T cells for the treatment of B-lineage malignancies Hum Gene Ther 21427–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.