

Abstract
The combination of an oncolytic virus, that directly destroys tumor cells and mediates an acute immune response, with an immune cell therapy, capable of further enlisting and enhancing the host immune response, has the potential to create a potent therapeutic effect. We have previously developed several strategies for optimizing the delivery of oncolytic vaccinia virus vectors to their tumor targets, including the use of immune cell-based carrier vehicles and the incorporation of mutations that increase production of the enveloped form of vaccinia (extracellular enveloped viral (EEV)) that is better adapted to spread within a host. Here, we initially combine these approaches to create a novel therapeutic, consisting of an immune cell (cytokine-induced killer, CIK) preloaded with an oncolytic virus that is EEV enhanced. This resulted in direct interaction between the viral and immune cell components with each assisting the other in directing the therapy to the tumor and so enhancing the antitumor effects. This effect could be further improved through CCL5 expression from the virus. The resulting multicomponent therapy displays the ability for synergistic crosstalk between components, so significantly enhancing tumor trafficking and antitumor effects.
Introduction
Although evasion of immune destruction represents an emerging hallmark of cancer,1 cancer's suppressive effects on the immune system are typically reversible. Biological therapies of cancer therefore have the potential to not only directly target the tumor, but also to reprogram the patient's immune response to help recognize malignant cells as foreign. However, to successfully achieve this goal will likely require the simultaneous targeting of multiple immune pathways, meaning that approaches that have a single mechanism of action are unlikely to succeed. Instead combinations of multi-mechanistic biological therapies represent the most promising approach.
It has been demonstrated in both preclinical and clinical studies that oncolytic viruses such as those based on vaccinia virus mediate an acute viral infection selectively within the tumor with lysis of tumor cells leading to release of tumor-associated antigens and other danger signals, localized transient reductions in immune suppressive cell types, and recruitment of natural killer (NK), dendritic cell, and T cells into the tumor environment.2,3,4,5,6,7,8,9,10,11,12,13,14,15,16 Further, the combination of oncolytic viruses with immune cell therapies can lead to even greater targeting of localized immune suppression and systemic immune activation.17,18,19,20
We have previously looked to improve the systemic delivery and intertumoral spread of oncolytic vaccinia through several distinct approaches. In one such approach, an immune cell therapy (such as cytokine-induced killer (CIK) cells) that can efficiently traffic to the tumor target was pre-infected with the viral therapy and used as a delivery vehicle in a “Trojan Horse” approach.17 It was further demonstrated that as the CIK-delivered vaccinia virus infected the tumor it induced increases in the levels of the stress response ligands MICA and MICB on the surface of the cancer cells. These ligands are recognized by NKG2D on the surface of the CIK cells17 and so this resulted in increased targeting of the tumor by the CIK cells, and synergy between the two therapies.
In an alternative approach, we have examined the role of the different forms of vaccinia virus that are produced naturally during its replication cycle, focusing particularly on the extracellular enveloped viral (EEV) form that is adapted for spread within a host, as this was felt likely to enhance the effectiveness of an oncolytic agent;6 EEV is released early after viral infection, meaning it can spread more rapidly within the tumor before immune-mediated removal; and it is shrouded in a host cell-derived membrane that incorporates host cell proteins, including complement control proteins and has relatively few viral antigens exposed on the outer surface, meaning that the EEV form is well adapted for systemic spread in the host (relative to the other viral forms such as the intracellular mature virus (IMV) form).21,22,23,24 Viral mutations that enhance the relative levels of the EEV form produced subsequent to infection resulted in more effective oncolytic vectors that are better able to spread within and between tumors.6
However, the EEV form of the virus is relatively unstable outside of a host and so needs to be primarily produced in situ. This was originally achieved entirely as a result of viral replication inside of the tumor.6 However, here we hypothesize that a susceptible cell pre-infected ex vivo with an EEV-enhanced oncolytic vector could act as a factory for production of the EEV form of the virus once returned to the host. As such, CIK cells pre-infected with EEV-enhanced oncolytic vaccinia might act both as a cell delivery vehicle and as an in situ EEV factory.
Further, because progeny EEV virus particles are released from an infected cell within 8–12 hours after infection, while CIK trafficking to the tumor takes greater than 24 hours after systemic delivery, EEV virus would be released before CIK infiltration into the tumor. This virus released as EEV from the pre-infected CIK cells might therefore be able to seed the tumor and alter the tumor microenvironment to further enhance subsequent CIK cell trafficking. The expression of the chemokine CCL5 (RANTES) from the virus was therefore also examined as we had previously shown CCL5 expression can enhance CIK tumor trafficking.10 Together, this combination therapy (CCL5-expressing and EEV-enhanced vaccinia pre-infected into CIK cells) was predicted to synergize in its crosstalk and ability to target the individual components to the tumor, and so to enhance overall antitumor effects.
Results
Complement inhibits systemic delivery of oncolytic vaccinia to the tumor
Even though systemic delivery of oncolytic vaccinia to the tumor has been demonstrated both in preclinical mouse models4 and in the clinical setting14 it is apparent that only a very small fraction of the viral inoculum infects the tumor, and that therapeutic effects depend on subsequent local amplification of the vector. This is true even in individuals that have not been previously immunized against the virus. It was therefore decided to examine the role of complement in limiting systemic viral delivery in the absence of neutralizing antibody. Initial in vitro experiments examined the effects of prior viral exposure to untreated human serum on the ability of virus to infect three-dimensional tumor spheroids in culture. Oncolytic vaccinia strain Western Reserve (WR).TK- expressing green fluorescent protein (GFP) (10,000 plaque-forming unit (PFU)) was mixed with serum (diluted 1:10 in phosphate-buffered saline (PBS)) for 5 minutes before addition to multicellular tumor spheroids (formed from the human breast cancer cell line MCF-7 using our previously described procedure).6 The TK deletion is known to create an oncolytic or tumor-targeting phenotype, and also allows the insertion of transgenes, such as the luciferase or GFP reporter genes.25 The serum was initially determined not to have measurable neutralizing antibody levels before its use. After 24 hours, spheroids were examined by fluorescence microscopy for the expression of GFP. GFP expression was also quantified in a fluorescence plate reader. It was seen (Figure 1a) that untreated serum was capable of effectively ablating viral infection. This was assumed to be due to complement-mediated inactivation of the virus. This was verified by both prior heat inactivation of the complement in the serum and by addition of the complement inhibitor cobra venom factor (CVF) to the serum, both treatments resulted in a return to full infectious potential for the virus.
Figure 1.
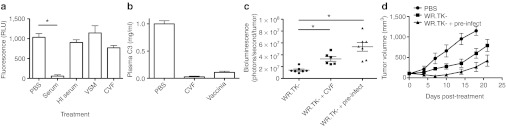
Complement severely inhibits systemic delivery of oncolytic vaccinia virus. (a) In vitro effects of serum on viral infection of multicellular tumor spheroids. Multicellular tumor spheroids were formed by growing MCF-7 tumor cells in non-coated tissue culture plates (as previously described). Virus (1 × 104 PFU of WR.TK- expressing GFP) was pre-mixed as indicated for 5 minutes at room temperature before being applied to the spheroids. GFP fluorescence was measured after 24 hours in a fluorescence plate reader. Conditions examined including (i) mixing 1:1 with PBS; (ii) mixing 1:1 with human serum from a non-immunized donor (as verified by a lack of neutralizing antibody); (iii) mixing 1:1 with the same serum after heat inactivation (56 °C for 30 minutes); (iv) mixing 1:1 with the same serum mixed with media collected from MCF-7 cells pre-infected with WR for 24 hours at an multiplicity of infection of 2.0, and filter sterilized (1 µm) to remove any viral particles (virus spent medium (VSM)); (v) mixing with serum treated with cobra venom factor (100 µl of serum with 2 µg CVF for 1 hour at 37 °C). Only serum alone displayed significant reduction in fluorescence relative light units (RLU) (*P = 0.0007). (b) In vivo effects of different treatments on C3 concentrations in plasma. BALB/c mice (n = 3) were treated with CVF (600 µg/kg delivered intraperitoneally 16 hours earlier) or vaccinia (1 × 106 PFU of strain WR delivered intradermally 8 hours earlier) before collection of plasma and measurement of C3 by ELISA. (c) In vivo effects of complement on systemic delivery of oncolytic vaccinia. Mice (BALB/c) bearing subcutaneous JC tumors (50–100 mm3) were injected with WR.TK- expressing luciferase (1 × 107 PFU via tail vein) and imaged after 7 hours (IVIS 200, PerkinElmer) to determine the viral gene expression levels from within a region of interest drawn around the tumor. Some mice were pretreated, either with intraperitoneal injection of CVF (600 µg/kg) 16 hours before viral injection; or with low dose (1 × 106 PFU) of wild-type vaccinia virus (WR) not expressing luciferase (pre-infect) (n = 7 per group). Inhibition of complement increased early viral gene expression from within the tumor. Both treatments significantly increase delivery of virus to the tumor relative to WR.TK- alone (*P < 0.001). Pre-infection with WR also significantly enhances delivery relative to CVF treatment (*P = 0.039). (d) Enhanced antitumor effect through inhibition of complement before viral delivery. In a repeat in vivo experiment (BALB/c mice with JC tumors), tumor volume was followed over time after treatment with PBS; WR.TK- alone (1 × 107 PFU) or WR.TK- after pre-treatment with low dose WR (n = 8 per group). Pre-treatment significantly enhanced therapeutic effect over WR.TK- alone at days 7 and 10 only (*P = 0.0013 and 0.02). Pre-treatment therefore enhanced therapeutic effect as well as initial delivery. GFP, green fluorescent protein; HI, heat inactivated; PBS, phosphate-buffered saline; PFU, plaque-forming unit; WR, Western Reserve.
However, vaccinia has naturally evolved several mechanisms to evade host complement, including expression of the vaccinia complement control protein (VCP),26,27 and production of the EEV form, that incorporates a panel of host cell-derived complement control factors into the outer envelope22 and so the potential of these mechanisms to enhance systemic delivery was examined. It was first demonstrated that spent media taken from vaccinia-infected cells and filter sterilized through a 1-µm filter (to remove viral particles but not secreted proteins such as VCP) was also capable of restoring viral infectious capability (Figure 1a). Although not directly measured, it is predicted this media would contain VCP (in addition to other secreted viral and cellular factors).
The inhibitory role of complement inactivation was further examined during in vivo systemic delivery of oncolytic vaccinia to the tumor using an immunocompetent mouse model. It was initially determined that both CVF treatment and vaccinia pre-infection (and VCP production) resulted in inhibition of complement activity (Figure 1b) in vivo. It was seen that vaccinia pre-infection (through an intradermal infection of the mouse with a low dose of wild-type vaccinia 8 hours before systemic delivery of oncolytic vaccinia) enhanced subsequent viral delivery (Figure 1c) and antitumor effects (Figure 1d). This approach might therefore allow for enhanced clinical delivery of oncolytic vaccinia. However, although it was predicted that VCP would be secreted from infected cells by the time of oncolytic viral delivery and may be mediating these effects, a variety of additional secreted viral virulence factors, or other interactions with the host immune response may also contribute to this enhanced delivery and antitumor effect. Therefore a second experiment was run utilizing CVF as a means to specifically inhibit complement before systemic viral delivery. In this situation, a small increase in viral delivery to the tumor was again seen, although not as great as for viral pre-infection, indicating other factors, such as desensitizing of the innate response, may also play a role, but that complement is clearly an important component of this effect (Figure 1c). Indeed, CVF treatment was not sufficient to increase antitumor effects to significant levels (data not shown) whereas viral pre-infection resulted in a small but significant increase in antitumor effects (Figure 1d).
Of note, for comparison, whereas 10 µl of either mouse or human serum was capable of inactivating ~90% of 5,000 PFU of virus, in the mouse model 2000X this amount of virus (1 × 107 PFU) was applied in vivo (to an animal with ~2 ml of blood).
The EEV form of vaccinia displays increased complement evasion
Because the EEV form of vaccinia is wrapped in a host cell-derived membrane that incorporates host cell complement control proteins, it was predicted it would also be capable of improved systemic delivery to tumors in naive mice. This was initially tested in culture, where it was confirmed that pure EEV (produced through exposure of a crude viral prep to anti-IMV antibody) was only marginally reduced in infectivity when pretreated with serum. However, pure IMV (produced through neutralization of EEV with anti-EEV antibody) demonstrated >90% reduction in viability following pre-mixing with serum (quantified through viral gene expression assays as before) (Figure 2a).
Figure 2.

The EEV form of vaccinia evades complement-mediated neutralization and accounts for a disproportionate percentage of the virus delivered systemically to the tumor. (a) The EEV form of vaccinia can evade complement in vitro. Virus (WR.TK- GFP+ as before) was pretreated with either anti-IMV (NR417) or anti-EEV (NR551) neutralizing antibody and then mixed with human serum (as before) or PBS before addition to MCF-7 multicellular tumor spheroids. Fluorescence was again determined after 24 hours. (b) Titering of EEV in viral preparations. WR.TK-.Luc+ and WI.TK-.Luc+ (EEV-enhanced) virus grown and purified by our standard procedures were treated with IMV-neutralizing antibody or EEV-neutralizing antibody (as before) before titering by plaque assay (addition of both antibodies together resulted in viral titers below the limits of detection, <100 PFU). (c) Relative importance of viral forms in systemic delivery. The WR.TK-.Luc+ preparation was again treated to remove IMV or EEV (as before) before intravenous injection of 1 × 107 PFU (of untreated preparation) into JC tumor bearing BALB/c mice. Viral gene expression from within the tumor was detected by bioluminescence imaging (BLI) after 24 hours. Ab, antibody; EEV, extracellular enveloped viral; GFP, green fluorescent protein; IMV, intracellular mature virus; PFU, plaque-forming unit; PBS, phosphate-buffered saline; WR, Western Reserve.
Many of the processes involved in purification of viral preparations for preclinical in vivo use or for clinical use, including freeze-thawing and centrifugation, tend to destroy the outer envelope of the EEV form of vaccinia, and so it was predicted that the level of EEV would be greatly reduced in the inoculum used for our in vivo work. We initially sought to verify this (through titering of viral preparations subsequent to exposure to high levels of anti-IMV– or anti-EEV–neutralizing antibody) and it was found that around 3% of a preparation of WR.TK- (grown and purified according to our standard procedures for obtaining virus for in vivo testing) was resistant to anti-IMV neutralization (Figure 2b). Over 95% of the viral preparation was resistant to similar treatment with an anti-EEV–neutralizing antibody, and no virus was recovered after treatment with both antibodies, implying that all virus in the prep was sensitive to neutralization with one or other of the antibodies.
Further studies looking at a viral preparation from a strain containing an EEV-enhancing mutation (WI.TK-) demonstrated that 12% of this prep was similarly resistant to anti-IMV–neutralizing antibody (Figure 2a). This indicated that in fact a significant amount of the enveloped virus could be retained after the downstream purification process.
We further looked to determine the relative importance of the EEV retained in the viral inoculum on the systemic delivery of oncolytic vaccinia to tumor targets. Once again the WR.TK- viral preparation was pretreated with (i) no antibody; (ii) anti-IMV; or (iii) anti-EEV–neutralizing antibody before intravenous delivery to tumor-bearing mice. Although antibody-neutralized virus will be delivered along with the remaining IMV or EEV in these groups, the human origin of the antibodies will minimize their interaction with the mouse immune system.
The relative bioluminescence (viral gene expression) from within the tumor was determined 16 hours later by bioluminescence imaging (BLI) (Figure 2c). Interestingly, the EEV within the inoculum appeared to contribute approximately one-third (36%) of the virus delivered to the tumor despite representing only 3% of the original inoculum. The small percentage of EEV within the initial preparation therefore appears to represent a disproportionate amount of the virus delivered to the tumor. This highlights a need to quantify the level of EEV present in clinical lots and indicates that any processes that might further stabilize or increase the level of EEV within the initial inoculum would benefit the therapy, especially as the clinical use of these viruses becomes more focused on achieving systemic activity.
EEV production in different cell lines
An alternative strategy that would lead to high systemic levels of EEV in vivo would be to pre-infect a susceptible cell line with an EEV-enhanced oncolytic strain and incorporate this as in situ EEV factory. This could be achieved subsequent to direct intratumoral injection of naked virus, however, we hypothesized that a susceptible pre-infected cell might act as an in situ EEV factory if delivered to a tumor-bearing host under situations where direct intratumoral injection may not be feasible. We therefore looked to compare the production of EEV after infection of different cells or cell lines with either WR.TK- or WI.TK- viruses. Initially a panel of murine cells was infected at a multiplicity of infection of 1.0 and supernatant taken 16 hours after infection (EEV is released directly into the supernatant at times early after infection). The supernatant was then treated with anti-IMV–neutralizing antibody to remove any non-enveloped viral particles that may have been released due to early lysis of infected cells or from the initial inoculum. The remaining (EEV only) virus was titered by plaque assay. It was seen (Figure 3a) that, whereas the WR.TK- strain produced very little EEV virus from any cell line (the WR strain is known to produce limited EEV), the WI.TK- strain produced significant quantities of EEV from many of the cell lines tested. These included tumor cell lines (4T1 and LLC) and therapeutic immune cell types (mouse CIK and activated NK cells). The mouse embryo fibroblasts replicated the viral strains poorly and little EEV was produced from these cells. This is expected as the TK-deleted oncolytic strains are attenuated in most normal cells, with limited cellular thymidine kinase produced to compensate for the loss of viral TK.28 In a further experiment, the 4T1 tumor cells were irradiated before infection in order to examine the possibility of using irradiated tumor cells as a potentially more clinically relevant therapeutic approach, however, it was seen that the irradiated cells were also very poor at replicating the virus (Figure 3a).
Figure 3.

In vivo production of EEV from cell-based factories enhances antitumor effects. (a) EEV production in different cell lines. WR and WI strains carrying a deletion in the viral thymidine kinase (TK) gene were each used to infect different cell lines at an MOI of 1.0. Supernatant was taken at 16 hours post-infection and treated with IMV-neutralizing antibody (NR417) before viral titering by plaque assay. Cell lines included mouse embryo fibroblasts (MEF), the murine tumor cell lines 4T1 and LLC, and the immune cell therapies, CIK and A-NK cells. In addition, the 4T1 cells were irradiated before infection (4T1-Irr). (b) Use of tumor cells as in situ EEV factories; 4T1 tumor cells were pre-infected with WR.TK-Luc+ or WI.TK-LUC+ at a MOI of 5.0 for 6 hours before 1 × 107 cells were injected into the peritoneal cavity of subcutaneous JC tumor-bearing BALB/c mice. The level of viral gene expression from the JC tumor was quantified at times after treatment by BLI. WI.TK- displays significantly enhanced gene expression from within the tumor at 8 and 24 hours post-treatment (P < 0.001). (c) Subsequent JC tumor growth was also followed by caliper measurement with mice killed (considered as the death endpoint) when tumors reached 1,400 mm3; 4T1+WI.TK- displayed a significantly enhanced survival benefit than all other treatments (P = 0.0166 relative to 4T1+WR.TK-). BLI, bioluminescence imaging; EEV, extracellular enveloped viral; IMV, intracellular mature virus; MOI, multiplicity of infection; PBS, phosphate-buffered saline; PFU, plaque-forming unit; WR, Western Reserve.
In situ production of EEV
Despite the presence of some EEV in the viral preparations used, the vast majority of the virus is still of the IMV form. In order to release a greater amount of EEV directly into the blood stream, we examined the use of a pre-infected cell as an in situ EEV factory in mouse models. We initially tested infected tumor cells in vivo, as these both produced the greatest levels of EEV (Figure 3a) and as they do not themselves traffic to the tumor or act as therapeutic agents, meaning that we could determine the antitumor effects of EEV production separately from any other therapeutic effects. We therefore pre-infected 4T1 tumor cells with different viruses (WR.TK- or WI.TK-, both expressing luciferase, multiplicity of infection of 5.0) and injected these cells into the peritoneal cavity of mice bearing subcutaneous JC tumors. We then examined the level of virus within the tumor (through BLI of luciferase transgene expression), and the survival of the treated animals over time (defined as the time until tumors reached a volume of 1,400 mm3) (Figure 3b,c). It was seen that when the 4T1 cells were pre-infected with WI.TK- this resulted in a more rapid and significantly higher level of viral delivery to the tumor over the crucial first 24 hours after treatment. Although the level of viral gene expression with both treatments was almost equivalent by 48 hours after treatment (Figure 3b), it should be noted that at this point much of the tumor had been destroyed with 4T1+WI.TK- treatment, and that the immune response began clearing virus from the tumor by 96 hours after initial treatment. The 4T1+WR.TK- treatment was therefore unable to deliver sufficient virus to the tumor to complete tumor destruction before immune-mediated clearance of the viral therapy. As a result, pre-infecting 4T1 cells with WI.TK- also resulted in dramatically improved survival of the animals (relative to 4T1 cells pre-infected with WR.TK-), with 9 of 10 mice treated displaying a complete response (Figure 3c).
Combination of EEV-enhancing mutation and CIK-based delivery
The one mouse that did not demonstrate a complete response after treatment with 4T1 cells pre-infected with WI.TK- virus (Figure 3c) was in fact killed due to peritoneal tumor burden, presumably due to some of the 4T1 cells used as a part of the treatment developing secondary tumors in this region. Although the 4T1 cells had been employed as a model system to differentiate between EEV production from a cell factory and tumor homing of a carrier cell, this emphasizes the fact that tumor cells used in this way are unlikely to become a viable treatment option. Alternatively, irradiated tumor cells and the normal cells tested (including mouse embryo fibroblasts) did not reliably replicate the viral vectors, meaning they also would not make suitable EEV factories. However, some cell types did produce EEV, even when infected with oncolytic vectors, including CIK cells and activated NK cells.
We have previously demonstrated that CIK cells can act as a useful carrier vehicle to deliver vaccinia to tumors,17 including for the treatment of minimal residual disease, and in the face of antiviral immunity.8 However, it was also noted that EEV production from CIK cells begun within 12 hours of infection, whereas initial detection of CIK cells in the tumor did not occur until after 24 hours. As a result EEV would be released into the blood stream before CIK cell infiltration into the tumor, and so it was hypothesized that this EEV may infect the tumor at early time points. We therefore initially looked to determine whether viral infection of the tumor would influence CIK cell homing (Figure 4a). It was seen that when CIK cells were delivered into mice bearing bilateral subcutaneous tumors, with one tumor treated with PBS, and that on the opposite flank treated with direct intratumoral injection of oncolytic vaccinia, the CIK cells preferentially homed to the tumor bearing a vaccinia infection. Of note, this experiment incorporated CIK cells labeled with luciferase obtained from a transgenic ubiquitous luciferase-expressing C57/BL6 mouse to permit noninvasive imaging of tumor trafficking by BLI. However, this required switching to a C57/BL6 mouse model, and so the mouse colorectal MC38 tumor model was incorporated in this and further studies (with the HCT 116 human colorectal cell line used for comparisons in a human cell line). It was also necessary to use these cell lines, as they are more resistant to viral therapy, in order to demonstrate differences in the effects of the additional components being tested.
Figure 4.

Combining EEV-enhanced (WI) oncolytic vaccinia with CIK cell-based delivery. (a) CIK cells preferentially home to tumors pre-infected with oncolytic vaccinia virus. Mice (C57/BL6) were implanted with bilateral MC38 tumors, with one flank tumor treated with direct intratumoral injection of 1 × 106 PFU of oncolytic vaccinia strain vvDD (WR.TK-.VGF-), and the other flank tumor injected with PBS. CIK cells expressing luciferase (obtained from transgenic luciferase-expressing C57/BL6 donor strain) were delivered intravenously and the level of CIK cells in each flank tumor was determined by BLI at times after treatment (P = 0.0016 at 120 hours after treatment). (b) CIK cells as EEV factories. Mice (C57/BL6 bearing MC38 tumors) were treated intravenously with 1 ×10 7 CIK cells pre-infected (MOI 1.0) with WR.TK-.Luc+ or WI.TK-.Luc+, viral gene expression (BLI) was measured in the tumor after 16 hours (*P < 0.001). (c) EEV seeding of the tumor enhances CIK delivery. Mice as before (MC38 tumor-bearing C57/BL6) were treated with 1 × 107 CIK cells expressing luciferase (expanded from C57/BL6-Luc+ transgenic mouse) pre-infected (MOI 1.0) with WR.TK-.GFP+ or WI.TK-.GFP+. This time CIK cell tumor homing was determined by BLI at 96 hours after treatment. WI.TK-/CIK treatment leads to significantly greater CIK delivery than CIK alone (P = 0.018) or WR.TK-/CIK treatment (P = 0.047). (d) Antitumor effects of EEV-enhanced vaccinia pre-infected into CIK cells. C57/BL6 mice implanted with subcutaneous MC38 tumors were treated intravenously with 1 × 107 CIK cells pre-infected with vaccinia (MOI 1.0), strains WR.TK-.Luc+ or WI.TK-.Luc+. Tumor growth was followed by caliper measurement and tumor volume of 1,400 mm3 was used as an endpoint for death. WI.TK-/CIK performed significantly better than WR.TK-/CIK (P = 0.049). BLI, bioluminescence imaging; CIK, cytokine-induced killer; EEV, extracellular enveloped viral; GFP, green fluorescent protein; MOI, multiplicity of infection; PBS, phosphate-buffered saline; PFU, plaque-forming unit.
Although we have previously shown that CIK cells pre-infected with WR.TK- retain the capability to traffic to the tumor,17 it was possible that the use of WI.TK- strains may result in reduced trafficking capability. Therefore, in the next step we examined the effects of pre-infecting CIK cells with the EEV-enhanced oncolytic vaccinia strain WI.TK-. In this way, we hypothesized that the CIK cells may act as both carrier vehicle and in situ EEV factory. In initial studies, viral strains expressing luciferase were used to determine viral gene expression and replication profiles (Figure 4b). It was found that there was a significant increase in the levels of virus delivered to the tumor (viral luciferase transgene expression) when CIK cells were pre-infected with the WI.TK-.Luc+ (relative to pre-infection with WR.TK-), with the most profound differences being at very early time points (<24 hours after intravenous delivery of pre-infected CIK cells). At this time after infection, we expect that the CIK cells will have begun releasing significant quantities of EEV into circulation when infected with WI.TK- (Figure 3a), but will not have begun to infiltrate the tumor in significant numbers. It therefore appears that the EEV produced from the infected CIK cells is indeed “seeding” the tumor with virus at time points before CIK homing. Because viral infection within the tumor was also shown to enhance CIK trafficking (Figure 4b), we looked to see if the EEV released early from pre-infected CIK cells and “seeding” the tumor could also act to enhance subsequent trafficking of the CIK cells (Figure 4c). This was indeed confirmed to be the case, with the level of CIK cells within the tumor at 72 hours after treatment found to be significantly greater when the CIK cells were pre-infected with WI.TK- (relative to either WR.TK- pre-infected CIK cells or CIK cells alone). It therefore appears that the use of CIK cells pre-infected with EEV-expressing virus created an additional advantage, with the EEV released from the CIK cells at early time points seeding and adapting the tumor microenvironment, making it a better target for CIK cell homing, and so subsequently attracting more of the therapeutic to the tumor itself.
Finally, we looked to determine whether this phenomenon of crosstalk between the viral and immune cell components of the therapy also led to a therapeutic advantage (Figure 4d). It was found that in a syngeneic MC38 tumor model the use of CIK cells pre-infected with WI.TK- resulted in significantly greater antitumor effect than CIK cells pre-infected with WR.TK-, with CIK cells pre-infected with WI.TK- resulting in over 50% complete responses in this model.
Use of viral chemokine expression to further enhance CIK cell trafficking
Because early viral infection in the tumor leads to modification of the tumor microenvironment and enhanced CIK cell trafficking, we looked to determine whether this effect could be further enhanced through the expression of transgenes from the virus. In particular, we have previously reported an oncolytic vaccinia vector expressing the chemokine CCL5, and have demonstrated that the expression of this chemokine can enhance attraction of cellular immunotherapies, including CIK cells, to an infected target tumor.10 We therefore looked to see whether expression of CCL5 from an oncolytic vaccinia in conjunction with incorporation of EEV-enhancing mutations (WI.TK-.CCL5+), and pre-infected into CIK cells could further enhance the therapy.
This was first tested in immunodeficient mice bearing HCT 116 tumors to separate any direct effects of the CCL5 expression on the host immune system from CCL5 effects on CIK cell attraction to the tumor (Figure 5). It was seen that the addition of CCL5 transgene expression into this system significantly enhanced antitumor effects in immunodeficient mice (Figure 5c). This was despite the fact that as expected, there was no further increase in the early delivery of virus to the tumor beyond that already seen with the use of EEV-enhanced strains (Figure 5a). However, although there was no increase in viral gene expression at early time points there was a significant further increase in the number of CIK cells trafficking to the tumor (Figure 5c). Therefore the addition of CCL5 transgene expression could further enhance trafficking of CIK cells, leading to further enhanced therapeutic benefit.
Figure 5.

CCL5 expression from EEV-enhanced vaccinia (WI) delivered in pre-infected CIK cells further enhances antitumor effects. Immunodeficient (CB17 SCID) mice bearing HCT 116 tumors (50–100 mm3 subcutaneous) were treated intravenously with 1 × 107 CIK cells (labeled with Cy5.5 NHS ester) pre-infected (MOI of 1.0 for 12 hours) with either WI.TK-Luc+ or WI.TK-CCL5+Luc+ (n = 10 per group). (a) After 16 hours, the level of viral luciferase gene expression from the tumor was determined (BLI), CCL5 expression did not enhance early viral delivery to the tumor (P = 0.97). (b) At 72 hours, the level of CIK cells in the tumor was determined by fluorescence imaging (FMT2500; Perkin Elmer) (P = 0.0025). (c) Tumor growth was also followed over time (WI-CCL5/CIK treatment was significantly better than all other treatments (P < 0.05) at days 28, 31, and 35). BLI, bioluminescence imaging; CIK, cytokine-induced killer; EEV, extracellular enveloped viral; MOI, multiplicity of infection; PBS, phosphate-buffered saline; SCID, severe combined immunodeficiency.
Furthermore, it was previously seen that CCL5 expression from oncolytic vaccinia virus could provide additional therapeutic benefits to the virus when used alone (in immunocompetent mice).10 We therefore examined the combination of CCL5 expression from an EEV-enhanced virus in the context of CIK delivery in two separate syngeneic tumor models in fully fully immune-competent mouse backgrounds (Figure 6a,b). In both cases, the expression of CCL5 was capable of significantly enhancing the therapeutic benefit of this approach. However, it was also observed that additional immune-activating effects were achieved with the expression of the CCL5 transgene (Figure 6c), as the level of interferon-γ (IFN-γ)–producing T cells as detected in an ex vivo ELISPOT assay was significantly increased at both day 3 (Figure 6c), and day 7 (data not shown). This was also demonstrated in our previous publication using vaccinia expressing CCL5 as a single agent.10
Figure 6.

CCL5 expression demonstrates therapeutic and immunotherapeutic advantages in immune-competent mice. (a) Mice (4T1-bearing BALB/c) were treated with 1 × 107 CIK cells pre-infected with the same viruses as before (n = 8 per group) and antitumor effects determined by caliper measurement over time (WI-CCL5/CIK performed significantly better than all other treatments (*P < 0.05) at all times after day 15). (b) In a repeat experiment, MC38 tumor-bearing C57/BL6 mice were treated as before (n = 10) and tumor growth followed. (c) In a satellite experiment, mice were killed 3 days after treatment (n = 4/group), and the level of T-cell response targeting tumor antigens was determined by IFN-γ ELISPOT. The CCL5 expression leads to a significant increase in the level of early immune activation (P = 0.024). CIK, cytokine-induced killer; IFN, interferon; PBS, phosphate-buffered saline; SFC, spot-forming cells.
Discussion
Recent clinical use of oncolytic vaccinia virus has highlighted the promise of this approach for the treatment of solid cancers,13,14 however, hurdles remain. In particular, although systemic delivery of oncolytic vaccinia to tumors can be reproducibly achieved in preclinical mouse models4 and has also been demonstrated in the clinic,14 it is evident that only a very small fraction of the original inoculum actually infects cells within the tumor with the majority being cleared by the host or forming non-productive infections in non-tumor tissue. Here, we demonstrate that complement-mediated removal, especially of the IMV form of the virus, is a critical factor in limiting the initial systemic delivery of the virus to tumors in naive mice.
It is apparent therefore that significant opportunity exists to enhance the therapeutic effects of these vectors through enhancing or targeting their systemic delivery. One possible approach would be to inhibit host complement. Indeed, vaccinia has developed several complement evasion strategies, including expression and secretion of a complement inhibiting factor (VCP),26 and production of the EEV form of the virus that incorporates host cell complement control proteins into its outer envelope.22 However, as VCP will not be expressed until after viral infection of the tumor occurs, and the standard approaches used to expand and purify vaccinia enrich the IMV form at the expense of EEV, these complement evasion strategies will probably not benefit initial delivery of oncolytic vectors to the tumor under normal conditions. We looked to examine the role of VCP in systemic complement evasion, and saw that pre-infection of mice with even low doses of wild-type virus allowed greatly increased subsequent systemic delivery of an oncolytic strain with concomitant enhanced therapeutic benefit. Although this demonstrates the therapeutic advantage of improving initial delivery to the tumor, the involvement of multiple other factors besides VCP may mediate this initial-enhanced delivery, and so further examination is needed to define the utility and safety of pre-treating with VCP. Indeed when a commonly used complement inhibitor (CVF) was used to reproduce this effect, only a small increase in initial delivery was noted, that did not lead to significant therapeutic advantage.
The other natural poxviral strategy for complement evasion, production of EEV, also appeared to have potential to enhance initial delivery of an oncolytic virus. It was demonstrated that even though only a very small percentage (3%) of an initial inoculum was EEV, this accounted for over one-third (36%) of the virus that was initially delivered to the tumor. This disproportionate importance of the EEV within the viral preparation needs to be carefully considered as the use of oncolytic vaccinia moves towards more routine systemic delivery in the clinic and raises the possibility that approaches to purify and stabilize the level of EEV during the manufacture of clinical viruses could lead to significantly improved viral delivery.
We have also previously shown that viral mutations that increase the relative amount of the EEV form of the virus that is produced in vivo can enhance the antitumor effects of oncolytic vectors by enhancing the spread of the virus within and between tumors.6 However, currently the most effective way of introducing high levels of the EEV form into a host is subsequent to infection of a susceptible cell type in situ. Alternatively, we have also demonstrated that the application of a pre-infected immune cell (CIK cell) as a therapeutic, and used as a cell carrier vehicle to improve viral delivery, also enhances subsequent therapeutic activity. This raises the possibility that pre-infected cells used as a therapy might also act as in vivo EEV-producing factories, releasing high levels of EEV directly into the blood stream in the host. This was initially demonstrated as a proof-of-concept using pre-infected tumor cells. However, despite profound antitumor effects, this was not considered a viable therapeutic approach as some mice apparently developed tumors from uninfected tumor cells implanted as a part of the therapy, while neither irradiated tumor cells or normal fibroblasts produced sufficient EEV to make them viable alternatives.
Alternatively, it was observed that at least some immune cells, including those previously used as tumor-targeting immune cell therapies (such as CIK cells or activated NK cells) could produce significant levels of EEV in vitro (when EEV-enhancing mutations were incorporated into the oncolytic vectors). We therefore looked to test CIK cells pre-infected with EEV-enhanced oncolytic viral strains as a therapeutic. Unlike the use of pre-infected tumor cells, these cells are themselves capable of tumor homing and independent therapeutic activity, which complicates their study, but was expected to further enhance the overall antitumor potential of this approach.
Interestingly, it was noted that EEV was released from the CIK cells at times before their expected infiltration into the tumor following systemic delivery. EEV was produced within 12 hours after infection, while the CIK cells typically take up to 48 hours to arrive in the tumor at significant levels. Seeing as the EEV was better able to traffic and infect the tumor systemically than other forms of the virus, and seeing as we also observed that CIK cells were more efficient at targeting tumors that had been pre-infected with oncolytic vaccinia, it seemed plausible that the combination of EEV-enhanced vaccinia and CIK cells would lead to (i) early release of EEV virus that might seed the tumor before CIK arrival; and (ii) increased subsequent tumor trafficking of the CIK cells towards the virus-infected tumor. Both of these observations were apparently confirmed in several mouse tumor models, along with the significantly enhanced antitumor effects of CIK cell delivery of EEV-enhanced oncolytic vaccinia virus.
This led to the further hypothesis that if the viral infection within the tumor was capable of modifying the tumor microenvironment so as to enhance CIK homing to the tumor, it might be possible to use selected viral transgene expression to further optimize CIK homing. To explore this possibility, we used the chemokine CCL5 as we had previously shown that its receptor is expressed on CIK cells, and that vaccinia expressing CCL5 could be used to enhance targeting of CIK cells to infected tumors.10 It was therefore decided to incorporate CCL5-expressing and EEV-enhanced oncolytic vaccinia delivered directly within pre-infected CIK cells. In this way, a single therapy might be used, so somewhat simplifying clinical translation of a complex therapy. The resultant combination of EEV-enhanced and CCL5-expressing oncolytic vaccinia delivered within pre-infected CIK cells was indeed shown to significantly enhance the therapeutic potential of this approach, even in immune-deficient mice, where the CCL5 expression would not be expected to have any effect within the host beyond increased attraction of CIK cells. When immune-competent mice were examined, the CCL5 expression demonstrated additional therapeutic benefits through production of increased levels of tumor-targeting and IFNγ-producing T cells within the host.
As a result of this combination approach, a single therapeutic can be applied that leads to in situ production of different biological agents that subsequently synergize in their ability to crosstalk, leading to mutual benefits in their delivery to the tumor. This leads to subsequent enhanced therapeutic benefit and improved interaction with the host immune response. As a result, up to 8 (of 10) mice demonstrated complete responses after a single systemic treatment. These effects might be further improved in human tumor models or cancer patients, as vaccinia only replicates weakly in mouse tumor cells. However, because the actions and interactions of this therapeutic are complex it is difficult to clearly define the relative contribution of the individual components. Further, because the therapy essentially involves application of a single initial agent, and because of the encouraging antitumor effects, this approach is felt to be both feasible and promising for clinical translation.
Materials and Methods
Cell lines, viruses, and neutralizing antibody. A variety of human and mouse tumor cell lines, including MCF-7, HCT 116, 4T1, JC, LLC, and MC38 were used in this study, all were obtained from ATCC (Manassas, VA) (expect JC, obtained from Cancer Research UK culture collection, London, UK) and were grown in Dulbecco's modified Eagle's medium with 10% fetal bovine serum. In some experiments MCF-7 were grown on nontreated tissue culture plates, to prevent cell attachment, and so induce formation of spheroids. In addition, several primary normal cell lines were used, including mouse embryo fibroblasts, CIK (human and mouse), and Activated NK cells were incorporated. These cells were expanded and used as previously described.29,30
Viral strains included the wild-type WR strain obtained from ATCC and the WR strain with the IHD-J A34R gene (WI) leading to increased EEV production, that was a gift from Prof Bernie Moss (National Institutes of Health).24 Both WR and WI were recombined with the pSC65 plasmid (obtained from Prof Bernie Moss) cloned to express either luciferase or GFP. The pSC65 recombination results in viral thymidine kinase gene insertion mutation. The WI strain was also cloned to express the CCL5 (RANTES) gene from within the TK locus along with the luciferase. In addition, strain vvDD (WR with deletions in viral TK and the viral growth factor genes) was used as a second model of oncolytic vaccinia.
Viral manufacture and purification for in vivo use involved growth of virus in HeLa cells, with cells collected and lysed by repeat freeze-thaw. Cellular debris was removed by slow (300g) centrifugation before virus was concentrated and purified with two ultracentifuge spins through sucrose cushions. Between these spins virus was treated with benzonase (SIGMA, St Louis, MO) to remove cellular DNA.
The anti-L1R IMV-neutralizing antibody (NR417) and the anti-B5R EEV-neutralizing antibody (NR551) were obtained form BEI Resources (Manassas, VA). These were used at a concentration of 1 µg/ml for 5 minutes to remove the different forms of the virus.
Complement studies. Human serum was obtained de-identified from the Pittsburgh Central Blood Bank. The level of neutralizing antibody present in the serum was determined using our standard assays.9 CVF was obtained from Quidel Corporation (San Diego, CA).
Concentrations of C3 in plasma samples collected from mice after different treatments were measured by enzyme-linked immunosorbent assays (Alpha Diagnostic International, San Antonio, TX).
Animal models. C57/BL6 and BALB/c mice (female 6–8 weeks old) were purchased from The Jackson Laboratory (Bar Harbor, ME). Tumor cell lines were implanted subcutaneously with 1 × 106 cells injected per mouse. Experiments began when tumors reached 50–100 mm3. Treatment doses and timings were as indicated, CIK cells were pre-infected with virus as indicated through simple mixing for 12 hours before infusion. Tumor growth was monitored by caliper measurement unless otherwise indicated. In some experiments BLI was used to image the animals. This was achieved using an IVIS 200 (Xenogen, part of PerkinElmer, Waltham, MA) after intraperitoneal injection of luciferin substrate with bioluminescence signal quantified using the Living Image software (Xenogen, part of PerkinElmer). In other experiments, CIK cells labeled with Cy5.5 NHS Ester (GE Healthcare, Waukesha, WI) were imaged in vivo using the FMT2500 fluorescence whole animal imaging system (VisEn, now part of PerkinElmer, Waltham, MA).
In survival assays, animals were considered to have reached a terminal endpoint and killed when tumors reached 1,400 mm3.
All experiments were performed according to Institutional Animal Care and Use Committee approved protocols.
ELISPOT assay. IFN-γ–producing splenocytes were quantified by ELISPOT assay. Splenocytes were prepared from mice bearing 4T1 tumors that were treated with different therapies 3 days earlier. Splenocytes and irradiated 4T1 cells were mixed (5:1) and seeded on plates, in triplicate, coated overnight with mIFN-γ antibody (100 µl of 15 µg/ml). They were incubated for 48 hours. The plate was then washed five times with PBS/0.05% Tween 20 and incubated with a biotinylated secondary antibody for 2 hours at room temperature. The plate was then washed a further five times, incubated for 1 hour with avidin-peroxidase complex (Vectastatin kit; Vector laboratories, Burlingame, CA), washed three times with wash buffer and two times with PBS. The plate was then developed by the addition of AEC (3-amino-9-ethylcarbazole) substrate. The spots were counted on a CTL-Immunospot analyzer (Cellular Technologies, Shaker Heights, OH). Spots from unstimulated splenocytes from each group was used to subtract the background
Statistical analysis. In most experiments, simple Student's t-test (non-weighted) were used, with significance considered to be P < 0.05. In animal “survival” studies Wilcoxon tests were used to determine P values.
Acknowledgments
This work was supported directly by National Institutes of Health grants, P01 CA132714 and R01 CA140215. In addition, core facilities used in this work were supported by P30 CA047904-21S3. The authors thank Julie Urban for assistance in growth of some cell lines and some immune assays and Rachel Sikorski for assistance in some imaging studies. The authors declared no conflict of interest.
References
- Hanahan D., and, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Kim JH, Oh JY, Park BH, Lee DE, Kim JS, Park HE.et al. (2006Systemic armed oncolytic and immunologic therapy for cancer with JX-594, a targeted poxvirus expressing GM-CSF Mol Ther 14361–370. [DOI] [PubMed] [Google Scholar]
- Kirn DH, Wang Y, Le Boeuf F, Bell J., and, Thorne SH. Targeting of interferon-beta to produce a specific, multi-mechanistic oncolytic vaccinia virus. PLoS Med. 2007;4:e353. doi: 10.1371/journal.pmed.0040353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorne SH, Hwang TH, O'Gorman WE, Bartlett DL, Sei S, Kanji F.et al. (2007Rational strain selection and engineering creates a broad-spectrum, systemically effective oncolytic poxvirus, JX-963 J Clin Invest 1173350–3358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo ZS, Thorne SH., and, Bartlett DL. Oncolytic virotherapy: molecular targets in tumor-selective replication and carrier cell-mediated delivery of oncolytic viruses. Biochim Biophys Acta. 2008;1785:217–231. doi: 10.1016/j.bbcan.2008.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirn DH, Wang Y, Liang W, Contag CH., and, Thorne SH. Enhancing poxvirus oncolytic effects through increased spread and immune evasion. Cancer Res. 2008;68:2071–2075. doi: 10.1158/0008-5472.CAN-07-6515. [DOI] [PubMed] [Google Scholar]
- Kirn DH., and, Thorne SH. Targeted and armed oncolytic poxviruses: a novel multi-mechanistic therapeutic class for cancer. Nat Rev Cancer. 2009;9:64–71. doi: 10.1038/nrc2545. [DOI] [PubMed] [Google Scholar]
- Contag CH, Sikorski R, Negrin RS, Schmidt T, Fan AC, Bachireddy P.et al. (2010Definition of an enhanced immune cell therapy in mice that can target stem-like lymphoma cells Cancer Res 709837–9845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorne SH, Liang W, Sampath P, Schmidt T, Sikorski R, Beilhack A.et al. (2010Targeting localized immune suppression within the tumor through repeat cycles of immune cell-oncolytic virus combination therapy Mol Ther 181698–1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, O'Malley M, Urban J, Sampath P, Guo ZS, Kalinski P.et al. (2011Chemokine expression from oncolytic vaccinia virus enhances vaccine therapies of cancer Mol Ther 19650–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang LC, Lynn RC, Cheng G, Alexander E, Kapoor V, Moon EK.et al. (2011Treating tumors with a vaccinia virus expressing IFNbeta illustrates the complex relationships between oncolytic ability and immunogenicity Mol Ther 20736–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu TC, Hwang T, Park BH, Bell J., and, Kirn DH. The targeted oncolytic poxvirus JX-594 demonstrates antitumoral, antivascular, and anti-HBV activities in patients with hepatocellular carcinoma. Mol Ther. 2008;16:1637–1642. doi: 10.1038/mt.2008.143. [DOI] [PubMed] [Google Scholar]
- Park BH, Hwang T, Liu TC, Sze DY, Kim JS, Kwon HC.et al. (2008Use of a targeted oncolytic poxvirus, JX-594, in patients with refractory primary or metastatic liver cancer: a phase I trial Lancet Oncol 9533–542. [DOI] [PubMed] [Google Scholar]
- Breitbach CJ, Burke J, Jonker D, Stephenson J, Haas AR, Chow LQ.et al. (2011Intravenous delivery of a multi-mechanistic cancer-targeted oncolytic poxvirus in humans Nature 47799–102. [DOI] [PubMed] [Google Scholar]
- Breitbach CJ, Thorne SH, Bell JC., and, Kirn DH. Targeted and armed oncolytic poxviruses for cancer: the lead example of JX-594. Curr Pharm Biotechnol. 2012;13:1768–1772. doi: 10.2174/138920112800958922. [DOI] [PubMed] [Google Scholar]
- Hwang TH, Moon A, Burke J, Ribas A, Stephenson J, Breitbach CJ.et al. (2011A mechanistic proof-of-concept clinical trial with JX-594, a targeted multi-mechanistic oncolytic poxvirus, in patients with metastatic melanoma Mol Ther 191913–1922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorne SH, Negrin RS., and, Contag CH. Synergistic antitumor effects of immune cell-viral biotherapy. Science. 2006;311:1780–1784. doi: 10.1126/science.1121411. [DOI] [PubMed] [Google Scholar]
- Thorne SH. Strategies to achieve systemic delivery of therapeutic cells and microbes to tumors. Expert Opin Biol Ther. 2007;7:41–51. doi: 10.1517/14712598.7.1.41. [DOI] [PubMed] [Google Scholar]
- Thorne SH., and, Contag CH. Combining immune cell and viral therapy for the treatment of cancer. Cell Mol Life Sci. 2007;64:1449–1451. doi: 10.1007/s00018-007-6550-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorne SH., and, Contag CH. Integrating the biological characteristics of oncolytic viruses and immune cells can optimize therapeutic benefits of cell-based delivery. Gene Ther. 2008;15:753–758. doi: 10.1038/gt.2008.42. [DOI] [PubMed] [Google Scholar]
- Vanderplasschen A, Hollinshead M., and, Smith GL. Antibodies against vaccinia virus do not neutralize extracellular enveloped virus but prevent virus release from infected cells and comet formation. J Gen Virol. 1997;78 (Pt 8):2041–2048. doi: 10.1099/0022-1317-78-8-2041. [DOI] [PubMed] [Google Scholar]
- Vanderplasschen A, Mathew E, Hollinshead M, Sim RB., and, Smith GL. Extracellular enveloped vaccinia virus is resistant to complement because of incorporation of host complement control proteins into its envelope. Proc Natl Acad Sci USA. 1998;95:7544–7549. doi: 10.1073/pnas.95.13.7544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law M, Hollinshead R., and, Smith GL. Antibody-sensitive and antibody-resistant cell-to-cell spread by vaccinia virus: role of the A33R protein in antibody-resistant spread. J Gen Virol. 2002;83 Pt 1:209–222. doi: 10.1099/0022-1317-83-1-209. [DOI] [PubMed] [Google Scholar]
- Blasco R, Sisler JR., and, Moss B. Dissociation of progeny vaccinia virus from the cell membrane is regulated by a viral envelope glycoprotein: effect of a point mutation in the lectin homology domain of the A34R gene. J Virol. 1993;67:3319–3325. doi: 10.1128/jvi.67.6.3319-3325.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puhlmann M, Brown CK, Gnant M, Huang J, Libutti SK, Alexander HR.et al. (2000Vaccinia as a vector for tumor-directed gene therapy: biodistribution of a thymidine kinase-deleted mutant Cancer Gene Ther 766–73. [DOI] [PubMed] [Google Scholar]
- Isaacs SN, Kotwal GJ., and, Moss B. Vaccinia virus complement-control protein prevents antibody-dependent complement-enhanced neutralization of infectivity and contributes to virulence. Proc Natl Acad Sci USA. 1992;89:628–632. doi: 10.1073/pnas.89.2.628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jha P., and, Kotwal GJ. Vaccinia complement control protein: multi-functional protein and a potential wonder drug. J Biosci. 2003;28:265–271. doi: 10.1007/BF02970146. [DOI] [PubMed] [Google Scholar]
- Hengstschläger M, Knöfler M, Müllner EW, Ogris E, Wintersberger E., and, Wawra E. Different regulation of thymidine kinase during the cell cycle of normal versus DNA tumor virus-transformed cells. J Biol Chem. 1994;269:13836–13842. [PubMed] [Google Scholar]
- Lu PH., and, Negrin RS. A novel population of expanded human CD3+CD56+ cells derived from T cells with potent in vivo antitumor activity in mice with severe combined immunodeficiency. J Immunol. 1994;153:1687–1696. [PubMed] [Google Scholar]
- Baker J, Verneris MR, Ito M, Shizuru JA., and, Negrin RS. Expansion of cytolytic CD8(+) natural killer T cells with limited capacity for graft-versus-host disease induction due to interferon gamma production. Blood. 2001;97:2923–2931. doi: 10.1182/blood.v97.10.2923. [DOI] [PubMed] [Google Scholar]