

Abstract
To determine if an ordered and repetitive display of an epitope promoted induction of superior antibody responses, we compared B-cell responses to an influenza A virus epitope that was either encoded as a transgene by an adenovirus (Ad) vector or expressed on the vector's surface. To this end, we constructed a panel of influenza A virus vaccines based on chimpanzee-derived replication-defective adenovirus (AdC) vectors of serotype SAd-V25 also called AdC68. AdC68 vectors were modified to express a linear B-cell epitope of the ectodomain of matrix 2 (M2e) within variable regions 1 (VR1) or 4 (VR4) of the adenovirus hexon. Additional vectors with wild-type or M2e-modified hexon encoded M2e fused to the influenza A virus nucleoprotein (NP) as a transgene product. Hexon-modified vectors were tested for immunogenicity and efficacy in mice in comparison to vectors with native hexon expressing the M2e-NP fusion protein. Upon priming, vectors expressing M2e within VR1 of hexon induced M2e-specific antibody responses of higher magnitude and avidity than those carrying M2e within VR4 or vectors expressing the M2e as part of a transgene product. CD8+ T-cell responses to the transgenic NP were comparable between vectors. M2e-specific antibody responses could be boosted by a second dose of the VR1 hexon-modified vector but not by repeated immunization with the VR4 hexon-modified vector.
Introduction
Viral vectors are being generated as second generation vaccines for pathogens for which traditional approaches of attenuated or inactivation have failed or are deemed unsafe. Numerous publications have demonstrated that recombinant viruses induce excellent cellular responses to foreign transgene products.1,2,3,4 They also induce humoral responses to the recombinant proteins.5,6 Antigen presentation for T and B cells fundamentally differs. T cells are stimulated by small peptides upon their cell surface displayed by major histocompatibility complex (MHC) molecules. B cells in turn recognize conformational or linear epitopes on commonly complex proteins and require cross-linkage of their immunoglobulin (Ig) receptors for activation of intracellular signaling events that together with help from CD4+ T cells leads to their maturation into antibody-secreting cells.7 Transgene products during their synthesis are in part misfolded and then upon degradation enter MHC presentation pathways for T-cell stimulation. Their structure upon secretion or expression on the surface of vector-transduced cells is less likely to serve optimal cross-linkage of B cell receptors.
To test whether we could improve B-cell responses to a recombinant viral vector based on a chimpanzee origin adenovirus (AdC) of serotype SAd-V25, also called AdC68,8 by developing a virus-like particle vaccine, we inserted a linear B-cell epitope of the ectodomain of matrix 2 protein (M2e) of influenza A virus into the AdC68 hexon. Hexon is the most abundant of the viral capsid proteins forming a total of 240 trimers on the surface of the icosahedral capsid. Hexon molecules contain a pseudohexagonal base that is anchored to the capsid, a conserved barrel domain followed by a tower on top of the molecule that contains flexible loops.9 Different serotypes of Ad show sequence variations mainly within these loops.9 AdC68 hexon, which has been characterized by X-ray crystallography,10 contains five variable regions (VR1-5) that form five distinctive loops on top of the molecule. The loop encoded by VR1 was defined as the dominant target of AdC68-neutralizing antibodies,11 suggesting that its localization allows easy access to the B cell receptors. Therefore, we inserted a linear B-cell epitope into VR1 and for comparison into VR4, which encodes another surface-exposed hexon loop.
Ad vectors derived from the common human serotype 5 (AdHu5) displaying B-cell epitopes from other pathogens within their hexon have been described previously and showed immunogenicity in mice.12,13 Neutralizing antibodies to AdHu5 virus are common in humans and dampen uptake of AdHu5 vectors and hence immune responses to vector encoded transgene products,14 although they would not necessarily be expected to affect B-cell responses to an epitope displayed within the viral hexon. It has been suggested that modification of the variable regions of Ad hexon prevents neutralization by antibodies to wild-type virus15 but such results remain debatable.16,17 Therefore, we opted to base the vaccine on an AdC vector to which most humans lack neutralizing antibodies.18
We selected a linear epitope from the M2e of influenza A virus as the vaccine insert, as this epitope elicits non-neutralizing but nevertheless protective antibodies that cross-react with most influenza A virus strains.19,20 We previously published on a novel universal influenza A vaccine candidate21 based on AdC68, and SAd-V23, also called AdC6, expressing in tandem a signal sequence linked to three different sequences of M2e and one sequence of the viral nucleoprotein (NP), which in mice induces a robust CD8+ T-cell response.22 The vaccines induced both antibodies to M2e and CD8+ T cells to NP, which together protected mice against different stains of influenza A virus.
To test the hypothesis that B-cell responses are best induced by antigen that is displayed in a repetitive and structured fashion thus allowing for cross-linkage of the B-cell receptors, we compared the previous vaccines to M2e hexon VR1- or VR4-modified AdC68 vectors that were in part further modified to express the influenza A virus NP together with M2e from a transgene placed into the deleted E1 domain. Our results show that vectors with wild-type or modified hexon induce comparable CD8+ T-cell responses in mice. Antibody responses to M2e were markedly higher upon immunization with the hexon-modified vectors that carried M2e within VR1. Furthermore M2e-specific B-cell responses could be boosted by a second immunization with the VR1- but not the VR4-modified vector. Vectors that carried only M2e within VR1 provided significant protection against influenza A virus challenge, which could be further improved by concomitant expression of the M2e-NP transgene.
Results
Construction of hexon-modified AdC68 vectors
The hexon was modified by direct cloning of the M2e sequence into a segment of the viral molecular clone as shown in Figure 1. Briefly, the AdC68 molecular clone was digested with Mlu I releasing a 5.1 kb fragment that contains most of the hexon sequence. The fragment was ligated into the Mlu I site of pcDNA3.1, resulting in plasmid pcDNA3.1-MM; 5′ oligonucleotides containing the Cla I site of hexon followed by the adjacent hexon sequences and the M2e sequence in positions 142-144 of hexon and 3′ primers containing the Nde I site of hexon were used to amplify a fragment that was then cut with Cla I and Nde I and cloned into the corresponding sites of pcDNA3.1-MM resulting in a plasmid containing M2e within VR1 of hexon. To construct the VR4-modified hexon, 5′ oligonucleotides containing the Nde I site of hexon followed by M2e in positions 253 to 254 of hexon and 3′ oligonucleotides containing the Sca I site and adjacent sequences of pcDNA3.1 were used to amplify a fragment of pcDNA3.1-MM. The amplicon was cut with Nde I and Sca I and cloned into the corresponding sites of pcDNA3.1-MM resulting in a plasmid that contained M2e within hexon VR4. Parts of the vectors were sequenced to ensure insertion of the M2e sequence. The hexon sequences were then cloned back into the viral molecular clone using Mlu I. The genomes of new VR1- or VR4-modified vectors, termed AdC68-Hx-M2e(R1) or AdC68-Hx-M2e(R4), were analyzed by Southern blotting to ensure correct insertion of the sequence. The molecular clones were further modified to express a previously described M2e(3)-NP fusion protein composed of three copies of M2e linked in tandem to NP21 from an expression cassette placed into the vectors' E1 domain resulting in vectors AdC68M2e(3)NP-Hx-M2e(R1) and AdC68 M2e(3)NP-Hx-M2e(R4). Table 1 provides a list of the different vectors. The sequence of the modified hexon molecules is show in Supplementary Data.
Figure 1.

Construction of hexon-modified vectors. The flowchart shows the cloning procedures for VR1 and VR4 hexon-modified E1-deleted AdC68 vectors. The upper part shows the entire sequence of the E1-deleted molecular clone of AdC68 including the Mlu I sites that were used to excise the gene encoding the hexon. The lower graph shows the pcDNA3.1 clone containing the viral hexon including the sites used for insertion of the M2e sequence into VR1 or VR4. Ad, adenovirus; ITR, inverted terminal repeat.
Table 1. A list of the new vectors and the nomenclature used throughout manuscript and pertinent growth characteristics.

Structure of M2e-modified AdC68 hexon
AdC68 hexon in its native structure forms trimers with the variable loops encoded by VR1-5 displayed on the top of the molecule. To assess the effect of the M2e insertion on hexon, we carried out western blotting with an M2e-specific monoclonal antibody and an antibody to a conserved part of hexon. As shown in Figure 2, the VR1- and VR4-modified hexons were shown to form trimers when probed with an antibody to M2e on a Blue Native Gel (Invitrogen, Carlsbad, CA). An antibody to the hexon termed “Adenovirus hexon protein Antibody 3G0” (Santa Cruz Biotechnology, Santa Cruz, CA) showed strong binding to unmodified hexon and markedly reduced binding to the loop-modified hexons, suggesting that the modification had affected the antibody's epitope. Upon boiling of the virus particles (vp) and their treatment with 2-mercaptoethanol, the antibody to hexon no longer bound, although the monoclonal antibody to M2e could detect VR1- and VR4-modified hexon as monomers.
Figure 2.

Western blotting of AdC68-modified hexon. Hexon was visualized upon gel electrophoresis of particles (a) Results of a blue native gel designed to visualize large proteins. (b) Results of an SDS-PAGE gel. Both types of gels were probed with a monoclonal antibody to M2e and a monoclonal antibody to adenovirus hexon. MW: molecular weight marker, 1: hexon of AdC68-Hx-Me2(R1), 2: hexon of AdC68-Hx-M2e(R4), 3: hexon of AdC68 with wild-type hexon. Ad, adenovirus; SDS-PAGE, sodium dodecyl sulfate-polyacrylamide gel electrophoresis.
Both types of hexon-modified vectors rescued easily and expanded yielding comparable amounts of vp and infectious units per batch (Table 2) supporting the notion that the insertions into the hexon loop had no detrimental effects on its structure.
Table 2. Hexon-modified vectors rescued and expanded yielding comparable amounts of virus particles and infectious units per batch.

Expression of M2e on hexon
We used two additional methods to measure expression of M2e within hexon on the vector surface or encoded by the transgene which also contains NP. To quantify expression of M2e on virions, we conducted ELISA on plates coated with hexon-modified or native hexon AdC68 vectors, which were probed with a monoclonal antibody to M2e. As shown in Figure 3a, the M2e antibody showed high reactivity to the capsid of VR1 hexon-modified vector and comparatively lower activity against the VR4-modified capsid. The higher binding of the M2e antibody to the VR1-modified hexon suggests that the loop encoded by VR1 is more accessible to antibodies, which is consistent with the finding that this region contains the dominant-neutralizing B-cell epitope of AdC68.11
Figure 3.

Expression of M2e. (a) Plates were coated with purified AdC68 vectors carrying native hexon or hexon carrying M2e within VR1 or VR4. Plates were blocked, treated with a monoclonal antibody to M2e followed by incubation with an alkaline phosphatase-conjugated antibody and the substrate. Color changes were measured in an ELISA reader. Graph shows mean adsorbance (± SD) of substrate in wells that received different dilutions of the monoclonal antibody to M2e. (b) HEK 293 cells were transduced with different concentrations of vectors expressing the M2e(3)NP fusion protein as a transgene product and analyzed for expression of the fusion protein by western blot with the monoclonal antibody to M2e. An antibody to β-actin was used as a loading control. Ad, adenovirus; NP, nucleoprotein; vp, virus particles.
To assess transgene product expression, cells were infected with the Ad vectors expressing the M2e(3)NP fusion protein (with or without hexon modifications). Cell lysates were tested with the M2e antibodies by western blot. As shown in Figure 3b, vectors expressed an M2e antibody-binding protein that had the predicted size of the transgene product.
Hexon VR1 modifications escape neutralization by antibodies to native AdC68
To test whether the VR1 or VR4 hexon modifications perturb binding of neutralizing antibodies to native hexon, mice were immunized with AdC68 vectors expressing native or M2e-modified hexon. Mouse sera were then tested for neutralization of an AdC68 vector expressing native hexon. As shown in Figure 4a, sera from mice immunized with AdC68 vector expressing native hexon and encoding the rabies virus glycoprotein (AdC68rab.gp) or carrying VR4-modified hexon readily neutralized wild-type AdC68 virus while sera from mice immunized with the VR1 hexon-modified vector failed to neutralize the vector with native hexon. To ensure that the VR1 hexon-modified vector could induce a neutralizing antibody response, mice were immunized with this vector and sera were tested for neutralization of the homologous vector and an AdC68 vector carrying wild-type hexon. Sera induced by the VR1 hexon-modified vector (Figure 4b) efficiently neutralized the vector used for immunization but only poorly neutralized the wild-type vector as was expected based on results shown in Figure 4a.
Figure 4.

Neutralization of AdC68 with native hexon by antibodies induced with hexon-modified vectors. Mice were immunized with vectors carrying M2e within VR1 or VR4 of hexon or with a vector with native hexon (AdC68rab.gp). Sera were tested for neutralization of an AdC68 vector expressing EGFP. (a) Shows neutralization of AdC68 with wild-type hexon and (b) shows neutralization of vectors with M2e within VR1. Graph shows the reciprocal neutralization titers. Ad, adenovirus; EGFP, enhanced green fluorescent protein; VNA, virus-neutralizing antibody; wt, wild-type.
M2e-specific antibody responses
To measure M2e-specific antibody responses elicited by the hexon-modified AdC vectors, groups of ICR mice were vaccinated with 1 × 1010 vp of recombinant AdC68 vectors and boosted 2 months later with same vector used at the same dose. For comparison, mice were vaccinated with the same dose of AdC68M2e(3)NP; these mice were boosted with the heterologous AdC6 vector expressing the same transgene product. A heterologous vector was used to prevent blunting of the recall response by vector-specific neutralizing antibodies induced upon priming. Sera were harvested from individual mice 5 weeks after the prime and the boost, respectively. Sera from mice immunized with vectors expressing the rabies virus glycoprotein served as controls. Sera were tested for antibodies to M2e by a peptide ELISA (Figure 5a). All vectors expressing M2e either within hexon or from a transgene product induced antibodies to M2e. Antibody titers increased markedly after the boost in mice immunized with the VR1 hexon-modified vector, whereas increases in mice immunized with the VR4 hexon-modified vector were modest. Immunization with the AdC68-Hx-M2e(R1) vector given twice resulted in higher antibody responses to the M2e peptide compared with the heterologous AdC68M2e(3)NP/AdC6M2e(3)NP vaccine regimen or two immunizations with the VR4-modified vector. It is of interest to note that presence of M2e within the transgene product did not, as we expected, increase antibody responses to M2e expressed on capsid. To ensure that the vaccine induced a response in a genetically distinct strain of mice, inbred C57BL/6 mice were tested using the same vaccine regimens. Results were similar (data not shown).
Figure 5.

Humoral responses to M2e. (a) An M2e-peptide ELISA was used to measure M2e-specific antibody titers in sera of ICR mice (n = 10). Sera were harvested 5 weeks after priming (black bars) or 5 weeks after the boost (gray bars). The following groups of mice were tested, group 1: mice immunized with AdC68-Hx-M2e(R1); group 2: mice immunized with AdC68M2e(3)NP-Hx-M2e(R1); group 3: mice immunized with AdC68-Hx-M2e(R4); group 4: mice immunized with AdC68M2e(3)NP-Hx-M2e(R4); group 5: mice immunized with AdC68M2e(3)NP; and group 6 (control group): mice immunized with AdC68rab.gp. The following differences were statistically significant by analysis of variance and Tukey's adjustment for the multiple pairwise comparisons. After priming, group 1 was significantly different from all other groups; group 2 was significantly different from groups 1 and 6; comparison of the other groups failed to reach significance. The second immunization caused a significant increase in titers of AdC68-Hx-M2e(R1), AdC68M2e(3)NP-Hx-M2e(R1), and AdC68M2e(3)NP primed mice as determined by t-tests. After booster immunization, the following differences between the following groups were significant. Group 1 differed from all groups except group 2; group 2 differed from groups 3, 4, 5, and 6; groups 3 and 4 differed from groups 1, 2, and 6; group 5 differed from groups 1, 2, and 6. The most pertinent differences are highlighted within the figure. (b) A cellular ELISA (black bars) and a peptide ELISA (gray bars) were used to measure antibodies from vaccinated C57BL/6 mice (n = 5). Sera were harvested 2 weeks after priming. The following groups were tested, group 1: AdC68M2e(3)NP-immunized mice; group 2: AdC68-Hx-M2e(R1)-immunized mice; group 3: AdC68M2e(3)NP-Hx-M2e(R1)-immunized mice; and group 4 (control group): AdC68rab.gp-immunized mice. The following differences were statistically significant by analysis of variance with Tukey's adjustment for multiple pairwise comparisons for the cellular ELISA: group 1 differed from groups 3 and 4; group 2 differed from group 4. For the peptide ELISA, the following groups showed significant differences: groups 1 and 2 differed from all groups; group 3 did not differ from group 4. Graphs in a and b show average titers ± SD normalized towards a monoclonal M2e-specific antibody. (c,d) Avidity was determined by testing one concentration of pooled vector-induced antisera from vector-immunized mice in comparison to the same concentration of pooled control sera on M2e peptide-coated plates by ELISA. Antibody was displaced by increasing concentrations of NaSCN. Background adsorbance in presence of control sera was subtracted from adsorbance of wells containing immune sera. Data were then normalized to adsorbance in wells that did not receive NaSCN (set at 100%). (c) Shows sera harvested after priming from ICR mice immunized with AdC68-Hx-M2e(R1) or AdC68M2e(3)NP. The difference was highly significant (P < 0.0001). (d) Shows sera harvested from the following groups of ICR mice that received prime boost regimens; sera were harvested 5 weeks after the boost. Group 1: mice immunized 2× with AdC68-Hx-M2e(R1); group 2: mice immunized 2× with AdC68M2e(3)NP-Hx-M2e(R1); group 3: mice immunized 2× with AdC68-Hx-M2e(R4); group 4: mice immunized 2× with AdC68M2e(3)NP-Hx-M2e(R4); group 5: mice immunized with AdC68M2e(3)NP/AdC6M2e(3)NP. After adjustment for multiparameter comparison, the following groups were significantly different from each other: group 1 differed from groups 2, 3, and 4; group 2 differed from groups 1, 3, and 4; group 3 differed from groups 1, 2, 4, and 5; and group 4 differed from groups 1, 2, 4, and 5. Ad, adenovirus; NP, nucleoprotein.
As was reported by Walter Gerhard, antibodies to M2e peptides may not necessarily bind native M2e as expressed by influenza virus or on influenza virus-infected cells.23 We therefore tested sera from C57BL/6 mice immunized 5 weeks earlier with 1010 vp of AdC68M2e(3)NP, AdC68-Hx-M2e(R1), AdC68-Hx-M2e(R4), or as a control AdC68rab.gp for antibodies in a cellular ELISA on cells transduced with the M2 protein, which more faithfully detect antibodies to M2e as expressed on influenza virus or on influenza virus-infected cells. The same sera were tested for comparison in a peptide ELISA (Figure 5b). For all samples, as expected, antibody titers were higher when tested by the peptide ELISA. Upon a single immunization, only the two hexon-modified vectors induced antibody responses that could be detected by the cellular ELISA; the response induced by the transgene product-expressing vector did not reach significance.
The avidity of sera of ICR mice immunized 5 weeks previously was tested by measuring the amount of antibodies that remained bound to an M2e peptide-coated ELISA plate after addition of increasing concentrations of sodium thiocyanate. As shown in Figure 5c, M2e-specific antibodies in sera from AdC68-Hx-M2e(R1)-primed mice showed markedly higher avidity compared with those from AdC68M2e(3)NP-immune mice. Avidity was also assessed from sera harvested at 5 weeks after a booster immunization either with the homologous vectors or the heterologous regimen for vectors expressing the M2e(3)NP transgene product (Figure 5d). Booster immunization markedly increased the avidity of M2e-specific antibodies induced by the AdC vectors expressing M2e only as part of the transgene product. Interestingly, M2e-specific antibodies from mice immunized twice with the AdC68M2e(3)NP-Hx-M2e(R1) vector had increased avidity to M2e compared with antibodies of mice immunized with AdC68-Hx-M2e(R1) vector. This was not seen upon priming (data not shown) and may suggest that the transgene product although it failed to increase M2e-specific antibody responses nevertheless drove maturation of memory B cells. Antibodies induced by the VR4 hexon-modified vector had low avidity to M2e.
NP-specific CD8+ T-cell responses
CD8+ T-cell responses to NP were tested at different timepoints after vaccination of C57BL/6 mice with the AdC68M2e(3)NP-Hx-M2e(R1) or AdC68M2e(3)NP-Hx-M2e(R4) vectors from blood (Figure 6). After priming, all of the mice developed detectable frequencies of NP-specific CD8+ T cells, which were comparable to those previously reported for mice immunized with the hexon unmodified AdC68M2e(3)NP vector.21 A booster immunization with the same vectors given 2 months after priming failed to increase circulating NP-specific CD8+ T-cell frequencies indicating that antibodies to the vaccine carrier impaired uptake of the vectors and thus expression of the transgene product.
Figure 6.

NP-specific CD8+ T-cell responses. Frequencies of NP-specific CD8+ T cells in blood were tested 5 weeks after priming or 5 weeks after boosting by tetramer staining. Graph shows mean frequencies of NP-specific CD8+ T cells of individual mice ± SD. NP, nucleoprotein.
Protection against A/PR8/34 challenge
We conducted two sets of experiments to determine vaccine efficacy. In the first inbred C57BL/6 mice were vaccinated with hexon-modified vectors with or without the M2e(3)NP fusion protein. In the second experiment, the same vectors were tested in ICR mice together with the AdC68M2e(3)NP vector carrying native hexon. In both experiments, mice vaccinated with the AdC68rab.gp vector were used as controls.
In the first experiment, vaccinated C57BL/6 mice (five per group) were infected 2 months after booster immunization with 10 LD50 of A/PR8/34 virus. Weight loss of vaccinated mice peaked by days 6–8 after challenge and then most mice began to gain weight and by 21 days after challenge most protected mice had returned to their prechallenge weight. Sham-vaccinated control mice continued to lose weight after challenge until they died or required euthanasia (Figure 7a). Upon challenge, 80% (P = 0.0238) of the mice vaccinated with AdC68M2e(3)NP-Hx-M2e(R1) survived, while 60% of the mice vaccinated with either AdC68-Hx-M2e(R1) or AdC68-M2e(3)NP-Hx-M2e(R4) survived (P > 0.05, Figure 7b); all mice in the AdC68-Hx-M2e(R4) vaccine and control groups died.
Figure 7.
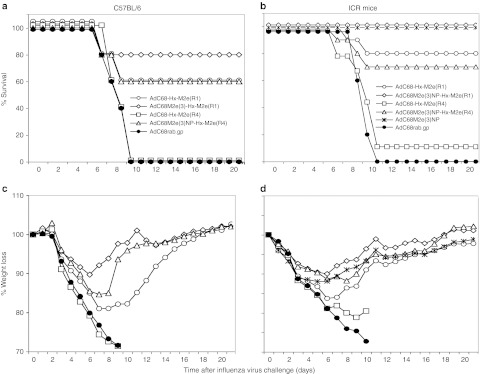
Protection against A/PR8/34 challenge. (a,c) C57BL/6 mice (n = 5) or (b,d) ICR mice (n = 10) were immunized 2× with the different vectors. Mice that were primed with AdC68M2e(3)NP were boosted with AdC6M2e(3)NP and control mice immunized with AdC68rab.gp were boosted with AdC6rab.gp; the other groups were boosted with the same vector used for priming. Mice were challenged with 10 LD50 of A/PR8/34 virus 2 months after boosting. (a,b) Survival and (c,d) mean weight loss after challenge. The following differences were statistically significant. (a) Using Fisher's exact test to determine significant differences in numbers of deaths between the groups showed significance between mice immunized with AdC68M2e(3)NP-Hx-M2e(R1) compared to those immunized with AdC68M2e(3)NP-Hx-M2e(R4) (P = 0.025) or AdC68rab.gp (P = 0.025). There was no significant difference in time to death using the Cox model. (b) Protection from death was highly significant for groups immunized with AdC68-Hx-M2e(R1), AdC68M2e(3)NP-Hx-M2e(R1) or AdC68M2e(3)NP-Hx-M2e(R4) (P < 0.001). There were also significant differences in time to death comparing group 1 with groups 5 and 6, groups 2 and 5 with groups 3, 4, and 6. Differences in weight loss on day 6 (the day before control mice started succumbing the infection) comparing vaccinated to control mice by t-tests were significant in (c) for mice immunized with AdC68M2e(3)NP-Hx-M2e(R1) (P = 0.025) and in (d) for mice immunized with AdC68M2(3)NP-Hx-M2e(R1) (P = 0.00004), AdC6M2e(3)NP (P = 0.0004) or AdC68M2e(3)NP-Hx-M2e(R4) (P = 0.03). Ad, adenovirus; NP, nucleoprotein.
The experiment was repeated with ICR mice (n = 10). For this experiment, a group receiving a previously described regimen composed of AdC68M2e(3)NP vector priming followed by a boost with the AdC6M2e(3)NP vector was included for comparison.21 Mice immunized with the AdC68M2e(3)NP-Hx-M2e(R1) vector twice or the AdC68M2e(3)NP/AdC6M2e(3)NP combination showed minimal weight loss of ~10% and all of the mice survived. The AdC68-Hx-M2e(R1) and AdC68M2e(3)NP-Hx-M2e(R4) vaccines also provided significant protection to 80 and 70% of mice, respectively. Only one of the AdC68-Hx-M2e(R4)-immunized mice survived and all of the mice of the control group succumbed the infection. Weight loss in general corresponded to level of protection against death but for mice immunized with the AdC68-Hx-M2e(R1) vector, which on average lost more weight than mice immunized with the AdC68M2e(3)NP-Hx-M2e(R1) vector. These results confirm as shown previously21 that concomitant stimulation of antibodies to M2e and T cells to NP results in superior protection than sole stimulation of M2e-specific antibodies; in both challenge experiments, animals that were vaccinated with hexon-modified vectors that also expressed the NP-containing fusion protein fared better than those that received the hexon-modified vectors that did not carry a transgene. This was especially pronounced for the VR4 hexon-modified vector that without the transgene failed to provide significant protection against a fatal influenza virus infection. In addition as expected from the immunological analyses, in absence of the M2e(3)NP transgene, the VR1 hexon-modified vector showed superior efficacy to the VR4 hexon-modified vector.
Discussion
Here we describe the construction, in vitro characterization, in vivo immunogenicity, and efficacy of two novel AdC vectors that were modified by insertion of a B-cell epitope from the influenza A M2e protein into two loops of hexon, which are exposed on the top of the molecule. Insertion of the epitope into VR1 or VR4 does not appear to alter the overall structure of hexon as modified hexons still form trimers on the virus capsid and bind antibodies to a conserved part of the molecule. The modification also does not affect vector fitness as hexon-modified vectors rescue easily and have growth characteristics similar to those of AdC vectors with wild-type hexon.
The M2e epitope within VR1 is more readily recognized by a monoclonal antibody to M2e and accordingly induces a more potent M2e-specific antibody response of higher avidity than the same sequence within VR4. We assume that this reflects that the loop encoded by VR1 is more accessible to antibodies compared with the loop encoded by VR4 as the former also carries the binding sites for the majority of neutralizing antibodies directed to native hexon.11 Nevertheless, we cannot rule out alternative explanations such as differences in the secondary structure of the M2e epitope placed into either loop.
Upon priming, the AdC68 vector carrying the M2e sequence within VR1 also induces higher antibody responses especially to the epitope's native structure within M2 as compared with a transgene product composed of a fusion protein of three M2e sequences and NP. One would assume that the amount of a transgene product that is produced for at least 7–10 days under the control of the potent cytomegalovirus promoter, until vector-transduced cells have been eliminated by the immune system,24 would be well in excess to that of an antigen present on the capsid that is not, or only at small amounts, synthesized in vivo by an E1-deleted Ad vector. The higher immunogenicity of M2e as displayed on the viral capsid may reflect that B-cell responses to rigidly arranged epitopes are less dependent on T helper cells as has been shown previously by Rolf M. Zinkernagel in the vesicular stromatitis virus system25 and that T helper cell is limited upon immunization with an AdC vector. Considering that AdC vectors carry a number of antigens with potential MHC class II epitopes,26 we favor the alternative explanation that the more structured display of antigen on a virus capsid favors B-cell stimulation compared to antigen primarily present non-repetitively within a transgene product.
Surprisingly, we were unable to further increase M2e-specific antibody responses by displaying M2e on hexon and within the same vectors encoding M2e as part of the transgene product. This was observed with both VR1 and VR4 hexon-modified vectors and is thus unlikely to reflect antigen saturation as the VR4 hexon-modified vector only induced low antibody responses to M2e.
B-cell responses induced by M2e within VR1 could easily be boosted by a second immunization with the same vector, which presumably reflects that M2e had replaced the main neutralizing B-cell epitope of AdC68. In contrast, the VR4 hexon-modified vector elicited only a marginal antibody recall response presumably due to interference by neutralizing antibodies to native parts of hexon. Upon booster immunization, the avidity of antibodies induced by AdC68/AdC6 vectors expressing M2e as a fusion protein with NP markedly increased. Interestingly upon booster immunization, but not upon priming, the VR1 hexon-modified vector, which in addition expressed the M2e fusion protein, elicited M2e-specific antibodies of higher avidity compared with the AdC68-Hx-M2e(R1) vector. The underlying mechanisms for this finding remain elusive. Antibody avidity increases are caused by Ig hypermutation followed by an antigen-driven selection process of B cells producing the best fitting antibodies. One could speculate that the transgenic M2e, which unlike the capsid-inserted M2e is not immediately available to the immune system, due to its suboptimal but protracted display over time may contribute to further selection of B cells producing avidity-matured antibodies. The selective effect on secondary immune responses, which in general arise more rapidly and require lower doses of antigen, is even more puzzling as we had expected that AdC transduction and hence transgene product production would become impaired in presence of AdC-neutralizing antibodies induced by priming. The increased avidity of antibodies by the AdC68M2e(3)NP-Hx-M2e vectors could potentially reflect differences in antigen presentation between naive mice which lack antibodies to Ad particles, and Ad-immune mice in which Ad-specific antibodies could have influenced presentation of the AdC vaccine.27 Alternatively, T helper cell responses to NP may have contributed.
AdC vectors expressing M2e within VR1 but not VR4 provided partial protection against A/PR8/34 challenge again stressing the superior immunogenicity of B-cell epitopes placed into the former domain. As reported previously,21 protection was improved by concomitant activation of NP-specific CD8+ T-cell responses through a transgene product. It should be pointed out that repeated immunization with VR1 hexon-modified vector encoding the M2e(3)NP fusion protein increased antibody titers to M2e but failed to augment NP-specific CD8+ T-cell responses presumably due to antibody-mediated inhibition of cell transduction by the Ad vector. Whether higher CD8+ T-cell responses by the use of heterologous display vectors would have further improved the efficacy of vaccines remains to be investigated.
Capsid-modified Ad vectors based in human serotype 5 have been tested previously as vaccine carriers in a number of systems. Modifications were incorporated as described here into viral hexon,12,13,15 the viral fiber28 that allows for the initial attachment to cellular receptors or they have been attached to protein IX,29 a small capsid protein that forms four tetramers on each facet on the icosahedral capsid of Ad virus. In experimental animals, all of these modified AdHu5 vectors induced humoral immune responses to the inserted sequence. We chose AdC vectors as vaccine carriers for the following reasons: such vectors can be constructed from molecular clones, they are in general genetically stable and can be scaled up for mass production. AdC vectors are highly immunogenic. They induce strong inflammatory responses30 thus not necessitating addition of adjuvants. They induce potent B- and T -cell responses in animals31 as well as in humans.32 Most importantly, responses are exceptionally sustained.4,32 In addition, Ad vectors due to deletion of E1, which renders such vectors replication-defective, are well tolerated in humans. We developed AdC vectors rather than Ad vectors based on human serotypes, as pre-existing neutralizing antibodies, which impair vaccine immunogenicity,8,14 are common in humans to the latter but not the former.18
We selected to assess the immunogenicity and efficacy of an epitope of influenza A virus as this virus remains a major threat to public health. Influenza A viruses cause severe illness in 3–5 million people worldwide and are linked to 250,000–500,000 deaths each year.33 The currently licensed influenza vaccines are annual vaccines that provide protective immunity mainly through induction of subtype-specific virus-neutralizing antibodies, and as such do not protect against new subtypes, antigenic variants or shift strains. Although the last pandemic caused by the so-called swine Flu in 2009 was comparatively benign, the speed with which the virus spread in spite of efforts of containment was remarkable. The 2009 swine flu was first identified in Mexico on 18 March 2009 and on 11 June 2009 was officially declared a pandemic virus by the WHO. A vaccine did not become available to the general US population till mid October. Previous pandemics such as the “Spanish flu” of 1918 had by far higher fatality rates and killed an estimated 50 million people. The current vaccines are based primarily on the viral surface proteins, the most variable of the viral proteins, which are the dominant targets for neutralizing antibodies. Other proteins are more conserved and have been shown to induce protective immunity such as M2e, which is the part of a tetrameric transmembrane protein of influenza A virus. M2e shows conservation among human influenza A virus strains and M2e-specific antibodies although they are not neutralizing have been shown to reduce the severity of influenza infection in animals against a wide range of influenza A virus strains.20,34 Influenza A NP, the major protein component of ribonucleoprotein complexes, is also relatively conserved making this protein an additional potential candidate for a universal influenza vaccine.22 Although the NP protein induces an antibody response, the role of such antibodies in providing protection remains controversial.35 NP induces both in mice and men a vigorous CD8+ T-cell response36,37 that, as epidemiological studies suggest, may contribute to resistance against severe disease following influenza A virus infection.38 More recently a segment within the hemagglutinin stalk that is conserved between several hemagglutinin subtypes was shown to contain conformational B-cell epitopes, which bind neutralizing antibodies39,40 and may thus provide the basis or a component of a universal influenza A virus vaccine.41
Influenza vaccines based on M2e, NP or both have been tested extensively in animal models where they showed sufficient promise that some of them advanced to clinical trials.34 We developed and tested several prototype universal influenza virus vaccines. Our initial universal influenza vaccines expressed only NP or M2e of A/PR8 as their transgene product. They provided very limited protection to a low dose A/PR8 challenge of young mice (data not shown). The subsequent vaccine which expressed a fusion protein composed of three M2e sequences from different strains of influenza A virus (H1N1, H5N1, and H7N2) fused to A/PR8/34 NP provided as published previously21 solid protection against challenge with different strains of influenza virus in young mice, which was shown to depend on both antibodies to M2e and CD8+ T cells to NP. Nevertheless, protection required prime boosting with two heterologous AdC vectors, a regimen that would be unduly costly and cumbersome for mass vaccination. The new vaccine, which expresses the M2e epitope within VR1 resulted in increased M2e-specific antibody responses and provided protection upon an homologous prime boost regimen.
This is the first report describing the immunogenicity of a foreign epitope placed into the capsid of an AdC vector demonstrating the versatility of such vectors not only as carriers of foreign transgenes but also as potent virus-like particle vaccines.
Materials and Methods
Ad vectors. AdC68 vectors expressing the M2e peptide within hexon were generated as follows: a fragment encoding most parts of the hexon sequence and flanked with Mlu I was released from the E1-deleted viral molecular clone of AdC68 and cloned into the pcDNA3.1 vector (Invitrogen). The part of the M2e sequence of A/PR8/34 virus encoding LTEVETPIRNEWG was cloned into VR1 of hexon. Namely, residues 142-144 (ETA) of hexon were deleted and replaced with the M2e sequence. To generate the VR4-modified vector, the same M2e sequence was inserted between residues of 253-254 of hexon. Upon verification of the correct insertion of the M2e-encoding base pairs by sequencing, the hexon sequence was excised from the pcDNA3.1 vector and cloned back into the viral molecular clone. For some vectors, an expression cassette containing the previously described M2e(3)NP sequence21 under the control of the cytomegalovirus early promoter was placed into E1. Recombinant viral molecular clones were used to rescue virus in HEK 293 cells. Virus was expanded on HEK 293 cells, purified by cesium chloride density-gradient centrifugation and vp content was determined by spectrophotometry at 260 nm. Vectors were titrated by a PCR-based method to determine infectious units. Table 1 shows a list of the new vectors and the nomenclature used throughout this manuscript as well as pertinent growth characteristics such as yields per 108 HEK 293 cells and vp to infectious unit ratios. Other Ad vectors such as the AdC68rab.gp vector, Ad vectors expressing green fluorescent protein (GFP) or AdC vectors expressing the M2e(3)NP fusion protein have been described previously21 or were generated using previously described cloning techniques.42
Western blot analysis. Purified AdC68 virus with modified hexon on VR1 and VR4 along with an AdC68 vector carrying wild-type hexon were tested by Blue Native polyacrylamide gel electrophoresis (PAGE) without denaturation on a 3–12% Bis Tris Native gel (Invitrogen). Purified virus was directly mixed with Native PAGE sample buffer containing digitonin (0.5%), n-dodecyl-β-d-maltoside (DDM; 0.25%), G-250 and loaded onto the 3–12% bis tris native gel for separation. Unstained Native PAGE marker (Invitrogen) was also loaded onto the gel and run at 150 V and transferred to polyvinylidene difluoride membrane. Alternatively, virions were boiled upon addition of β-mercaptoethanol. Samples were loaded onto 8% sodium dodecyl sulfate-PAGE. The proteins were then transferred to a polyvinylidene difluoride membrane and blocked with 5% non-fat dry milk for 60 minutes at room temperature. The membrane was then incubated with 1:1,000 dilution of mouse monoclonal antibody to hexon termed 3G0 (Santa Cruz Biotechnology) and M2e (14C2-S1-4.2) overnight at 4 °C and washed thrice with 0.1% phosphate-buffered saline (PBS) with Tween. The membrane was probed with 1:20,000 dilution of anti-mouse horseradish peroxidase-conjugated IgG secondary antibody for 1 hour, washed and developed using west pico chemiluminescent substrate (Thermo Scientific, Waltham, MA).
For analysis by ELISA, Nunc 96-well plates were coated with 1010 vp of Ad vectors per well in 100 µl of coating buffer (15 mmol/l Na2CO3, 35 mmol/l NaHCO3, and 3 mmol/l NaN3, pH 9.6) at 4 °C overnight. Plates were blocked with PBS containing 5% bovine serum albumin at room temperature for 1 hour. Plates were then treated for 1 hour with serially diluted M2e monoclonal antibody (14C2-S1-4.2) at room temperature followed by incubation with the alkaline phosphatase-conjugated goat anti-mouse Ig and then with the substrate.
Identification of the encoded transgene product. To identify the presence of M2e(3)NP fusion protein, cell lysates from virus-infected HEK 293 were prepared and proteins were separated by gel electrophoresis and transferred to a membrane, which was then blotted with a monoclonal antibody to M2e (14C2-S1-4.2).
Influenza virus. Influenza viruses A/PR/8/34 was grown in the chorioallantoic fluid of embryonated chicken eggs and titrated in adult mice upon their intranasal infection to determine the mean lethal dose (LD50).
Mice. Female C57BL/6 and ICR mice were purchased at age 6–8 weeks from ACE Animals (Boyertown, PA). Mice were housed at the Wistar Institute Animal Facility and all animal procedures in this study were based on approved institutional protocols.
Immunization of mice. Groups of 5–10 mice were vaccinated intramuscularly with a total of 1 × 1010 vp of the Ad vectors. Two months later, some groups of mice were boosted intramuscularly with the same or a heterologous vector given intramuscularly at the same dose.
Antibody responses to M2e. Antibody responses specific to M2e were measured from sera of individual mice by an M2e-peptide ELISA using a previously published procedure.23 Briefly, the multiple antigenic peptide consisting of a Cys-(Gly-Lys)3-Ala backbone with two attached M2e(2–24) peptides was used to coat wells of Nunc 96-well plates (Thermo Fisher Scientific, Rochester, NY) by incubating 50 µl of the peptide dilution at 85 nmol/l in 0.02 mol/l NaCl at 4 °C overnight. Plates were blocked for 2–18 hours with PBS containing 5% bovine serum albumin. After washing, they were incubated for 1 hour with serial dilutions of sera in PBS + 5% bovine serum albumin followed by incubation with 1:200 dilution of alkaline phosphatase-conjugated goat anti-mouse Ig (Cappel, Irvine, CA) for 1 hour at room temperature. After being washed, plates were incubated for 20 minutes with substrate (10 mg d-nitrophenyl phosphate disodium dissolved in 10 ml of 1 mmol/l MgCl2, 3 mmol/l NaN3, and 0.9 mol/l diethanolamine, pH 9.8) and then read in an automated ELISA reader at 405 nm. The assay was standardized with the monoclonal antibody to M2e (14C2-S1-4.2).
Alternatively, antibody titers were determined by a previously described cellular ELISA.21 Briefly, 293T cells were infected with a lentivirus expressing the full-length M2 sequence of A/PR8/34 virus to generate stable M2+ cell lines. A control cell line was generated by infection of 293T cells with empty lentivirus. These cell lines were used as immunosorbents in an ELISA as described.23 The assay was standardized with the 14C2-S1-4.2 antibody.
Avidity of antibodies to M2e was assessed by sodium thiocyanate-displacement ELISA. Briefly, pooled serum samples from each group of immunized mice were diluted to a level calculated to have a remaining titer of 1:100 and incubated on ELISA plates coated with M2e peptide for 2 hours at room temperature. Then, ascending concentrations of the chaotropic agent sodium thiocyanate were added to the wells (0–4 mol/l) and plates were incubated for 15 minutes at room temperature before washing. The ELISA was then completed as described above.
Antibody responses to Ad vectors. Ad-specific neutralization titers were measured on HEK 293 cells infected with AdC68 vectors expressing enhanced GFP (AdC68EGFP), as described previously.18 Briefly, a dose of AdC68EGFP (with or without hexon modifications) that caused enhanced GFP expression in 70–90% of the cells within 24 hours was chosen. Sera from mice vaccinated with 1 × 1010 vp of vectors were harvested 5 weeks after vaccination, and inactivated at 55 °C for 30 minutes. Serially diluted sera were then mixed with appropriate doses of AdC68EGFP and incubated for 60 minutes at room temperature. The vector-serum mixture was mixed with an equal volume of HEK 293 cells at 106 cells/ml and the mixture was transferred into flat-bottom 96-well plates. The plates were incubated overnight at 37 °C and then screened visually for green fluorescent cells under a ultraviolet microscope. The titer was determined as the reciprocal serum dilution that caused 50% reduction of fluorescent cells in comparison to that seen in control wells infected with vector only.
Tetramer staining of T cell. MHC class I NP peptide tetramer (ASNENTETM) conjugated with APC was provided by the Tetramer Core Facility (Emory University, Atlanta, GA). Lymphocytes were stained with the NP tetramer, a PerCP-Cy5.5-labeled antibody to CD8 and a live cell stains (both from BD Biosciences, San Jose, CA) for 30 minutes at 4 °C. Flow cytometric acquisition and analysis of samples was performed on at least 500,000 events. The post-acquisition data were processed using FlowJo 7.1.1 (TreeStar, Ashland, OR).
Influenza virus challenge. Two months after vaccination, mice were anesthetized and then challenged intranasally with 10 LD50 of influenza A/PR/8/34 virus diluted in 30 µl PBS. Mice were monitored daily for weight loss and survival after challenge. Mice were euthanized once they lost in excess of 30% of their prechallenge weight.
Statistical analyses. Student's t-tests were used for pairwise comparisons. Antibody titers were compared using analysis of variance, with Tukey's adjustment for multiple pairwise comparisons. General linear regression models adjusting for the molarity and the interactions between vaccine and molarity were used to examine the different effect between the vaccine on antibody avidity with Bonferroni adjustment for multiple pairwise comparisons. Fisher's exact test was used to compare the number of deaths across the groups. Differences in time to death were assessed via Cox models with Sidak adjustment for pairwise comparisons.
SUPPLEMENTARY MATERIAL Supplementary Data.
Acknowledgments
This work was funded by a Broad Agency Announcement contract from National Institute of Allergy and Infectious Diseases and by a grant from the State of Pennsylvania. S.T. is the recipient of a Howard Hughes Medical Institute Gilliam Fellowship. The authors declared no conflict of interest.
Supplementary Material
REFERENCES
- Colloca S, Barnes E, Folgori A, Ammendola V, Capone S, Cirillo A.et al. (2012Vaccine vectors derived from a large collection of simian adenoviruses induce potent cellular immunity across multiple species Sci Transl Med 4115ra2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen SG, Ford JC, Lewis MS, Ventura AB, Hughes CM, Coyne-Johnson L.et al. (2011Profound early control of highly pathogenic SIV by an effector memory T-cell vaccine Nature 473523–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahn JS, Roberts A, Weibel C, Buonocore L., and, Rose JK. Replication-competent or attenuated, nonpropagating vesicular stomatitis viruses expressing respiratory syncytial virus (RSV) antigens protect mice against RSV challenge. J Virol. 2001;75:11079–11087. doi: 10.1128/JVI.75.22.11079-11087.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatsis N, Fitzgerald JC, Reyes-Sandoval A, Harris-McCoy KC, Hensley SE, Zhou D.et al. (2007Adenoviral vectors persist in vivo and maintain activated CD8+ T cells: implications for their use as vaccines Blood 1101916–1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rupprecht CE, Wiktor TJ, Johnston DH, Hamir AN, Dietzschold B, Wunner WH.et al. (1986Oral immunization and protection of raccoons (Procyon lotor) with a vaccinia-rabies glycoprotein recombinant virus vaccine Proc Natl Acad Sci USA 837947–7950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang ZQ, Yang Y, Wilson JM., and, Ertl HC. A replication-defective human adenovirus recombinant serves as a highly efficacious vaccine carrier. Virology. 1996;219:220–227. doi: 10.1006/viro.1996.0239. [DOI] [PubMed] [Google Scholar]
- Dintzis HM, Dintzis RZ., and, Vogelstein B. Molecular determinants of immunogenicity: the immunon model of immune response. Proc Natl Acad Sci USA. 1976;73:3671–3675. doi: 10.1073/pnas.73.10.3671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang Z, Gao G, Reyes-Sandoval A, Cohen CJ, Li Y, Bergelson JM.et al. (2002Novel, chimpanzee serotype 68-based adenoviral vaccine carrier for induction of antibodies to a transgene product J Virol 762667–2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rux JJ, Kuser PR., and, Burnett RM. Structural and phylogenetic analysis of adenovirus hexons by use of high-resolution x-ray crystallographic, molecular modeling, and sequence-based methods. J Virol. 2003;77:9553–9566. doi: 10.1128/JVI.77.17.9553-9566.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue F., and, Burnett RM. Capsid-like arrays in crystals of chimpanzee adenovirus hexon. J Struct Biol. 2006;154:217–221. doi: 10.1016/j.jsb.2005.12.006. [DOI] [PubMed] [Google Scholar]
- Pichla-Gollon SL, Drinker M, Zhou X, Xue F, Rux JJ, Gao GP.et al. (2007Structure-based identification of a major neutralizing site in an adenovirus hexon J Virol 811680–1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews QL, Fatima A, Tang Y, Perry BA, Tsuruta Y, Komarova S.et al. (2010HIV antigen incorporation within adenovirus hexon hypervariable 2 for a novel HIV vaccine approach PLoS ONE 5e11815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worgall S, Krause A, Rivara M, Hee KK, Vintayen EV, Hackett NR.et al. (2005Protection against P. aeruginosa with an adenovirus vector containing an OprF epitope in the capsid J Clin Invest 1151281–1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzgerald JC, Gao GP, Reyes-Sandoval A, Pavlakis GN, Xiang ZQ, Wlazlo AP.et al. (2003A simian replication-defective adenoviral recombinant vaccine to HIV-1 gag J Immunol 1701416–1422. [DOI] [PubMed] [Google Scholar]
- Abe S, Okuda K, Ura T, Kondo A, Yoshida A, Yoshizaki S.et al. (2009Adenovirus type 5 with modified hexons induces robust transgene-specific immune responses in mice with pre-existing immunity against adenovirus type 5 J Gene Med 11570–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley RR, Lynch DM, Iampietro MJ, Borducchi EN., and, Barouch DH. Adenovirus serotype 5 neutralizing antibodies target both hexon and fiber following vaccination and natural infection. J Virol. 2012;86:625–629. doi: 10.1128/JVI.06254-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pichla-Gollon SL, Lin SW, Hensley SE, Lasaro MO, Herkenhoff-Haut L, Drinker M.et al. (2009Effect of preexisting immunity on an adenovirus vaccine vector: in vitro neutralization assays fail to predict inhibition by antiviral antibody in vivo J Virol 835567–5573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang Z, Li Y, Cun A, Yang W, Ellenberg S, Switzer WM.et al. (2006Chimpanzee adenovirus antibodies in humans, sub-Saharan Africa Emerging Infect Dis 121596–1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Zou P., and, Chen YH. Monoclonal antibodies recognizing EVETPIRN epitope of influenza A virus M2 protein could protect mice from lethal influenza A virus challenge. Immunol Lett. 2004;93:131–136. doi: 10.1016/j.imlet.2004.03.003. [DOI] [PubMed] [Google Scholar]
- Neirynck S, Deroo T, Saelens X, Vanlandschoot P, Jou WM., and, Fiers W. A universal influenza A vaccine based on the extracellular domain of the M2 protein. Nat Med. 1999;5:1157–1163. doi: 10.1038/13484. [DOI] [PubMed] [Google Scholar]
- Zhou D, Wu TL, Lasaro MO, Latimer BP, Parzych EM, Bian A.et al. (2010A universal influenza A vaccine based on adenovirus expressing matrix-2 ectodomain and nucleoprotein protects mice from lethal challenge Mol Ther 182182–2189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulmer JB, Donnelly JJ, Parker SE, Rhodes GH, Felgner PL, Dwarki VJ.et al. (1993Heterologous protection against influenza by injection of DNA encoding a viral protein Science 2591745–1749. [DOI] [PubMed] [Google Scholar]
- Mozdzanowska K, Feng J, Eid M, Kragol G, Cudic M, Otvos L., Jret al. (2003Induction of influenza type A virus-specific resistance by immunization of mice with a synthetic multiple antigenic peptide vaccine that contains ectodomains of matrix protein 2 Vaccine 212616–2626. [DOI] [PubMed] [Google Scholar]
- Yang Y, Li Q, Ertl HC., and, Wilson JM. Cellular and humoral immune responses to viral antigens create barriers to lung-directed gene therapy with recombinant adenoviruses. J Virol. 1995;69:2004–2015. doi: 10.1128/jvi.69.4.2004-2015.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachmann MF, Hengartner H., and, Zinkernagel RM. T helper cell-independent neutralizing B cell response against vesicular stomatitis virus: role of antigen patterns in B cell induction. Eur J Immunol. 1995;25:3445–3451. doi: 10.1002/eji.1830251236. [DOI] [PubMed] [Google Scholar]
- Veltrop-Duits LA, Heemskerk B, Sombroek CC, van Vreeswijk T, Gubbels S, Toes RE.et al. (2006Human CD4+ T cells stimulated by conserved adenovirus 5 hexon peptides recognize cells infected with different species of human adenovirus Eur J Immunol 362410–2423. [DOI] [PubMed] [Google Scholar]
- Choi JH, Dekker J, Schafer SC, John J, Whitfill CE, Petty CS.et al. (2012Optimized adenovirus-antibody complexes stimulate strong cellular and humoral immune responses against an encoded antigen in naive mice and those with preexisting immunity Clin Vaccine Immunol 1984–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krause A, Joh JH, Hackett NR, Roelvink PW, Bruder JT, Wickham TJ.et al. (2006Epitopes expressed in different adenovirus capsid proteins induce different levels of epitope-specific immunity J Virol 805523–5530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayer W, Tenbusch M, Lietz R, Johrden L, Schimmer S, Uberla K.et al. (2010Vaccination with an adenoviral vector that encodes and displays a retroviral antigen induces improved neutralizing antibody and CD4+ T-cell responses and confers enhanced protection J Virol 841967–1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hensley SE, Giles-Davis W, McCoy KC, Weninger W., and, Ertl HC. Dendritic cell maturation, but not CD8+ T cell induction, is dependent on type I IFN signaling during vaccination with adenovirus vectors. J Immunol. 2005;175:6032–6041. doi: 10.4049/jimmunol.175.9.6032. [DOI] [PubMed] [Google Scholar]
- Reyes-Sandoval A, Fitzgerald JC, Grant R, Roy S, Xiang ZQ, Li Y.et al. (2004Human immunodeficiency virus type 1-specific immune responses in primates upon sequential immunization with adenoviral vaccine carriers of human and simian serotypes J Virol 787392–7399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes E, Folgori A, Capone S, Swadling L, Aston S, Kurioka A.et al. (2012Novel adenovirus-based vaccines induce broad and sustained T cell responses to HCV in man Sci Transl Med 4115ra1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson WW, Comanor L., and, Shay DK. Epidemiology of seasonal influenza: use of surveillance data and statistical models to estimate the burden of disease. J Infect Dis. 2006;194 suppl. 2:S82–S91. doi: 10.1086/507558. [DOI] [PubMed] [Google Scholar]
- Fan J, Liang X, Horton MS, Perry HC, Citron MP, Heidecker GJ.et al. (2004Preclinical study of influenza virus A M2 peptide conjugate vaccines in mice, ferrets, and rhesus monkeys Vaccine 222993–3003. [DOI] [PubMed] [Google Scholar]
- Carragher DM, Kaminski DA, Moquin A, Hartson L., and, Randall TD. A novel role for non-neutralizing antibodies against nucleoprotein in facilitating resistance to influenza virus. J Immunol. 2008;181:4168–4176. doi: 10.4049/jimmunol.181.6.4168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gschoesser C, Almanzar G, Hainz U, Ortin J, Schonitzer D, Schild H.et al. (2002CD4+ and CD8+ mediated cellular immune response to recombinant influenza nucleoprotein Vaccine 203731–3738. [DOI] [PubMed] [Google Scholar]
- McMichael AJ, Michie CA, Gotch FM, Smith GL., and, Moss B. Recognition of influenza A virus nucleoprotein by human cytotoxic T lymphocytes. J Gen Virol. 1986;67 Pt 4:719–726. doi: 10.1099/0022-1317-67-4-719. [DOI] [PubMed] [Google Scholar]
- Epstein SL. Prior H1N1 influenza infection and susceptibility of Cleveland Family Study participants during the H2N2 pandemic of 1957: an experiment of nature. J Infect Dis. 2006;193:49–53. doi: 10.1086/498980. [DOI] [PubMed] [Google Scholar]
- Ekiert DC, Bhabha G, Elsliger MA, Friesen RH, Jongeneelen M, Throsby M.et al. (2009Antibody recognition of a highly conserved influenza virus epitope Science 324246–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekiert DC, Friesen RH, Bhabha G, Kwaks T, Jongeneelen M, Yu W.et al. (2011A highly conserved neutralizing epitope on group 2 influenza A viruses Science 333843–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang TT, Tan GS, Hai R, Pica N, Ngai L, Ekiert DC.et al. (2010Vaccination with a synthetic peptide from the influenza virus hemagglutinin provides protection against distinct viral subtypes Proc Natl Acad Sci USA 10718979–18984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou D, Zhou X, Bian A, Li H, Chen H, Small JC.et al. (2010An efficient method of directly cloning chimpanzee adenovirus as a vaccine vector Nat Protoc 51775–1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.