

Abstract
Despite treatments combining surgery, radiation-, and chemotherapy, patients affected by glioblastoma (GBM) have a limited prognosis. Addition of temozolomide (TMZ) to radiation therapy is the standard therapy in clinical application, but effectiveness of TMZ is limited by the tumor's overexpression of the DNA repair protein O6-methylguanine-DNA methyltransferase (MGMT). The goal of this study was to use the highly specific and efficient RNA interference (RNAi) pathway to modulate MGMT expression to increase TMZ efficiency in chemotherapy resistant GBM. Using lentiviral-based anti-MGMT small hairpin RNA (shRNA) technology we observed a specific inhibition of the MGMT expression in GBM cell lines as well as in subcutaneous tumors. Tumor growth inhibition was observed following TMZ treatment of xenografts with low MGMT expression in contrast to xenografts with high MGMT expression. Bioluminescence imaging (BLI) measurements indicated that luciferase and shRNA-expressing lentiviruses were able to efficiently transduce the GBM xenografts in vivo. Treatment combining injection of a lentivirus expressing an anti-MGMT shRNA and TMZ induced a reduction of the size of the tumors, in contrast with treatment combining the lentivirus expressing the control shRNA and TMZ. Our data suggest that anti-MGMT shRNA therapy could be used in combination with TMZ chemotherapy in order to improve the treatment of resistant GBM.
Introduction
The World Health Organization (WHO) classifies the primary brain tumors in four categories.1 WHO grade I and II are low-grade gliomas, whereas anaplastic astrocytomas, anaplastic oligoastrocytomas, and anaplastic oligodendrogliomas (WHO grade III), as well as glioblastomas (GBMs) (WHO grade IV), are collectively referred to as malignant gliomas. Primary malignant central nervous system (CNS) tumors represent ~2% of all cancers but account for a disproportionate rate of morbidity and mortality. They are the leading cause of death from solid tumors in children and the third leading cause of death from cancer in adolescents and adults aged 15–34 years.2 Among the malignant gliomas, GBMs are the most common and fatal neoplasms, representing ~50% of all gliomas and 24% of all primary intracranial tumors.3 Despite aggressive treatment strategies, including surgery, radiotherapy and chemotherapy, median survival of patients with GBM is still limited to 1–3 years. Therapeutic challenges for GBM include its invasive nature, the proximity of the tumor to vital brain structures often preventing total resection and efficient radiotherapy, and the high heterogeneity of GBM leading to resistance to conventional chemotherapy.4,5
Resection followed by combined radiotherapy and temozolomide (TMZ) chemotherapy is currently the standard of care for most patients suffering from GBM.6,7 TMZ belongs to the class of alkylating agents. The principal mechanism of cell killing by this class of therapeutical molecules is initiated by abnormal methylations of DNA bases, and in particular the formation of O6-alkylguanine in DNA.8 The DNA repair enzyme -methylguanine-DNA methyltransferase (MGMT) antagonizes the genotoxic effects of alkylating agents and MGMT gene silencing through promoter methylation is a favorable prognostic marker, predicting benefits from this sort of chemotherapy in GBM.6,9 Clinical studies in malignant glioma confirmed a strong correlation between MGMT promoter methylation and improved response to alkylating agent chemotherapy as well as improved survival of the patients.10,11 GBM expressing a high level of MGMT protein are resistant to TMZ chemotherapy and alternative treatments for patients affected by such GBM are limited. Small molecule inhibitors of MGMT exist, but their use in combination with TMZ is limited by toxicity, due to MGMT inhibition in peripheral organs.12
Inhibition of MGMT using a specific targeting could therefore be of interest in order to improve the treatment of resistant gliomas. RNA interference (RNAi) is one of the most powerful tools to specifically inhibit a gene at the post-transcriptional level. In vivo, however, this approach is difficult to achieve because of the poor pharmacological properties of this class of oligonucleotides.13 Replication-deficient lentiviral vectors have been derived from HIV14 and can be used to express small hairpin RNA (shRNA) in mammalian cells.15 These vectors have been validated in clinically relevant GBM animal models and are therefore attractive candidates for the treatment of brain cancer.16
The goal of our study was therefore to use the high specific and efficient RNAi pathway in order to modulate MGMT expression and increase TMZ efficiency in resistant GBM. We first characterized three human GBM cell lines regarding MGMT expression and resistance to TMZ. We then used lentiviral vectors containing anti-MGMT shRNA to produce the corresponding cell lines stably depleted in MGMT. Inhibition of MGMT led to an increase of TMZ sensitivity in culture as well as in vivo in nude mice xenografts. Finally, the anti-MGMT shRNA delivering lentiviral vector was able to induce a reduction of the tumor sizes in combination with TMZ treatment after direct injection of the virus into TMZ resistant xenografts in vivo.
Results
Construction of lentiviral-based anti-MGMT shRNA vectors
DNA sequences of two shRNAs targeting MGMT and of one control shRNA were introduced into the backbone of the pLKO.1 vector (Figure 1a). Before in vivo application, the sequence of the firefly luciferase reporter gene under the control of the cytomegalovirus (CMV) minimal promoter was introduced into the pLKO.1-shRNA backbones (Figure 1b).
Figure 1.

O6-Methylguanine-DNA methyltransferase (MGMT) inhibiting lentiviral vectors. The pLKO.1 vectors contain all necessary cis-elements for packaging, reverse transcription and integration, which are required to genetically modify infected cells. Elements required for the production of the capsid and envelope proteins are deleted making the produced viral particles replication-incompetent. (a) Two small hairpin RNA (shRNA) sequence targeting the MGMT mRNA and one control sequence were cloned into the pLKO.1 lentiviral backbone after the human U6 promoter allowing expression of shRNAs in target cells. (b) For intratumoral applications, the luciferase reporter gene under the control of a cytomegalovirus (CMV) promoter was introduced subsequently (pLKO.1-CMVLuc-shRNA). cPPT, central polypurine tract; hPGK, human phosphoglycerate kinase promoter; RSV, rous sarcoma virus promoter; RRE, rev responsive element; Psi, lentiviral packaging signal.
LV-shMGMT vectors significantly alter MGMT expression and function in cell culture
The MGMT protein expression of three different GBM cell lines was analyzed by western blot using actin as reference (Figure 2). Blots were quantified and normalized to the value of the LN18 MGMT protein level (Figure 2a). MGMT expression was detected in all three cell lines, with LN18 showing the highest protein expression. The MGMT protein content in T98 and VU28 cells was 24% and 71% less than in LN18 cells, respectively. The three human glioma cell lines were then examined for TMZ EC50 values in acute growth inhibition and clonogenic survival assays (Figure 2a). LN18 cells were found to be the most resistant cells to TMZ with EC50 values of 740 and 345 µmol/l TMZ in growth and clonogenic assays, respectively. T98 cells characterized by a lower MGMT expression compared to LN18 cells were less resistant to TMZ than the LN18 cells, with EC50 values of 500 and 217 µmol/l TMZ in growth and clonogenic assay, respectively. VU28 cells showed a high resistance toward TMZ with EC50 values comparable with those of LN18 (697 and 243 µmol/l TMZ in growth and clonogenic assay, respectively) despite a lower expression of MGMT (Figure 2a,b).
Figure 2.

O6-Methylguanine-DNA methyltransferase (MGMT) inhibition enhances the sensitivity of glioma cells toward temozolomide (TMZ). (a) Determination of the MGMT protein level in human LN18, T98, and VU28 glioma cells was performed by western blot using actin as reference. The western blots were quantified and normalized to the value of the LN18 MGMT protein level. (b) TMZ sensitivity was assessed using growth and clonogenic assay. For this, two small hairpin RNA (shRNA) sequence targeting the MGMT mRNA and one control sequence were cloned into the pLKO.1 lentiviral backbone. Lentiviruses were used to infect LN18, T98, and VU28 glioma cell lines. The determination of the MGMT protein level was performed by western blot using actin as reference. The western blots were quantified and normalized to the MGMT protein level in the corresponding wild-type cell. (c,d) Wild-type LN18, T98, and VU28 glioma cell lines, as well as in corresponding cells modified with the pLKO.1-shRNA vectors were examined for EC50 values in (c) acute growth inhibition and (d) clonogenic survival assays (mean ± SD, n = 3). EC50 values measured in the different modified cells were normalized to the EC50 values of the corresponding wild-type glioblastoma (GBM) line. All data are presented as mean + SD. Differences between the obtained values for LN18 and the values obtained for T98 and VU28 cells or differences between wild-type LN18, T98, and VU28 cells and the corresponding modified cells were tested for significance using t-tests (*P < 0.05, **P < 0.01).
The three cell lines were modified through infection with the pLKO.1-shControl, pLKO.1-shMGMT1, and pLKO.1-shMGMT2 lentiviral vectors. MGMT protein expression in cells depleted in MGMT through the expression of the shRNAs was measured by western blot (Figure 2b) and the effect of the MGMT inhibition on the cells TMZ resistance was examined in acute growth inhibition and clonogenic survival assays (Figure 2c,d). The shMGMT1 and shMGMT2 were able to induce a reduction of MGMT expression in the three cell lines, not observed with the shControl sequence. The strongest inhibition was observed in LN18 and T98 cells displaying a high-basal MGMT expression level. The shMGMT1 sequence induced a reduction of 71% and 65% of the protein level in LN18 and T98 cells, respectively. The shMGMT2 sequence was found to be even more efficient with an inhibition of 82% and 80%, respectively. The strong inhibition of the MGMT protein was associated with an increase of TMZ sensitivity in the LN18 and T98 cells (Figure 2c,d). LN18-shMGMT1 cells were characterized by TMZ EC50 values 60% and 58% lower than the values of the wild-type cells, as assessed using growth and clonogenic assays, respectively (P < 0.01). Reduction of TMZ resistance was found to be induced in the LN18-shMGMT2 more effectively than in the LN18-shMGMT1 cells with a reduction of 86% and 72% of the TMZ EC50 values in the growth and clonogenic assay, respectively (P < 0.01). The same trends were observed for the T98 cells, with TMZ EC50 values in growth and clonogenic assays 48% and 49% lower for the T98-shMGMT1 cells compared to the T98 wild-type cells (P < 0.05), and 55% and 85% lower for the T98-shMGMT2 cells (P < 0.05). In the VU28 cells, characterized by a basal level of MGMT much smaller than in the two other GBM lines, inhibition of the protein level by the shRNAs was limited (31% and 34% of protein expression in the wild-type cells for the shMGMT1 and shMGMT2 sequences, respectively, Figure 2a). The increase of TMZ sensitivity of the VU28-shMGMT1 and VU28-shMGMT2 compared to VU28 wild-type cells was only of 43% and 33%, respectively, as assessed by growth assay and of 49% and 34%, respectively, as assessed by clonogenic assay, which did not reach statistical significance (Figure 2c,d).
Altered MGMT expression in human LN18 glioma cells increases their sensitivity toward TMZ in vivo
In order to determine whether inhibition of MGMT would lead to increased TMZ sensitivity in vivo as observed in vitro, LN18 wild-type cells, as well as LN18 cells stably expressing the shMGMT1, shMGMT2, and shControl shRNA sequences were xenografted in nude mice (Figure 3). When tumors reached a size of ~200 mm3, mice were treated with TMZ diluted in dimethyl sulfoxide (DMSO) or with DMSO alone as control treatment, and tumor size evolution was followed by caliper measurement (Figure 3a) before MGMT expression was analyzed using immunohistochemistry (Figure 3b). Despite TMZ treatment, the size of the LN18 wild-type xenografts doubled between day 0 and day 12. During the same period, under control treatment, LN18 wild-type tumors increased by a factor of 3.3. Similarly, no significant differences were observed between the treated and nontreated LN18-shControl xenografts. In contrast, for LN18-shMGMT1 and LN18-shMGMT2 xenografts with a low expression of MGMT (Figure 3b) a significant difference (two-way ANOVA, P < 0.01) was observed between treated and nontreated tumors. Upon TMZ treatment the size of the LN18-shMGMT1 xenografts was stabilized after 7 days, and after 12 days tumor sizes were only 23% larger than at day 0 (n.s.). LN18-shMGMT2 xenografts were even more sensitive, and at day 12 the mean LN18-shMGMT2 tumor size was reduced to 46% of that at day 0 (P < 0.01). Immunohistochemistry confirmed that inhibition of the tumor growth was only observed in tumors depleted in MGMT (Figure 3b).
Figure 3.

O6-Methylguanine-DNA methyltransferase (MGMT) inhibition enhanced the sensitivity of human LN18 glioma xenografts toward temozolomide (TMZ) in vivo. (a) Nude mice were xenografted (two xenografts per animal) with LN18 wild-type cells (obtained: ntumor = 16 in nmice = 15), as well as LN18 cells stably expressing the shMGMT1 (obtained: ntumor = 13 in nmice = 7), shMGMT2 (obtained: ntumor = 12 in nmice = 8) and shControl RNA sequences (obtained: ntumor = 17 in nmice = 9). When tumors reached a size of ~200 mm3, mice were divided in two groups and treated with TMZ or dimethyl sulfoxide (DMSO). Tumor sizes were evaluated by caliper measurements and size variations upon treatment were plotted against the duration of treatment (a). At day 12, mice were sacrificed and the presence of MGMT in the xenografts was controlled by immunohistochemistry (b). All data are presented as mean + SD. Two-way ANOVA tests were used to analyze the differences between DMSO- and TMZ-treated tumors. Pair wise multiple comparisons were analyzed using the Holm–Sidak method (*P < 0.05, **P < 0.01). Bars = 200 µm.
Direct in vivo transduction of LV-CMVLuc-shMGMT vectors confer decreased MGMT expression and increased sensitivity toward TMZ
Increase of TMZ sensitivity by direct intratumoral injection of lentiviral vectors delivering anti-MGMT shRNAs was tested in the TMZ resistant LN18 wild-type xenografts. In order to measure the in vivo tumor cell infection, bioluminescence emission was measured after injection of lentiviral particles containing the pLKO.1-CMVLuc-shControl plasmid (LV-CMVLuc-shControl) or the pLKO.1-CMVLuc-shMGMT2 plasmid (LV-CMVLuc-shMGMT2). The luciferase transgene expression was detectable as early as 6 hours post-injection and increased during the first 2 days post-injection (Figure 4a). No significant differences regarding the luminescence expression were observed between xenografts injected with the lentivirus vector expressing the luciferase reporter gene and the anti-MGMT shRNA or the shRNA control sequence (Figure 4b). MGMT protein levels in the xenografts were assessed 2 days after virus injection using immunohistochemistry (Figure 4c). Injection of the LV-CMVLuc-shMGMT2 induced a reduction of the MGMT protein level in the LN18 wild-type xenografts, which was not observed for the xenografts injected with the LV-CMVLuc-shControl.
Figure 4.

In vivo inhibition of O6-methylguanine-DNA methyltransferase (MGMT) protein expression in human LN18 glioma xenografts through lentivirus delivery of small hairpin RNA (shRNA) targeting MGMT. (a) LN18 wild-type cells were xenografted in nude mice (four per animal). The lentiviral vector expressing the luciferase reporter gene and the shMGMT2 sequence (LV-CMVLuc-shMGMT2) was injected into the left tumors (ntumor = 4 in nmouse = 4). The lentiviral vector expressing the luciferase reporter gene and the control shRNA sequence (LV-CMVLuc-shControl) was injected into the right tumors (ntumor = 4 in nmouse = 4). Bioluminescence emission was measured from 6 to 48 hours after virus intratumoral injection. (b) The bioluminescence emitted by the xenografts was quantified (n = 4 per group). (c) Two days after virus injection, mice were sacrificed and MGMT expression was measured by immunostaining. All data are presented as mean + SD. Bars = 200 µm. CMV, cytomegalovirus.
Two days after the viral vector injection, mice were treated with TMZ or with DMSO as control treatment (Figure 5). Bioluminescence emission was measured after 7 and 12 days of TMZ or DMSO treatment (i.e., 9 and 14 days after lentivirus injection). The bioluminescence imaging (BLI) emission of the xenografts from mice receiving combined TMZ and LV-CMVLuc-shMGMT2 treatment decreased as early as after 7 days of TMZ therapy, before a stabilization (Figure 5a,c). In contrast, the BLI emission of the xenografts from mice receiving combined TMZ and LV-CMVLuc-shControl injections increased between day 2 and day 9, before showing various responses, with some tumors showing increasing and some decreasing LUC activity between day 9 and 14 (Figure 5a,c). The bioluminescence emission of the xenografts from mice receiving combined DMSO and LV-CMVLuc-shControl or DMSO and LV-CMVLuc-shMGMT2 treatment increased between day 2 and day 9 (Figure 5b,c). Between day 9 and day 14 the BLI signal was decreased, especially in big tumors. Two-way ANOVA test indicated a significant difference between mice receiving combined DMSO/LV-CMVLuc-shMGMT2 and mice receiving combined TMZ/LV-CMVLuc-shMGMT2 (P < 0.001). A significant difference was also observed between mice receiving combined TMZ/LV-CMVLuc-shMGMT2 and mice receiving combined TMZ/LV-CMVLuc-shControl (P < 0.05). In contrast, no significant difference was observed between mice receiving combined DMSO/LV-CMVLuc-shControl and mice receiving combined DMSO/LV-CMVLuc-shMGMT2. No significant difference was observed between mice receiving combined DMSO/LV-CMVLuc-shControl and mice receiving combined TMZ/LV-CMVLuc-shControl.
Figure 5.
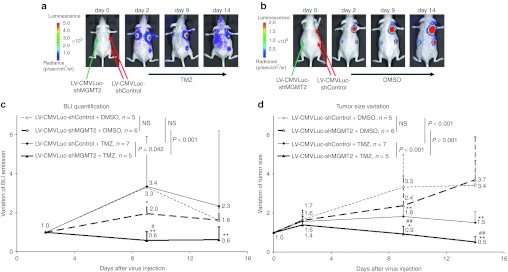
Combined anti-O6-methylguanine-DNA methyltransferase (MGMT) gene therapy and temozolomide (TMZ) chemotherapy induces an inhibition of the tumor growth of resistant glioblastoma (GBM) xenografts. LN18 wild-type cells were xenografted in nude mice (four per animal). The lentiviral vector expressing the luciferase reporter gene and the shMGMT2 sequence (LV-CMVLuc-shMGMT2) was injected into the left tumors (obtained: ntumor = 11 in nmouse = 10). The lentiviral vector expressing the luciferase reporter gene and the control small hairpin RNA (shRNA) sequence (LV-CMVLuc-shControl) was injected into the right tumors (obtained: ntumor = 12 in nmouse = 10). Two days after virus injection, mice were divided in two groups and treated with daily intraperitoneal injections of (a) dimethyl sulfoxide (DMSO) or 50 mg/kg TMZ (b). At day 0, 2, 9, and 14 the bioluminescence emitted by the xenografts was (a,b) measured and (c) quantified. Tumor sizes were evaluated by caliper measurements and size variations upon treatment were plotted against the (d) duration of treatment. All data are presented as mean + SD. Two-way ANOVA tests were used to analyze the differences between DMSO- and TMZ-treated tumors, and between tumors injected with the LV-CMVLuc-shControl and the LV-CMVLuc-shMGMT2. Pair wise multiple comparisons were analyzed using Holm–Sidak method. CMV, cytomegalovirus.
Tumor sizes were measured in order to evaluate the effect of the combined gene- and chemotherapy (Figure 5d). Combined treatment with LV-CMVLuc-shMGMT2 and TMZ induced a reduction of the size of the tumors. At day 2, when TMZ treatment starts, tumors were 1.4 times larger than at day 0, whereas at day 14 tumors were 50% smaller than at day 0. In contrast, combined treatment of LV-CMVLuc-shControl and TMZ only induced a stabilization of the tumor size. At day 2, tumors were 1.6 times larger than at day 0, whereas at day 14, tumors were 1.5 times larger than at day 0. Combined treatment of LV-CMVLuc-shControl or LV-CMVLuc-shMGMT2 and DMSO did not induce a reduction of the tumor size. On the contrary, mean tumor size was multiplied by 3.7 and 3.4 for the shMGMT2-expressing lentivirus and the shControl-expressing lentivirus, respectively (n.s.). Two-way ANOVA test indicated a significant difference between mice receiving combined TMZ/LV-CMVLuc-shMGMT2 and mice receiving combined DMSO/LV-CMVLuc-shMGMT2 (P < 0.001). A significant difference was also observed between mice receiving combined TMZ/LV-CMVLuc-shMGMT2 and mice receiving combined TMZ/LV-CMVLuc-shControl (P < 0.001). MGMT protein levels in the xenografts were assessed after the end of the experiment (day 14) using immunohistochemistry (Figure 6). Tumors receiving the LV-CMVLuc-shControl virus were characterized by a high and homogeneous MGMT staining (Figure 6a,c). On the other hand tumors receiving the LV-CMVLuc-shMGMT2 virus were composed of a mix population of tumoral cells displaying a low or a high MGMT expression (Figure 6b,d). In DMSO-treated tumors as well as in tumors receiving combined TMZ and LV-CMVLuc-shControl injections necrotic areas, in particular in the center of the xenografts, were observed (dashed line).
Figure 6.

O6-Methylguanine-DNA methyltransferase (MGMT) tumor expression after combined treatment. After the last in vivo measurement (day 14), mice were sacrificed. MGMT expression in tumor receiving combined (a) DMSO/LV-CMVLuc-shControl, (b) DMSO/LV-CMVLuc-shMGMT2, (c) TMZ/LV-CMVLuc-shControl, and (d) TMZ/LV-CMVLuc-shMGMT2 was determined by immunostaining. Hematoxylin–eosin stainings (H&E) revealed necrotic areas (dashed line). Bars = 200 µm. CMV, cytomegalovirus; DMSO, dimethyl sulfoxide; TMZ, temozolomide.
Discussion
TMZ is the standard therapy against GBM in clinical application, but overall survival of patient is strongly limited by the GBM ability to overexpress the DNA repair protein MGMT. Our results clearly indicate that employing vectors able to sufficiently alter in vivo expression of genes involved in mechanisms of resistance to therapy, such as MGMT, may improve significantly the response to chemotherapy. Anti-MGMT shRNA gene therapy could be used in combination with TMZ chemotherapy in order to improve treatment of resistant GBM. It should be pointed out, that treatment of tumors with the lentiviral vector alone did not show any toxicity, and the use of a control shRNA sequence showed the specificity of the combined approach for GBM overexpressing MGMT.
Treatment of TMZ resistant primary brain tumors or recurrent GBM remains problematic. Different molecular markers such 1p/19q codeletion, isocitrate dehydrogenase mutation or MGMT promoter methylation have been investigated in order to provide important diagnostic and prognostic information for the care of patients affected by GBM.6 As MGMT tumoral expression can provide resistance to alkylating agents by DNA damage reversal, inhibition of this protein is of clinical interest.17 Different strategies are being tested to improve TMZ efficacy in resistant GBM such as depletion of MGMT by the alkylating drug itself, by pseudo-substrates, or by RNAi.8,12 Since the DNA repair by MGMT provokes inactivation and degradation of the protein, it has been suggested that modulating the schedule of TMZ administration, as for example administration of high doses of TMZ or on the contrary repeated low doses of TMZ, could lead to an improvement of the chemotherapy efficacy.18,19 However, the correct balance between tumor suppression and TMZ-induced toxicity still remains to be determined. Another approach consists of the use of MGMT inhibitors, with O6-benzylguanine being so far the most extensively studied compound.20 Tumor growth inhibition could be demonstrated through the combination of an alkylating agent and O6-benzylguanine in different human tumor cell lines and xenografts, and this compound is now included in clinical trials. However, if MGMT inhibitors are not toxic themselves, inhibition of MGMT in peripheral organs in combination with the chemotherapeutic drug induced collateral toxicity, in particular a consistent myelo-suppressive effect.21 Therefore, the RNAi technology may be another attractive approach to silence DNA repair genes. In our study, a specific inhibition of the MGMT protein was obtained through the use of shRNA delivered by lentiviral vectors. Inhibition of MGMT in LN18 and T98 cells characterized by a high expression of MGMT and a high resistance to TMZ led to an increased sensitivity in culture as well as in vivo confirming the relation between MGMT and TMZ resistance shown in previous studies.22,23 The level of MGMT protein was found to be lower in human VU28 glioma cells than in human LN18 glioma cells, despite similar EC50 values observed in the growth and clonogenic assays. In van Nifterik et al.23 the MGMT protein level in VU28 cells seemed to be higher that in T98 cells as assessed using western blot. In our experiment, on the contrary, the level of MGMT protein as assessed using western blot was higher in T98 than in VU28 cells. This may come from a change of the cell genotype/phenotype upon passaging in cell culture. Other factors in addition to MGMT may explain the TMZ resistance of VU28 cells, such as non functional mismatch repair system or p53 function.22 Indeed, the pathway leading to apoptosis after DNA methylation is complex and involves among others the mismatch repair system, the DNA double-strand break repair system and the apoptosis cascade activated by p53.8 Even if no direct correlation between TMZ resistance and the mismatch repair system or p53 protein expressions is observed, modulation of p53 and mismatch repair system function lead to modulation of TMZ resistance.22
RNAi is one of the most powerful tools to specifically inhibit a gene at the post-transcriptional level, and this cellular mechanism has been used to render a variety of cancer cell lines sensitive to chemotherapeutic agents.24 However, the use of shRNA or siRNA is limited in vivo due to the poor pharmacological properties of RNA.13 Different viral vectors, as for example HSV-based vectors now in clinical development for the treatment of GBM,25 can be used for transient or persistent expression of shRNA in vivo.26,27 Lentivirus-derived vectors in particular are appealing due to (i) the lower risk of insertional mutagenesis compared to standard gamma retrovirus vectors, (ii) ability to efficiently transduce primary and nondividing cells and (iii) the high capacity of transgene insertion and have shown efficiency in preclinical models.16 In our study, we locally delivered the lentiviral vector into the GBM xenografts. Local delivery of viruses is used in nearly all clinical trials in glioma patients, in order to increase the viral load in the tumor and therefore the transduction efficiency, to overcome the problem of crossing the blood brain barrier, and to reduce toxicity to peripheral organs.25
In our approach local delivery, use of shRNA and low MGMT expression in normal brain tissue9 should support low toxicity even at relatively high viral load. However, the correct balance between efficacy and safety of gene therapy approaches still remains to be established,28 and development of technologies for noninvasive monitoring of the distribution and kinetics of vector-mediated gene expression is one critical issue for ensuring success of gene-based therapies.28 Molecular imaging could play a central role in optimizing gene therapy by quantitative imaging of reporter gene expression for the assessment of the location, magnitude, and duration of transgene expression.29 In the present study, the extent of the virus infection was followed by BLI. For the xenografts receiving combined LV-CMVLuc-shMGMT2/TMZ the measured luminescence signal decreased between day 2 and 9, showing that the infected cells were responding to the treatment in contrast to xenografts receiving control treatments. After 9 days, the BLI signals decreased also for the xenografts receiving the control treatments, even if the size of the tumors was not reduced. The observed BLI signal reduction originates most likely from central necrosis formation in larger tumors.30 Another explanation might be the fact that not all cells within the xenografts are transduced after a single virus injection. Noninfected luciferase-negative cells may in some tumors overgrow luciferase-positive tumors cells.
Our results show that MGMT inhibition using gene therapy can lead to an improvement of the therapeutic effect of TMZ in resistant GMB. Although the best routine clinical method to assess the MGMT status of patients with gliomas is still controversial (e.g., promoter methylation analysis; mRNA hybridization; determination of protein expression by immunohistochemistry; enzyme activity measurement), the MGMT status itself is currently recognized as an essential molecular marker in patients with gliomas.9 MGMT overexpression by GBM cells leads to resistance to the standard clinical chemotherapy and alternative treatments are limited for patients suffering of GBM presenting this characteristic. The possibility to specifically inhibit the MGMT protein using lentiviral vectors as demonstrated in our study represents therefore an attractive clinical option and could pave the way for personalized medicine. Furthermore, the RNAi approach can be adapted to any protein of interest for which the mRNA sequence is known. It should therefore be possible to not only deliver the anti-MGMT shRNA by the lentiviral vector but also other shRNAs targeting proteins with a key role in GBM resistance to chemotherapy.
Materials and Methods
Cell culture. Human LN18 (CRL-2610; ATCC, Wesel, Germany), T98 and VU28 (kind gift of Dr P. Sminia, Department of Radiation Oncology, Vrije Universiteit Medical Center, Faculty of Medicine, Amsterdam, the Netherlands)31 glioma cells and human embryonic kidney 293 cells (kind gift of Dr R. Thomas, MPI for Neurological Research, Cologne, Germany) were grown as monolayers in Dulbecco's modified Eagle's medium high glucose GlutaMAX (DMEM; Gibco, Darmstadt, Germany) supplemented with 5% fetal bovine serum (Invitrogen) for LN18 or 10% FBS (Invitrogen, Darmstadt, Germany) for T98, VU28, and human embryonic kidney 293, and 1% Penicillin/Streptomycin (P/S; PAA Laboratories, Cölbe, Germany) at 37 °C in a 5% CO2/95% air atmosphere.
Generation of reporter vectors. The schematic structures of the lentiviral vectors used in this study are presented in Figure 1 and are derived from the vector backbone pLKO.1-TRC containing the puromycin resistance gene (kind gift of Dr R. Thomas, MPI for Neurological Research, Cologne, Germany).14,15
The original pLKO.1-TRC cloning vector had a 1.9 kb insert that was released by digestion with AgeI and EcoRI (Fermentas, Villebon sur Yvette, France). In order to obtain the pLKO.1-shRNA vector the following sense oligonucleotides and their corresponding antisense sequences were chemically synthesized (Eurofins MWG Operon, Ebersberg, Germany), annealed and cloned into the AgeI and EcoRI sites (in bold: sequence of the shRNA):
shMGMT1-fDNA (sense) 5′-CCGGAAGCTGGAGCTGTCTGGTTGTTCAAGAGAACAACCAGACAGCTCCAGCTTTTTTTG-3′
shMGMT1-rDNA (antisense) 5′-AATTCAAAAAAAGCTGGAGCTGTCTGGTTGTTCTCTTGAAACAACCAGACAGCTCCAGCTT-3′
shMGMT2-fDNA (sense) 5′-CCGGAAGCTGCTGAAGGTTGTGAAATTCAAGAGATTTCACAACCTTCAGCAGCTTTTTTTG-3′
shMGMT2-rDNA (antisense) 5′-AATTCAAAAAAAGCTGCTGAAGGTTGTGAAATCTCTTGAATTTCACAACCTTCAGCAGCTT-3′
shControl-fDNA (sense) 5′-CCGGAAACTACCGTTGTTATAGGTGTTCAAGAGACACCTATAACAACGGTAGTTTTTTTTG-3′
shControl-rDNA (antisense) 5′-AATTCAAAAAAAACTACCGTTGTTATAGGTGTCTCTTGAACACCTATAACAACGGTAGTTT-3′
The sequence of the shMGMT1 shRNA was presented by Kuo et al.32 The sequence of the shMGMT2 and the shControl shRNA were designed with the BLOCK-iT RNAi Designer software from Invitrogen (https://rnaidesigner.invitrogen.com/rnaiexpress; Invitrogen, Germany). Transcription of the shRNA is driven by the RNA Polymerase III U6 promoter. The shRNA contains 21 sense bases that are identical to the target gene, a loop containing an XhoI restriction site, and 21 antisense bases that are complementary to the sense bases. The shRNA is followed by a polyT termination sequence for RNA Polymerase III. In order to follow the in vivo lentiviral infection using BLI the firefly luciferase reporter gene under the control of the CMV minimal promoter was introduced into the pLKO.1-shRNA backbones. The CMV-Luciferase sequence was amplified by PCR from the plasmid pHSV-fireLuc-IRES-TK39(gly)EGFP33,34 and introduced before the U6 promoter using the ClaI restriction site (Fermentas).
Lentiviral vector particle production. Lentiviral vectors were produced by a standardized three-plasmid transient transfection protocol. One day before transfection, 1.5 × 106 human embryonic kidney 293 cells were plated in a 10-cm tissue culture dish in 8 ml DMEM supplemented with 10% FBS and 1% P/S. Just before transfection the medium was removed and 8 ml of prewarmed DMEM without serum and antibiotics (plain DMEM) was added. 1.7 µg pLKO.1 (pLKO.1-shControl, pLKO.1-shMGMT1, pLKO.1-shMGMT2, pLKO.1-CMVLuc-shControl, pLKO.1-CMVLuc-shMGMT1, or pLKO.1-CMVLuc-shMGMT2) were mixed with 2.8 µg of a second-generation packaging plasmid (pCMV-dR8.2 dvpr) and 0.5 µg of a plasmid encoding the glycoprotein G of vesicular stomatitis virus (pCMV-VSV-G) in 500 µl of OptiMEM I Reduced Serum Media (Gibco). 15.0 µl of Fugene HD (Roche Applied Biosciences, Mannheim, Germany) were added to the DNA mix. The solution was incubated for 15 minutes at room temperature and added drop wise to the cells. After 24 hours, the medium was replaced with 8.0 ml DMEM supplemented with 30% FBS. The two following days, virus supernatants were harvested. Supernatants were cleared through a 0.45 µm filter and stored at –80 °C.
Transduction of tumor cell lines. Human LN18, T98, and VU28 glioma cells were plated each in 6-cm tissue culture dishes in order to be ~70% confluent at the day of the virus infection. The cell media was changed to fresh culture media containing 8.0 µg/ml of polybrene (Hexadimethrine bromide; Sigma-Aldrich, Germany), and 1.0 ml of lentiviral particle solution was added drop wise. Twenty four hours after infection, the virus-containing media was changed to fresh DMEM containing 10% FBS and 1% P/S. Thirty hours after infection, 2.0 µg/ml of puromycin (Roth, Lauterbourg, France) was added to the cells in order to select stable modified cell lines.
Western blot analysis. The expression of MGMT in the wild type and modified cell lines was evaluated using western blotting. Cells were lysed with a commercial lysis buffer (Cell Signaling, Frankfurt am Main, Germany) complemented with phosphatase inhibitors (Phosphatase Inhibitor Cocktail; Calbiochem, Darmstadt, Germany), and the amount of protein present in each sample was quantified using a Bio-Rad protein assay (Bio-Rad Laboratories, Marnes-la-Coquette, France). Equal amounts of denatured (95 °C; 5 minutes) protein were loaded onto a 12% Tris-glycin sodium dodecyl-sulfate polyacrylamide gel. After separation (2 hours; 140 V), proteins were blotted to a nitrocellulose membrane (Protran; Whatman, Münster, Germany) using a semi-dry blotting system (1 hour, 120 mA; Biometra, Goettingen, Germany). After blocking nonspecific binding with 3% bovine serum albumin fraction V (BSA; PPA Laboratories) dissolved in Tris-buffered saline containing 0.1% Tween 20 (TBST) for 1 hour, the membrane was washed with TBST and probed with primary antibodies. After washing, the membrane was incubated with horseradish peroxidase conjugated secondary antibody in TBST for 1 hour. After three washing steps (TBST, 15 minutes), protein detection was achieved through chemiluminescent reaction using the Pierce ECL Plus Western Blotting Substrate (Pierce Biotechnology, Bonn, Germany).
The following antibodies were used: primary monoclonal mouse anti-MGMT (1:250 dilution in 3% TBST-BSA solution; MT3.1; Abcam, Cambridge, UK), primary monoclonal mouse anti-actin clone C4 (1:1,000 dilution in 3% TBST-BSA solution; MP Biomedicals, Eschwege, Germany), secondary polyclonal rabbit anti-mouse immunoglobulins (1:2,000 dilution in TBST solution; DAKO, Hamburg, Germany).
Clonogenic and growth assay. Acute growth inhibition/cytotoxicity assays involved the exposure of the glioma cells seeded at 5.0 × 104 cells/well in 24-well plates to increasing concentrations (0–1,000 µmol/l) of TMZ (Sigma-Aldrich) for 72 hours. Cells were counted using a Z2 coulter particle count and size analyzer (Beckman Coulter, Krefeld, Germany).
Clonogenic survival assays were performed by seeding 2,000 cells in six-well plates and exposing them to increasing concentrations of TMZ (0–1,000 µmol/l), followed by further observation for 7–14 days. The number of colonies was assessed using crystal violet (Roth) staining.
Animal experiments. All animal procedures were in accordance with the German Laws for Animal Protection and were approved by the LANUV NRW (Landesamt für Natur, Umwelt und Verbraucherschutz, North Rhine-Westphalia; Düsseldorf). NMRI nude mice (Janvier, Saint Berthevin, France) were housed at constant temperature (23 °C) and relative humidity (40%), under a regular light/dark schedule. Food and water were freely available.
Nude mice were injected subcutaneously with 2.0 × 106 LN18, LN18-shControl, LN18-shMGMT1, and LN18-shMGMT2 cells suspended in 250 µl of a solution containing 125 µl DMEM without supplement and 125 µl BD Matrigel Basement Membrane Matrix (BD Biosciences, Heidelberg, Germany). About 2 months after cell injection, growing tumors were explanted and cut in small pieces, which were subsequently subcutaneously reimplanted into nude mice.
Nude mice xenografted with cells depleted or not in MGMT were treated with daily injections of 50 mg/kg of TMZ in the intraperitoneal cavity, when xenografts reached a size of ~200 mm3. Tumor sizes were measured using a caliper before treatment and at days 7 and 12 after treatment.
Intratumoral injection of lentiviral particules. For further in vivo experiments, lentivirus vectors expressing the shRNA sequences and the firefly luciferase reporter gene were injected directly into the LN18 wild-type tumors 2 days before starting the treatment with TMZ. Lentiviral supernatants were concentrated 100-fold using the LentiX Concentrator Kit (Clontech, Saint-Germain-en-Laye, France) and re-suspended in DMEM plain media. In order to normalize the quantity of virus injected into the tumors, viral supernatants were titrated using bioluminescence emission measurements. LN18 cells were seeded at 5.0 × 104 cells/well in 24-well plates. The following day, cells were infected using serially diluted lentiviral particle solution in 500 µl of fresh culture media containing 8 µg/ml of polybrene. Twenty four hours after infection the virus-containing medium was replaced with fresh medium. After 48 hours, the medium was replaced with a solution containing 0.33 µg/µl D-luciferin (Synchem, Felsberg/Altenburg, Germany) in DMEM supplemented with FCS and P/S. Bioluminescence emission was measured using the Perkin Elmer Fusion Plate Reader (Perkin Elmer, Rodgau, Germany) and expressed as relative light unit (RLU). After the BLI measurement, the luciferin solution was removed and an MTT assay was performed in order to control the cell number.
Lentiviral solution concentrations were expressed in (RLU/cell)/µl. Before the intratumoral vector injection, tumor sizes were measured by caliper measurements in order to inject a virus titer of 4.6 × 10–7 (RLU/cell)/mm3-of-tumor. Between 25 and 100 µl of lentiviral solution, depending of the tumor size and the lentivirus titer, was injected directly into the tumors using insulin syringes. Two days after virus injection mice were treated with daily intraperitoneal injections of 50 mg/kg TMZ. BLI was used to follow the virus infection at 6 hours, 12 hours, 24 hours, 2 days, 9 days, and 14 days after virus injection, and tumor size evolution was evaluated at day 0, 2, 9, and 14 by caliper measurements.
BLI. Bioluminescence acquisition and analysis were performed using the IVIS Spectrum Imaging System and Living Image 4.0 software (Caliper Life Sciences, Hopkinton, MA). Mice were narcotized with isoflurane (2% isoflurane, 0.5 l/minute oxygen) and injected intraperitoneally with D-luciferin (150 mg/kg, i.e., around 200 µl of a 20 mg/ml luciferin solution diluted in PBS; Synchem). Images were acquired 6–20 minutes after substrate injection at 2-minute time intervals (f/stop: 1, binning: 8, exposure time: auto). Grayscale photographic images and bioluminescence color images were superimposed. Regions of interest were drawn over each tumor to determine the signal intensity (total flux, p/s).
MGMT immunostaining of paraffin sections. Depending on the experimental setup, mice were sacrificed 2 days after virus injection or after the 12 days of TMZ treatment (14 days after virus injection). The tumors were excised, fixed in 4% PFA, embedded in paraffin, cut in 5 µm sections and prepared for immunohistological localization of MGMT. In brief, after rehydrating and heat-induced epitope retrieval for 30 minutes in citrate buffer (pH 6.0), sections were incubated in peroxidase-blocking solution (S3022; DAKO) for 5 minutes and treated with serum-blocking solution for 15 minutes. Then, sections were incubated overnight at 4 °C with monoclonal mouse anti-MGMT antibody (dilution 1:25; MT3.1, ab39253; Abcam). Labeling of the primary antibody was performed using a commercial avidin-biotin complex detection kit based on a biotinylated anti-mouse antibody (PK-6102; Vector Laboratories, Loerrach, Germany) according to the manufacturers manual, followed by treatment with 3,3′-diaminobenzidine (DAB, D-5637; Sigma-Aldrich) for 5 minutes. Sections were counterstained with hematoxylin, dehydrated, and mounted using Entellan (Merck, Darmstadt, Germany).
Histological analysis was performed using a Nikon Eclipse 90i light microscope (Nikon, Düsseldorf, Germany) and the NIS-Elements software package (Nikon).
Statistical analysis. Student's t-test and Two-way ANOVA test were performed using SigmaStat 3.0 (SPSS, Inc., Chicago, IL).
Acknowledgments
The authors thank Gabriele Schneider (MPI, Cologne), Irmgard Hoppe (EIMI, Münster), Christa Möllmann (EIMI, Münster), Claudia Gräf (EIMI, Münster), Melanie Becker (EIMI, Münster), Wiebke Gottschlich (EIMI, Münster), Nicole Sponer (MPI, Cologne), Selman öztürk (MPI, Cologne), Isabell Wienpahl (MPI, Cologne), Sara Rapic (MPI, Cologne), and Gerhard Jungwirth (MPI, Cologne) for technical support throughout the project. This work was supported by Joint Translational Research Program on Cancer (DAAD-INCa, PKZ: D/0811485), the BMBF funding MoBiMed, and CLINIGENE NoE (LSHB-CT-2006–018933). The authors declared no conflict of interest.
References
- Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A.et al. (2007The 2007 WHO classification of tumours of the central nervous system Acta Neuropathol 11497–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckner JC, Brown PD, O'Neill BP, Meyer FB, Wetmore CJ., and, Uhm JH. Central nervous system tumors. Mayo Clin Proc. 2007;82:1271–1286. doi: 10.4065/82.10.1271. [DOI] [PubMed] [Google Scholar]
- Jemal A, Siegel R, Ward E, Hao Y, Xu J., and, Thun MJ. Cancer statistics, 2009. CA Cancer J Clin. 2009;59:225–249. doi: 10.3322/caac.20006. [DOI] [PubMed] [Google Scholar]
- Candolfi M, Kroeger KM, Muhammad AK, Yagiz K, Farrokhi C, Pechnick RN.et al. (2009Gene therapy for brain cancer: combination therapies provide enhanced efficacy and safety Curr Gene Ther 9409–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viel T, Talasila KM, Monfared P, Wang J, Jikeli JF, Waerzeggers Y.et al. (2012Analysis of the growth dynamics of angiogenesis-dependent and -independent experimental glioblastomas by multimodal small-animal PET and MRI J Nucl Med 531135–1145. [DOI] [PubMed] [Google Scholar]
- Weller M. Novel diagnostic and therapeutic approaches to malignant glioma. Swiss Med Wkly. 2011;24:141–148. doi: 10.4414/smw.2011.13210. [DOI] [PubMed] [Google Scholar]
- Silber JR, Bobola MS, Blank A., and, Chamberlain MC. O(6)-Methylguanine-DNA methyltransferase in glioma therapy: Promise and problems. Biochim Biophys Acta. 2012;1826:71–82. doi: 10.1016/j.bbcan.2011.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verbeek B, Southgate TD, Gilham DE., and, Margison GP. O6-Methylguanine-DNA methyltransferase inactivation and chemotherapy. Br Med Bull. 2008;85:17–33. doi: 10.1093/bmb/ldm036. [DOI] [PubMed] [Google Scholar]
- Weller M, Stupp R, Reifenberger G, Brandes AA, van den Bent MJ, Wick W.et al. (2010MGMT promoter methylation in malignant gliomas: ready for personalized medicine Nat Rev Neurol 639–51. [DOI] [PubMed] [Google Scholar]
- Hegi ME, Diserens AC, Gorlia T, Hamou MF, de Tribolet N, Weller M.et al. (2005MGMT gene silencing and benefit from temozolomide in glioblastoma N Engl J Med 352997–1003. [DOI] [PubMed] [Google Scholar]
- Paz MF, Yaya-Tur R, Rojas-Marcos I, Reynes G, Pollan M, Aguirre-Cruz L.et al. (2004CpG island hypermethylation of the DNA repair enzyme methyltransferase predicts response to temozolomide in primary gliomas Clin Cancer Res 104933–4938. [DOI] [PubMed] [Google Scholar]
- Sabharwal A., and, Middleton MR. Exploiting the role of O6-methylguanine-DNA-methyltransferase (MGMT) in cancer therapy. Curr Opin Pharmacol. 2006;6:355–363. doi: 10.1016/j.coph.2006.03.011. [DOI] [PubMed] [Google Scholar]
- Viel T, Boisgard R, Kuhnast B, Jego B, Siquier-Pernet K, Hinnen F.et al. (2008Molecular imaging study on in vivo distribution and pharmacokinetics of modified small interfering RNAs (siRNAs) Oligonucleotides 18201–212. [DOI] [PubMed] [Google Scholar]
- Zufferey R, Nagy D, Mandel RJ, Naldini L., and, Trono D. Multiply attenuated lentiviral vector achieves efficient gene delivery in vivo. Nat Biotechnol. 1997;15:871–875. doi: 10.1038/nbt0997-871. [DOI] [PubMed] [Google Scholar]
- Moffat J, Grueneberg DA, Yang X, Kim SY, Kloepfer AM, Hinkle G.et al. (2006A lentiviral RNAi library for human and mouse genes applied to an arrayed viral high-content screen Cell 1241283–1298. [DOI] [PubMed] [Google Scholar]
- Huszthy PC, Giroglou T, Tsinkalovsky O, Euskirchen P, Skaftnesmo KO, Bjerkvig R.et al. (2009Remission of invasive, cancer stem-like glioblastoma xenografts using lentiviral vector-mediated suicide gene therapy PLoS ONE 4e6314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villano JL, Seery TE., and, Bressler LR. Temozolomide in malignant gliomas: current use and future targets. Cancer Chemother Pharmacol. 2009;64:647–655. doi: 10.1007/s00280-009-1050-5. [DOI] [PubMed] [Google Scholar]
- Wick W, Platten M., and, Weller M. New (alternative) temozolomide regimens for the treatment of glioma. Neuro-oncology. 2009;11:69–79. doi: 10.1215/15228517-2008-078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasparini G. Metronomic scheduling: the future of chemotherapy. Lancet Oncol. 2001;2:733–740. doi: 10.1016/S1470-2045(01)00587-3. [DOI] [PubMed] [Google Scholar]
- Rabik CA, Njoku MC., and, Dolan ME. Inactivation of O6-alkylguanine DNA alkyltransferase as a means to enhance chemotherapy. Cancer Treat Rev. 2006;32:261–276. doi: 10.1016/j.ctrv.2006.03.004. [DOI] [PubMed] [Google Scholar]
- Schilsky RL, Dolan ME, Bertucci D, Ewesuedo RB, Vogelzang NJ, Mani S.et al. (2000Phase I clinical and pharmacological study of O6-benzylguanine followed by carmustine in patients with advanced cancer Clin Cancer Res 63025–3031. [PubMed] [Google Scholar]
- Hermisson M, Klumpp A, Wick W, Wischhusen J, Nagel G, Roos W.et al. (2006O6-methylguanine DNA methyltransferase and p53 status predict temozolomide sensitivity in human malignant glioma cells J Neurochem 96766–776. [DOI] [PubMed] [Google Scholar]
- van Nifterik KA, van den Berg J, van der Meide WF, Ameziane N, Wedekind LE, Steenbergen RD.et al. (2010Absence of the MGMT protein as well as methylation of the MGMT promoter predict the sensitivity for temozolomide Br J Cancer 10329–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pai SI, Lin YY, Macaes B, Meneshian A, Hung CF., and, Wu TC. Prospects of RNA interference therapy for cancer. Gene Ther. 2006;13:464–477. doi: 10.1038/sj.gt.3302694. [DOI] [PubMed] [Google Scholar]
- Mohyeldin A., and, Chiocca EA. Gene and viral therapy for glioblastoma: a review of clinical trials and future directions. Cancer J. 2012;18:82–88. doi: 10.1097/PPO.0b013e3182458b13. [DOI] [PubMed] [Google Scholar]
- Li CX, Parker A, Menocal E, Xiang S, Borodyansky L., and, Fruehauf JH. Delivery of RNA interference. Cell Cycle. 2006;5:2103–2109. doi: 10.4161/cc.5.18.3192. [DOI] [PubMed] [Google Scholar]
- Saydam O, Glauser DL, Heid I, Turkeri G, Hilbe M, Jacobs AH.et al. (2005Herpes simplex virus 1 amplicon vector-mediated siRNA targeting epidermal growth factor receptor inhibits growth of human glioma cells in vivo Mol Ther 12803–812. [DOI] [PubMed] [Google Scholar]
- Waerzeggers Y, Monfared P, Viel T, Winkeler A, Voges J., and, Jacobs AH. Methods to monitor gene therapy with molecular imaging. Methods. 2009;48:146–160. doi: 10.1016/j.ymeth.2009.03.007. [DOI] [PubMed] [Google Scholar]
- Jacobs A, Voges J, Reszka R, Lercher M, Gossmann A, Kracht L.et al. (2001Positron-emission tomography of vector-mediated gene expression in gene therapy for gliomas Lancet 358727–729. [DOI] [PubMed] [Google Scholar]
- Jacobs AH, Rueger MA, Winkeler A, Li H, Vollmar S, Waerzeggers Y.et al. (2007Imaging-guided gene therapy of experimental gliomas Cancer Res 671706–1715. [DOI] [PubMed] [Google Scholar]
- van Nifterik KA, Elkhuizen PH, van Andel RJ, Stalpers LJ, Leenstra S, Lafleur MV.et al. (2006Genetic profiling of a distant second glioblastoma multiforme after radiotherapy: Recurrence or second primary tumor J Neurosurg 105739–744. [DOI] [PubMed] [Google Scholar]
- Kuo CC, Liu JF., and, Chang JY. DNA repair enzyme, O6-methylguanine DNA methyltransferase, modulates cytotoxicity of camptothecin-derived topoisomerase I inhibitors. J Pharmacol Exp Ther. 2006;316:946–954. doi: 10.1124/jpet.105.095919. [DOI] [PubMed] [Google Scholar]
- Jacobs AH, Winkeler A, Hartung M, Slack M, Dittmar C, Kummer C.et al. (2003Improved herpes simplex virus type 1 amplicon vectors for proportional coexpression of positron emission tomography marker and therapeutic genes Hum Gene Ther 14277–297. [DOI] [PubMed] [Google Scholar]
- Monfared P, Winkeler A, Klein M, Li H, Klose A, Hoesel M.et al. (2008Noninvasive assessment of E2F-1-mediated transcriptional regulation in vivo Cancer Res 685932–5940. [DOI] [PubMed] [Google Scholar]