

Abstract
Adenoviruses harboring the herpes simplex virus thymidine kinase (HSVtk) gene under the regulation of a trans-splicing ribozyme targeting human telomerase reverse transcriptase (hTERT-TR) show marked and specific antitumor activity. In addition to inducing tumor cell death by direct cytotoxicity, it is becoming clear that HSVtk also induces antitumor immunity. Programmed death ligand 1 (PD-L1) expressed on tumor cell surfaces mediates tumor-induced immunoresistance by inhibiting PD1-expressing tumor-infiltrating T cells. Here, we explored whether a soluble form of PD1 (sPD1-Ig), which blocks PD-L1, could synergize with TERT-TR-regulated HSVtk to enhance the adenoviral therapeutic efficacy by boosting antitumor immunity. Tumor antigen released by HSVtk-transduced tumors successfully primed tumor antigen–specific CD8 T cells via dendritic cells (DC). Regression of murine tumors was markedly enhanced when sPD1-Ig was incorporated into the adenovirus as compared with a single-module adenovirus expressing only HSVtk. This effect was abolished by CD8 T-cell depletion. Consistent with this, following adoptive transfer of tumor antigen–specific CD8 T cells into tumor-bearing Rag1−/− mice, dual-module adenovirus significantly enhanced CD8 T cell–mediated tumor rejection. In addition, secondary tumor challenge at a distal site was completely suppressed in mice treated with a dual-module adenovirus. These results suggest that a dual-targeting strategy to elicit both tumor antigen priming and tumor-induced immunoresistance enhances CD8 T cell–mediated antitumor immunity.
Introduction
Adenoviruses harboring herpes simplex virus thymidine kinase (HSVtk) have been proposed as a therapeutic approach to the treatment of various cancers. Using a human telomerase reverse transcriptase (hTERT)-targeting ribozyme (hTERT-TR) to control tumor-specific expression of HSVtk, we successfully achieved HSVtk expression in primary liver and colorectal cancer cells.1,2 When HSVtk-expressing cells are exposed to ganciclovir (GCV), a prodrug, HSVtk phosphorylates GCV at a single site. This monophosphorylated form of GCV is trapped inside the cell and converted into tri-phosphorylated GCV by cellular kinases. The tri-phosphorylated GCV in turn inhibits DNA polymerase, causing single-strand DNA breaks, eventually leading to apoptosis.3,4,5,6 Although this direct cytotoxic effect is thought to be the main mechanism of HSVtk antitumor activity, it has become clear from recent reports that HSVtk-mediated tumor cell lysis elicits antitumor immunity.7,8,9 New strategies to potentiate this antitumor immunity by combining various immune modulators such as FMS-like tyrosine kinase 3 ligand and granulocyte-macrophage-colony stimulating factor with adenoviruses harboring HSVtk have shown great promise as immunotherapeutic vaccines.10,11,12 Most of these studies focused on tumor antigen priming and enhancing the recruitment or activation of antigen presenting cells such as dendritic cells (DCs). However, to maximize HSVtk-based antitumor immunity, tumor immunoresistance mechanisms such as immunosuppressive cytokines, major histocompatibility complex downregulation, and immunosuppressive cell-surface molecules expressed on the cancer cell surface must be overcome.13,14,15,16 To date, these issues have not been addressed in the context of HSVtk-based gene therapy.
Programmed death ligand 1 (PD-L1) is a cell-surface glycoprotein and a member of the B7 family of T-cell costimulatory molecules.17 Although PD-L1 mRNA is ubiquitously expressed in humans, cell-surface expression of PD-L1 is restricted to cells of the macrophage lineage.17 Of note, PD-L1 is also expressed on the surface of many human tumor cells.18,19 The PD-L1 receptor (programmed death-1, PD-1) is expressed on T cells.20 Binding of PD-L1 transduces negative regulatory signals via the PD-1 inhibitory cytoplasmic domain.21,22 PD-L1 expressed on the surface of tumor cells abrogates cell-mediated antitumor immunity by inducing T-cell apoptosis and inhibiting cytokine production and tumor-killing effect of activated T cells.23,24 Therefore, PD-L1 expression on the tumor cell surface appears to mediate tumor-induced immunoresistance. Consistent with this, neutralizing antibodies against either PD-1 or PD-L1 overcome this resistance and enhance antitumor immunity in preclinical mouse models.25,26,27 Therapeutic antibodies targeting both molecules are currently being tested in phase I clinical trials.23,28 However, systemic downregulation of the PD-L1/PD-1 axis can also trigger systemic autoimmunity via uncontrolled activation of autoreactive T cells, as was shown for antibodies to cytotoxic T lymphocyte–associated antigen 4, another negative regulator of T cells.29,30 Thus, localized inhibition of the PD-L1/PD-1 axis in the tumor microenvironment may be a more preferable and safer strategy for overcoming tumor-induced T-cell tolerance.
In the current study, as a strategy for blocking PD-L1 in the tumor microenvironment, we generated recombinant soluble PD-1 in which the extracellular domain of PD-1 was fused to the Fc portion of mouse IgG2a (sPD1-Ig). The sPD1-Ig cDNA was integrated into a replication-deficient adenovirus harboring HSVtk under the regulation of mouse TERT-TR to achieve the dual goal of enhancing tumor antigen priming and blocking local immune resistance. In addition to the direct cytotoxic effects of HSVtk, the dual-module adenovirus was designed to enhance antitumor vaccine efficiency by maximizing antitumor immunity. Our experimental evidence suggests that the dual-module adenovirus enhances antitumor immunity by amplifying the CD8 T-cell response.
Results
Construction of an adenovirus harboring mouse TERT-TR- HSVtk
We previously developed a novel strategy for tumor-specific adenoviral expression of HSVtk using a well-known tumor marker, TERT. In this strategy, a ribozyme that recognizes and digests mRNA sequence of hTERT is delivered to tumor cells using a recombinant adenovirus. In addition, this ribozyme is designed to ligate HSVtk mRNA to the digested 3′ end of hTERT mRNA, which leads to new translation of HSVtk mRNA in the tumor cells. This kind of ribozyme is called trans-splicing ribozyme.1,2 (see also Supplementary Figure S1). In theory, trans-splicing ribozyme–controlled HSVtk expression is restricted to cells that express high levels of hTERT mRNA, which include tumor cells. Thus, this system represents an efficient way to deliver HSVtk into tumor cells without affecting cells in the surrounding normal tissues, which express low levels of hTERT. In fact, this strategy was highly effective in human colorectal cancer cells in xenograft mouse models. One drawback of this system in immunological analysis is that experiments with human cancer cells have to be performed in immune-deficient mice to prevent xenograft rejection. Hence, this system is not appropriate for evaluating the effect of HSVtk on antitumor immunity.
To overcome this limitation, we designed a similar system using a trans-splicing ribozyme that targets mouse TERT mRNA (mTERT-TR) to evaluate the immunological effects of TERT-TR-regulated HSVtk using syngeneic mouse tumor models. The TERT recognition sequence was placed proximal to the HSVtk coding sequence such that HSVtk expression was dependent on the presence of mTERT transcripts (Supplementary Figure S1).31 This entire expression cassette, under the control of the cytomegalovirus (CMV) promoter, was integrated into an E1/E3-deficient adenovirus genome to generate an adenovirus (Ad5mTR) harboring mTERT-TR-regulated HSVtk (mTERT-TR-HSVtk) (Figure 1a). An adenovirus (Ad5MOCK) lacking the expression cassette was also generated as a control.
Figure 1.
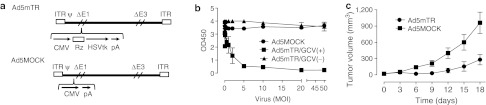
Regulation of adenoviral HSVtk gene expression by mouse telomerase reverse transcriptase–targeting ribozyme (TERT-TR). (a) Ad5mTR, an E1/E3-deficient adenovirus, expressing HSVtk under the regulation of mouse TERT-TR. Ad5MOCK is a control E1/E3-deficient adenovirus. (b) CT26 cells (3 × 103) were seeded in 96-well plates and then exposed to Ad5MOCK (closed circles) or Ad5mTR adenovirus at the indicated multiplicity of infections in the presence (closed squares) or absence of 100 µmol/l ganciclovir (GCV) (closed triangles). On day 3 postinfection, cytotoxicity was measured using a cell proliferation assay kit. Data represent the mean ± SD of triplicate assays. (c) BALB/c mice were inoculated subcutaneously in both flanks with CT26 cells (1 × 106) followed by intratumoral injection of 5 × 108 plaque forming units of either Ad5MOCK (closed squares) or Ad5mTR (closed circles), and GCV (75 mg/kg) was administered intraperitoneally twice per day. Data represent the mean ±SEM (n = 5). ITR, inverted terminal repeats; CMV, cytomegalovirus promoter; HSVtk, herpes simplex virus thymidine kinase; pA, poly A; Rz, mouse TERT targeting trans-splicing ribozyme; δE1 and δE3, deletion of E1 and E3; ψ, packaging signal.
To determine whether HSVtk expression mediated by Ad5mTR was cytotoxic to murine tumor cells, we used CT26 cells, a colon cancer cell line derived from a BALB/c mouse, because this cell line expresses high levels of mTERT mRNA (data not shown). CT26 cells were exposed to Ad5mTR at varying multiplicity of infection (MOIs) in the presence or absence of GCV. An MOI of 2.5 in the presence of GCV was sufficient to kill most of the cells, whereas Ad5MOCK exhibited no cytotoxicity up to an MOI of 50 (Figure 1b). Consistent with this in vitro cytotoxicity, tumor growth was significantly inhibited when Ad5mTR was injected intratumorally into subcutaneous CT26 tumors in BALB/c mice (Figure 1c). Thus, Ad5mTR showed similar antitumor effects to those of adenoviruses harboring human TERT-TR-HSVtk, which indicated that it would be appropriate for studies of antitumor immunity in mice.
Enhanced DC-mediated tumor antigen presentation by HSVtk
Some reports suggest that HSVtk expression in tumor cells, either by DNA transfection or adenovirus infection, enhances antigen priming of cytotoxic CD8 T cells.9,32 Because HSVtk expression in the presence of GCV elicits cell death in a significant proportion, if not all, of the tumor mass in vivo, tumor antigens derived from dead cells may be captured by APCs such as DCs, and these cells would then migrate to draining lymph nodes to prime tumor antigen–specific T cells. Although this model was suggested in the literature,33 we decided to test this possibility using a defined antigen-specific T cells. For this experiment, we used the murine tumor cell line E.G7 (a derivative of EL4 cells that stably expresses ovalbumin; OVA) as a model tumor antigen, and T cells purified from OVA-specific T-cell receptor transgenic mice (OT-I mice) as the model tumor-antigen (OVA)-specific CD8 T-cell population. Most of CD8 T cells purified from OT-1 mice are OVA-specific naive cytotoxic T cells.
We first tested the ability of Ad5mTR to induce cell death in subcutaneous E.G7 tumors in syngeneic B6 mice. When tumors were isolated and examined histologically following intratumoral injection of Ad5mTR, there was a significant degree of cell death, which suggested that tumor antigen release was likely following viral infection (Figure 2a). Next, we purified DCs from the tumor-draining lymph nodes and examined whether these cells were loaded with OVA using a DC/CD8 T-cell coculture assay for interferon-γ (IFN-γ) production. Purified OT-I T cells cultured with DCs from Ad5mTR-treated tumor-bearing mice produced much more IFN-γ than OT-I T cells cultured with DCs from control virus–treated tumor-bearing mice. These results indicated that DCs from Ad5mTR-treated mice presented a sufficient level of OVA antigen to stimulate OVA-specific T cells (Figure 2b). Thus, expression of HSVtk in tumors and subsequent GCV-induced cell death resulted in the release of tumor antigens, and these antigens were efficiently captured by DCs capable of stimulating tumor antigen–specific cytotoxic T cells. Thus, tumor-specific expression of HSVtk can effectively stimulate antitumor T-cell activity via DCs, in addition to inducing direct tumor cell cytotoxicity.
Figure 2.

Enhanced dendritic cells (DC)-mediated antigen presentation elicited by HSVtk. (a) E.G7 tumors in C57/BL6 mice were injected intratumorally with either Ad5MOCK or Ad5mTR. Tumors were harvested 4 days later for histological analysis by hematoxylin and eosin staining. (b) Two days after the injection of adenoviruses, DCs (5 × 104) were harvested from draining lymph nodes around E.G7 tumors by MACS chromatography using anti-CD11c microbeads and then cocultured with ovalbumin-specific CD8 T cells (1 × 106) from OT-1 transgenic mice. After 72 hours, culture supernatants were collected and the amount of interferon-γ was measured. DC-Ad5MOCK, DCs from Ad5MOCK-injected mice; CD-Ad5mTR, DCs from Ad5mTR-injected mice were also measured. Data represent the mean ±SD. P, unpaired t-test.
Construction of an adenovirus harboring both mTERT-TR-HSVtk and sPD1-Ig
Ad5mTR stimulated antitumor CD8 T-cell reactivity, raising the intriguing possibility that combining this approach with another strategy for inactivating tumor-induced immune resistance could further enhance antitumor T-cell reactivity. PD-L1 is a well-known immune suppressor expressed on tumor cell surfaces. We generated a soluble form of the PD-L1 receptor, sPD1, which neutralizes PD-L1 and abrogates PD-L1-mediated T-cell inhibition. To increase the stability of sPD1 in vivo, sPD1 was fused to the Fc portion of IgG2a to generate sPD1-Ig. We constructed a dual-module adenovirus (Ad5mTR.sPD1) containing HSVtk under the regulation of mTERT-TR in the E1 region and sPD1-Ig in the E3 region of the adenoviral genome (Figure 3a).
Figure 3.

Construction of a dual-module adenovirus expressing mouse telomerase reverse transcriptase-targeting ribozyme (mTERT-TR)-regulated HSVtk and sPD1-Ig. (a) Ad5mTR.sPD1 encodes mTERT-TR-HSVtk under the control of the CMV promoter in the E1 region and sPD1-Ig under control of the EF1α promoter in the E3 region. Details of the construction are described in Material and Methods section. (b) HEK293 cells (4 × 105) were infected with the indicated adenovirus as an multiplicity of infection of 10 for 24 hours, followed by lysis in RIPA buffer and immunoblot analysis. sPD1-Ig expression by Ad5mTR.sPD1 (total of 80 µg protein per lane) was confirmed using an anti-PD1 antibody. (c) The enzymatic activity of HSVtk in phosphorylating ganciclovir was evaluated in CT26 cells by measuring the accumulation of radio-labeled [H3]PCV (1 µCi/ml) as described in Materials and Methods section. Data represent the mean ± SD of triplicate assays. (d) Culture supernatant harvested from cells infected with adenovirus (5 or 10 MOI) was mixed with cocultures of ovalbumin (OVA)-specific CD8 T cells (1 × 106) purified from OT-1 mice and MC38/OVA cells (1 × 104) in 6-well plates. After 72 hours, culture supernatants were collected and interferon-γ levels were measured. Data represent the mean ± SD. P, unpaired t-test. ψ, packaging signal; δE1 and δE3, deletion of E1 and E3; CMV, cytomegalovirus promoter; EF1α, elongation factor 1α promoter; HSVtk, herpes simplex virus thymidine kinase; ITR, inverted terminal repeats; MOI, multiplicity of infection; Rz, mouse TERT targeting trans-splicing ribozyme; pA, poly A; PCV, penciclovir.
HEK293 cells infected with Ad5mTR.sPD1 expressed and secreted sPD1-Ig into the extracellular milieu, as confirmed by immunoblot analysis (Figure 3b). The expression level of HSVtk in Ad5mTR.sPD1-infected cells was comparable with that of Ad5mTR-infected cells as measured by enzymatic activity (Figure 3c).34 We also tested whether secreted sPD1-Ig could enhance antitumor T-cell reactivity by neutralizing PD-L1 on the tumor cell surface in vitro. Culture supernatant from Ad5EF1α.sPD1-infected 293HEK cells was added to cocultures of OVA-expressing tumor cells and OVA-specific OT-I T cells, and T-cell reactivity was measured according to the amount of IFN-γ secretion. As expected, IFN-γ secretion by antigen-specific T cells in response to cancer cell challenge was enhanced by sPD1-Ig-containing supernatant (Figure 3d). The expression of PD-L1 on the cell surface of cancer cells and the binding of sPD1-Ig in the culture supernatant to the cancer cell surface were confirmed by flow cytometry (Supplementary Figure S2). These results suggest that the dual-module adenovirus may enhance antitumor T-cell reactivity within the tumor microenvironment in vivo.
Effective suppression of tumor growth by Ad5mTR.sPD1
To examine whether sPD1-Ig enhanced the antitumor activity of HSVtk in vivo, we used a CT26 colon cancer model (see Figure 1c). This model was chosen in part because CT26 cells express high levels of PD-L1 on their cell surface (Supplementary Figure S2). CT26 cells infected with Ad5mTR.sPD1 showed a similar degree of in vitro cytotoxicity to those infected with Ad5mTR in the presence of GCV, which suggested that sPD1-Ig does not contribute to direct cytotoxicity by HSVtk (Figure 4a). By contrast, Ad5mTR.sPD1 significantly enhanced tumor regression as compared with Ad5mTR following intratumoral injection into CT26 tumors in BALB/c mice; indeed, near-complete tumor regression was observed (Figure 4b). A control adenovirus harboring sPD1-Ig alone (Ad5EF1.sPD1) did not show any antitumor effects in this model, which suggests that sPD1-Ig expression in the tumor tissue is not sufficient to induce tumor regression, and that antigen priming by HSVtk in vivo is required for the effects of sPD1-Ig.
Figure 4.

In vitro and in vivo anticancer activity of Ad5mTR.sPD1. (a) CT26 cells were infected with Ad5MOCK (inverted closed triangle), Ad5mTR (closed triangle), Ad5EF1.sPD1 (closed square), or Ad5mTR.sPD1 (closed circle, closed diamond) at various MOIs together with 100 µmol/l ganciclovir (GCV) (closed triangle, closed circle), or without GCV (closed triangle, closed square, closed diamond). After 3 days, cell cytotoxicity was measured as described for Figure 1b. Data represent the mean ± SD of triplicate assays. (b) BALB/c mice were inoculated subcutaneously in both flanks with CT26 cells (1 × 106). Tumors were treated with 5 × 108 plaque forming units of Ad5MOCK (closed inverted triangle), Ad5EF1.sPD1 (closed square), Ad5mTR (closed triangle), or Ad5mTR.sPD1 (closed circle). GCV (75 mg/kg) was administered intraperitoneally twice a day. Tumor volumes were calculated and plotted at the indicated time points. Data represent the mean ±SEM (n = 10). P, unpaired t-test. GCV, ganciclovir; MOI, multiplicity of infection.
Enhanced suppression of tumor growth by sPD1-Ig is mediated by CD8 T cells
HSVtk enhanced antigen-specific CD8 T-cell priming by DCs (Figure 2b) and sPD1-Ig enhanced antitumor CD8 T cell reactivity in vitro (Figure 3d). These observations suggest that enhanced CD8 T-cell reactivity is responsible for the enhanced antitumor effects of Ad5mTR.sPD1 in vivo. To test this possibility, CT26 tumor–bearing mice were treated with anti-CD8 antibody to deplete the CD8 T-cell population. Enhanced tumor growth suppression by dual-module Ad5mTR.sPD1 was nearly abolished in CD8 T cell–depleted mice (Figure 5a). Of note, the antitumor activity of Ad5mTR was also decreased, suggesting that the effect of Ad5mTR is partially dependent on an antitumor immune responses mediated by CD8 T cells in the CT26 mouse tumor model. However, the extent to which CD8 depletion affected antitumor activity was far greater for Ad5mTR.sPD1 than for Ad5mTR (4.76-fold versus 1.9-fold), underscoring the importance of CD8 T cells in antitumor activity induced by the former versus the latter (Figure 5b).
Figure 5.

Tumor growth suppression by sPD1-Ig is mediated by CD8 T cells. (a) Subcutaneous CT26 tumors in BALB/c mice were injected intratumorally with Ad5MOCK (closed triangle), Ad5mTR (closed square), or Ad5mTR.sPD1 (closed circle) (left panel). To determine the involvement of CD8 T cells in the antitumor activity of sPD1-Ig, subcutaneous CT26 tumors in C57/BL6 mice were injected intratumorally with Ad5MOCK (closed triangle), Ad5mTR (closed square), or Ad5mTR.sPD1 (closed square) (right panel). Two days before virus treatment and every 5 days after treatment, mice were injected intravenously with the 2.43 anti-CD8 antibody (500 µg) to deplete CD8 T cells (right panel). Arrow indicates the time of antibody injection. Tumor growth was calculated and plotted at the indicated time points. The arrows indicate the time when the antibody was injected. (b) Reduction of tumor growth in the presence (closed square) or absence (open square) of 2.43 anti-CD8 antibody was calculated relative to Ad5MOCK on day 12. Data represent the mean ± SEM. P, unpaired t-test. Abs, antibodies.
To examine the role of “tumor-specific ” CD8 T cells more directly, we used the E.G7 tumor model and OVA-specific OT-I T cells. Intratumoral injection of subcutaneous E.G7 tumors in syngeneic B6 mice with Ad5mTR.sPD1 resulted in enhanced tumor regression, similar to that observed in the CT26 model. By contrast, when the same experiment was performed in syngeneic lymphocyte-deficient Rag1−/− mice, the antitumor effect of both viruses was markedly reduced (Figure 6a). The degree of reduction was again far stronger for Ad5mTR.sPD1 than for Ad5mTR (6.58-fold versus 1.61-fold), consistent with the effects of CD8 T-cell depletion in the CT26 model (Figure 6b). When antigen-specific T-cell response was reconstituted by introducing OT-I T cells intravenously into E.G7-bearing Rag1−/− mice, the antitumor effect of intratumoral injection of adenovirus was restored, and the effect was more prominent following injection of Ad5mTR.sPD1 than following injection of Ad5mTR (Figure 6c). Notably, the number of OT-I T cells in the blood was greatly increased in mice treated with Ad5mTR.sPD1 as compared with Ad5mTR (Figure 6d). These results suggest that Ad5mTR.sPD1 enhances tumor regression by directly enhancing tumor-specific CD8 T-cell function.
Figure 6.
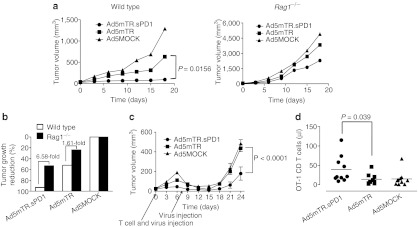
Tumor growth suppression by sPD1-Ig in T and B cell–deficient Rag1−/− mice. (a) Subcutaneous E.G7 tumors in C57/BL6 mice were injected intratumorally with Ad5MOCK (closed triangle), Ad5mTR (closed square), or Ad5mTR.sPD1 (closed circle) (left panel). To measure lymphocyte involvement in the antitumor activity of sPD1-Ig, subcutaneous E.G7 tumors in Rag1−/− mice were injected intratumorally with Ad5MOCK (closed triangle), Ad5mTR (closed square), or Ad5mTR.sPD1 (closed circle) (right panel). Tumor growth was calculated and plotted at the indicated time points. P, unpaired t-test. (b) Reduction of tumor growth in Rag1−/− (closed square) or wild type mice (open square) was calculated relative to Ad5MOCK on day 12. (c) E.G7 tumors (1 × 106 cells) in Rag1−/− mice were injected intratumorally twice at 7 day intervals with 5 × 108 plaque forming units of Ad5MOCK (closed triangle), Ad5mTR (closed square), or Ad5mTR.sPD1 (closed circle). CD8 T cells (5 × 104) from OT-1 mice were injected intravenously at the same time as the first adenovirus injection. Tumor growth was calculated and plotted at the indicated time points. Data represent the mean ± SEM (n = 6). P, unpaired t-test. (d) For ex vivo analysis of immune cells, E.G7 tumors in Rag1−/− mice were cotreated with CD8 T cells from OT-1 mice and the indicated adenovirus. Eight days after the first adenovirus injection, peripheral blood mononuclear cells were harvested from the mouse eye vein. Absolute numbers of OT-1 CD8 T cells were calculated from total numbers of live cells and percentages of TCR Vα2+, Vβ5+, T cells (OT-1 cells) in flow cytometry profile. P, Wilcoxon matched-pairs signed rank test.
Systemic antitumor effects of localized Ad5mTR.sPD1 treatment
There was a systemic increase in tumor-specific T cells in the blood of Ad5mTR.sPD1-treated mice, which raised the possibility that localized injection of virus at the primary tumor site may inhibit secondary tumor growth at a distal site in the same mouse. CT26 tumors in BALB/c mice were treated with Ad5mTR.sPD1 three times at 3-day intervals. Fourteen days after the initial treatment, secondary CT26 tumor challenge was performed in the opposite flank of the same mouse. At this time, there was minimal growth of the primary tumor in Ad5mTR.sPD1-treated mice, whereas Ad5MOCK-treated mice had developed large primary tumors (>600 mm3), which hampered secondary tumor injection in these mice. Therefore, a new cohort of normal BALB/c mice was used as a control group for secondary tumor challenge. Secondary tumors grew well in the control group, but did not grow at all in Ad5mTR.sPD1-treated mice (Figure 7a,c). The same result was observed in the E.G7 tumor model in B6 mice (Figure 7b,c). For the more rigorous comparison, when the secondary tumor was challenged to the mice bearing a small primary tumor for the control group, similar results were obtained (Supplementary Figure S3). Thus, localized injection of a dual-module adenovirus elicits systemic antitumor effects by enhancing antitumor immunity.
Figure 7.

Inhibition of secondary tumors by Ad5mTR.sPD1 treatment. (a) CT26 tumors were implanted subcutaneously in BALB/c mice and then injected intratumorally with 5 × 108 plaque forming units of Ad5mTR.sPD1 three times at 3-day intervals. Two weeks after the initial treatment, the opposite flank was challenged with 1 × 106 tumor cells. Ganciclovir (75 mg/kg) was administered intraperitoneally during virus treatment starting 1 day after the first virus injection and continuing for 12 days. Tumor size was calculated every 3 days. (b) E.G7 tumors were implanted subcutaneously in C57/BL6 mice and then treated as described for (a). (c) On day 7 after the second secondary challenge, tumor size at the challenge site in mice previously treated with Ad5mTR.sPD1 or in untreated mice was measured. Data represent the mean ± SD. Avg, average.
Discussion
As a favored strategy for delivering transgenes into cells in situ, adenoviruses are generally considered both efficient and safe. However, there has been limited success in transferring suicide genes, such as HSVtk, into tumor cells via replication-incompetent adenoviruses due to low rates of infection and the consequent low potency in terms of clinical application.35 Nonetheless, it is now clear that, in addition to direct cytotoxicity, partial tumor cell death induced by adenoviral HSVtk in the infected area can efficiently potentiate antitumor immunity and achieve indirect killing of tumors. Therefore, HSVtk-based adenoviruses could be considered for use as tumor vaccines as well as cytotoxic therapeutics. In addition, adenoviral vectors have the advantage of being able to accommodate multiple genes within a single vehicle, which enables delivery of combination therapeutics as a single agent. The efficacy of HSVtk-based adenoviruses as tumor vaccines could be greatly improved by incorporating other immune modulators within the same vector.
Tumor vaccines aimed at boosting antitumor immunity generally face one major barrier to maximum efficacy; namely, immunoresistance, aimed at avoiding recognition and attack by the immune system. This is most likely one of the main reasons why numerous attempts to develop efficient therapeutic vaccines have failed. The role of the PD-L1/PD1 signaling axis in tumor protection has been well demonstrated in several studies, making it a good target for disrupting tumor immunoresistance. PD1 knockout mice exhibit spontaneous autoimmune responses due to enhanced T-cell reactivity.36,37 Consistent with this, implanted tumors are efficiently rejected by PD1 knockout mice.25 Likewise, anti-PD-L1 antibody treatment of tumor-bearing mice efficiently abrogates tumor grafts.25,27 However, data from PD1 knockout mice also indicate that systemic blockade of the PD1/PD-L1 axis may induce adverse autoimmune side effects. In this sense, localized disruption of PD-L1 may be more advantageous in terms of safety.
The concept of soluble PD1 as a neutralizing agent for PD-L1 has been demonstrated by several groups.38,39 However, these studies used primarily plasmid DNA to express sPD1 systemically in an effort to augment other therapeutic DNA or adenoviral antigenic vaccines. Here, we demonstrated an alternative method which delivered sPD1 directly into the tumor microenvironment using an adenoviral vector containing HSVtk. In this system, tumor cells infected with the virus release tumor antigens on HSVtk-mediated cell death. These antigens are then processed by local DCs, which then efficiently prime tumor antigen–specific cytotoxic T cells. Once primed, antitumor cytotoxic T cells infiltrate the tumor tissue and encounter virus-infected tumor cells expressing sPD1. Within the tumor microenvironment, sPD1 can enhance the T-cell antitumor reactivity with minimal side effects.
In the current study, this type of dual-module adenovirus harboring HSVtk and sPD1-Ig efficiently suppressed established tumor growth in two different murine tumor models. This effect was mediated by tumor-specific CD8 T cells, as demonstrated by both CD8 T-cell depletion experiments (loss-of-function experiments) and tumor-specific T-cell reconstitution experiments (gain-of-function experiments). Although previous studies reported the systemic T-cell activation during HSVtk/GCV treatment,40,41 to our knowledge, this is the first demonstration that CD8 T cells specific to “a defined tumor antigen ” can directly mediate the antitumor effects of HSVtk-based adenoviral therapy.
Upon CD8 T-cell depletion, or in Rag1−/− mice, the observation that treatment with Ad5mTR.sPD1 still resulted in mildly enhanced antitumor activity was intriguing. This suggests that although CD8 T cells play a major role in the antitumor activity of sPD1, other immune mechanisms may also be involved. sPD1 may have a direct effect on other immune cells, such as natural killer cells,42 or, alternatively, antibody-dependent cell cytotoxicity mediated by the Fc portion of immunoglobulin fused to sPD1 may play a role in this phenomenon.43
A number of previous studies demonstrate the synergistic effect of adenovirus-mediated expression of HSVtk and other cancer therapies such as radiation, surgery, and some chemotherapeutic agents.44 Such synergistic effects resulted from an increased level of phosphorylated GCV incorporated into the DNA during DNA-repair processes, leading to enhanced cell death. This raises the possibility that Ad5mTR.sPD1 could also be combined with standard therapy, because it is likely to potentiate tumor-infiltrating immune cells, which are primed by surgery or radiation-induced antigen release and inflammation. Alternatively, AdmTR.sPD1 could be modified to attract or activate antigen presenting cells via incorporation of a third immune modulator, such as FMS-like tyrosine kinase 3 ligand or granulocyte-macrophage-colony stimulating factor. This approach may provide an additional DC “boost ” in our immune activation axis of Ad5mTR.sPD1 including tumor cell lysis, dendritic cell priming, and T-cell activation. In fact, a treatment based on an HSVtk adenoviral vector in conjugation with a FMS-like tyrosine kinase 3 ligand-expressing adenovirus is very close to entering phase I clinical trials.10 Thus, adenoviral vectors harboring HSVtk, FMS-like tyrosine kinase 3 ligand, and sPD1 within a single vector may soon be realized as the next-generation of adenoviral tumor vaccines. This avenue is currently under active investigation.
For clinical applications, although adenoviral gene therapy has generally been considered a localized therapeutic, our results suggest that Ad5mTR.sPD1 could be used to treat tumors with multiple metastatic foci. As suggested by Figure 7, treatment of a primary tumor, even a sizeable one, with Ad5mTR.sPD1 may elicit a systemic antitumor response to other small metastatic foci. We propose that the dual-module adenovirus described herein can both enhance antigen priming and overcome tumor immune resistance. It, therefore, represents a promising strategy for strengthening the clinical applications of HSVtk-based gene therapy.
Materials and Methods
Cells and mice. All cells were cultured in RPMI supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin. Six-week-old female BALB/c and C57/BL6 mice were purchased from SLC (Japan). OT-1 (B6 background) and Rag1−/− mice (B6 background) were from the Jackson laboratory (Bar Harbor, ME). All animal experiments were performed in accordance with the Guidelines for the Care and Use of Laboratory Animals of the National Cancer Center (Korea).
Adenovirus construction. The AdenoZAPTM and Adeno QuickTM systems (OD260; Boise, ID) were used to generate Ad5mTR harboring mouse TERT-TR-HSVtk under the control of the CMV promoter (CMV.mTR.HSVtk). A DNA fragment containing CMV.mTR.HSVtk was obtained by digesting pAVQ-CMV-mTERT AS100 Rib (+67) TK31 with SpeI/SacII and then inserted the resulting fragment into the SpeI/EcoRV sites of pZAP1.1 (OD260) to generate pZAP1.1.CMV.mTR.HSVtk. pZAP1.1.CMV.mTR.HSVtk was digested with PacI/DraIII, ligated with RightZAP1.2, and then transfected into HEK293 cells. To generate sPD1-Ig, the extracellular domain of PD-1 was amplified by PCR using the primers: 5′-CCG CTC GAG CTC ACC ATG TGG GTC CGG CAG GTA CCC TGG-3′ and 5′-AGA TCT TCC TCC TCC TCC TTG AAA CCG GCC TTC TGG TTT GGG-3′. The amplified product was inserted into the XhoI/BglII sites of pFUSE-mIgG2A.Fc1 (Invitrogen, San Diego, CA) to generate pFUSE-mIgG2A.Fc1.EF1α.sPD-1. EF1.sPD1-Ig from pFUSE-mIgG2A.Fc1.EF1.sPD1-Ig was subcloned into the EcoRI/SwaI sites of pE3.1 (OD260) to generate pE3.1.EF1.sPD1-Ig. The CMV.mTR.HSVtk fragment of pZAP1.1.CMV.mTR.HSVtk was ligated into the BamHI/SpeI sites of pE1.2 (OD260) to generate pE1.2.CMV.mTR.HSVtk. To generate Ad5mTR.sPD1, pE1.2.CMV.mTR.HSVtk and pE3.1.EF1.sPD1-Ig were digested with DraIII/PflMI, ligated with AdenoQuick13.1, and then transfected into HEK293 cells. For Ad5EF1.sPD1, pE3.1.EF1.sPD1-Ig and pE1.2 empty vector were digested with DraIII/PflMI and then ligated with AdenoQuick13.1, followed by transfection into HEK293 cells as described for Ad5mTR.sPD1. Construction of Ad5MOCK was performed as previously described.1,2
In vitro GCV uptake and cell cytotoxicity assay. HSVtk enzyme activity was determined by measuring accumulated phosphorylated GCV in cells.1 A cell proliferation assay (Dojindo Laboratories, Rockville, MD) was performed to evaluate adenovirus cytotoxicity using the standard protocol, with some modifications. Briefly, cells (3 × 103) were seeded in 96-well plates and incubated overnight at 37 °C. Cells were exposed to adenovirus at the indicated MOI. One day later, GCV was added to a final concentration of 100 µmol/l and then cell proliferation was measured on day 3. All experiments were performed in triplicate.
Antibodies and reagents. To confirm sPD1-Ig protein expression from Ad5CMV.mTR.sPD1, HEK293 cells (4 × 105) were infected with adenovirus at an MOI of 10. Twenty-four hour postinfection, culture supernatants and cell lysates (80 µg) were analyzed by immunoblot using an anti-Pdcd-1 antibody (Santa Cruz Biotechnology, Santa Cruz, CA). The concentration of sPD1-Ig in the culture supernatant was at least 580 ng/ml, which was calculated from the purified sPD1-Ig using protein G affinity purification. The yield was 5.23 µg out of 9 ml of culture supernatant. For CD8 T-cell depletion, 500 µg of 2.43 anti-CD8 antibody was injected intraperitoneally 2 days before adenovirus treatment and then every 5 days thereafter for 15 days.
CD8 T cell/DC coculture assay for IFN-γ production. Draining lymph node–derived DCs were enriched using a 17.5% Nycodenze gradient and then further purified on a MACS column using anti-CD11c microbeads (Miltenyi Biotec, Auburn, CA). Naive CD8 T cells from OT-1 mice were isolated using anti-CD8 microbeads. Lymph node–derived DCs (5 × 104) were cultured together with naive CD8 T cells (1 × 105) in 96 well-plates for 72 hours. Culture supernatants were collected after 72 hours and the amount of IFN-γ was measured by ELISA (eBioscience, San Diego, CA).
In vitro OT-1 T-cell activation assay. MC38/OVA cells, which are murine colorectal cancer cells (MC38, B6 background) stably expressing OVA, were prepared on 6-well plates. sPD1-Ig was obtained from culture supernatants of HEK293 cells infected with adenovirus at an MOI of 10 for 24 hours and then incubated with MC38/OVA cells (1 × 104) plus OT-1 CD8 T lymphocytes (1 × 106) for 72 hours. Naive CD8+ T lymphocytes from OT-1 mice were isolated using a MACS column as described above. IFN-γ produced by CD8 T lymphocytes was measured using a mouse IFN-γ CBA assay (BD bioscience, San Jose, CA).
In vivo animal studies and ex vivo analysis. Female BALB/c mice (6-weeks-old) were inoculated subcutaneously with 1 × 106 CT26 cells. When tumors were palpable (around day 7), 5 × 108 plaque forming units of adenovirus were injected intratumorally and GCV (75 mg/kg) was administered intravenously twice a day for 14 days. The adenovirus was administered twice 7 days apart, unless indicated otherwise. Tumor volume was determined using the following formula: length × width2 × 0.5236. Similar procedures for subcutaneous tumor formation and virus injection were performed in the CD8 T cell–depletion experiment. E.G7 cells (C57/BL6 background; 1 × 106) were injected into Rag1−/− or C57/BL6 mice, and adenovirus and GCV treatment was performed as described for CT26 cells. For ex vivo analysis, E.G7 tumors in Rag1−/− mice were treated with Ad5mTR.sPD1 along with intravenous infusion of CD8 T cells isolated from an OT-1 mouse. Eight-day postinfection, peripheral blood mononuclear cells were harvested from the blood and analyzed by flow cytometry.
SUPPLEMENTARY MATERIAL Figure S1. Schematic diagram of the mouse TERT-targeting trans-splicing ribozyme (mTERT-TR)-HSVtk construct. Figure S2. PD-L1 expression on the surface of mouse cancer cells. Figure S3. Inhibition of secondary tumors by Ad5mTR.sPD1 treatment.
Acknowledgments
This work was supported by grants from the Innovative Research Institute for Cell Therapy (A062260) and the Global Core Research Center grant (No.20110030678)), Republic of Korea.
Supplementary Material
REFERENCES
- Hong SH, Jeong JS, Lee YJ, Jung HI, Cho KS, Kim CM.et al. (2008In vivo reprogramming of hTERT by trans-splicing ribozyme to target tumor cells Mol Ther 1674–80. [DOI] [PubMed] [Google Scholar]
- Jeong JS, Lee SW, Hong SH, Lee YJ, Jung HI, Cho KS.et al. (2008Antitumor effects of systemically delivered adenovirus harboring trans-splicing ribozyme in intrahepatic colon cancer mouse model Clin Cancer Res 14281–290. [DOI] [PubMed] [Google Scholar]
- Abate-Daga D, Garcia-Rodríguez L, Sumoy L., and, Fillat C. Cell cycle control pathways act as conditioning factors for TK/GCV sensitivity in pancreatic cancer cells. Biochim Biophys Acta. 2010;1803:1175–1185. doi: 10.1016/j.bbamcr.2010.06.009. [DOI] [PubMed] [Google Scholar]
- Chen SH, Shine HD, Goodman JC, Grossman RG., and, Woo SL. Gene therapy for brain tumors: regression of experimental gliomas by adenovirus-mediated gene transfer in vivo. Proc Natl Acad Sci USA. 1994;91:3054–3057. doi: 10.1073/pnas.91.8.3054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smythe WR, Hwang HC, Amin KM, Eck SL, Davidson BL, Wilson JM.et al. (1994Use of recombinant adenovirus to transfer the herpes simplex virus thymidine kinase (HSVtk) gene to thoracic neoplasms: an effective in vitro drug sensitization system Cancer Res 542055–2059. [PubMed] [Google Scholar]
- Tomicic MT, Thust R., and, Kaina B. Ganciclovir-induced apoptosis in HSV-1 thymidine kinase expressing cells: critical role of DNA breaks, Bcl-2 decline and caspase-9 activation. Oncogene. 2002;21:2141–2153. doi: 10.1038/sj.onc.1205280. [DOI] [PubMed] [Google Scholar]
- Barba D, Hardin J, Sadelain M., and, Gage FH. Development of anti-tumor immunity following thymidine kinase-mediated killing of experimental brain tumors. Proc Natl Acad Sci USA. 1994;91:4348–4352. doi: 10.1073/pnas.91.10.4348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuriyama S, Kikukawa M, Masui K, Okuda H, Nakatani T, Akahane T.et al. (1999Cancer gene therapy with HSV-tk/GCV system depends on T-cell-mediated immune responses and causes apoptotic death of tumor cells in vivo Int J Cancer 83374–380. [DOI] [PubMed] [Google Scholar]
- Okada T, Shah M, Higginbotham JN, Li Q, Wildner O, Walbridge S.et al. (2001AV.TK-mediated killing of subcutaneous tumors in situ results in effective immunization against established secondary intracranial tumor deposits Gene Ther 81315–1322. [DOI] [PubMed] [Google Scholar]
- Ghulam Muhammad AK, Candolfi M, King GD, Yagiz K, Foulad D, Mineharu Y.et al. (2009Antiglioma immunological memory in response to conditional cytotoxic/immune-stimulatory gene therapy: humoral and cellular immunity lead to tumor regression Clin Cancer Res 156113–6127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King GD, Muhammad AK, Larocque D, Kelson KR, Xiong W, Liu C.et al. (2011Combined Flt3L/TK gene therapy induces immunological surveillance which mediates an immune response against a surrogate brain tumor neoantigen Mol Ther 191793–1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brockstedt DG, Diagana M, Zhang Y, Tran K, Belmar N, Meier M.et al. (2002Development of anti-tumor immunity against a non-immunogenic mammary carcinoma through in vivo somatic GM-CSF, IL-2, and HSVtk combination gene therapy Mol Ther 6627–636. [PubMed] [Google Scholar]
- Rabinovich GA, Gabrilovich D., and, Sotomayor EM. Immunosuppressive strategies that are mediated by tumor cells. Annu Rev Immunol. 2007;25:267–296. doi: 10.1146/annurev.immunol.25.022106.141609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li MO., and, Flavell RA. TGF-beta: a master of all T cell trades. Cell. 2008;134:392–404. doi: 10.1016/j.cell.2008.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phan GQ, Yang JC, Sherry RM, Hwu P, Topalian SL, Schwartzentruber DJ.et al. (2003Cancer regression and autoimmunity induced by cytotoxic T lymphocyte-associated antigen 4 blockade in patients with metastatic melanoma Proc Natl Acad Sci USA 1008372–8377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cromme FV, Airey J, Heemels MT, Ploegh HL, Keating PJ, Stern PL.et al. (1994Loss of transporter protein, encoded by the TAP-1 gene, is highly correlated with loss of HLA expression in cervical carcinomas J Exp Med 179335–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenwald RJ, Freeman GJ., and, Sharpe AH. The B7 family revisited. Annu Rev Immunol. 2005;23:515–548. doi: 10.1146/annurev.immunol.23.021704.115611. [DOI] [PubMed] [Google Scholar]
- Zou W., and, Chen L. Inhibitory B7-family molecules in the tumour microenvironment. Nat Rev Immunol. 2008;8:467–477. doi: 10.1038/nri2326. [DOI] [PubMed] [Google Scholar]
- Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB.et al. (2002Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion Nat Med 8793–800. [DOI] [PubMed] [Google Scholar]
- Ishida Y, Agata Y, Shibahara K., and, Honjo T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. 1992;11:3887–3895. doi: 10.1002/j.1460-2075.1992.tb05481.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zha Y, Blank C., and, Gajewski TF. Negative regulation of T-cell function by PD-1. Crit Rev Immunol. 2004;24:229–237. doi: 10.1615/critrevimmunol.v24.i4.10. [DOI] [PubMed] [Google Scholar]
- Keir ME, Butte MJ, Freeman GJ., and, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol. 2008;26:677–704. doi: 10.1146/annurev.immunol.26.021607.090331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topalian SL, Drake CG., and, Pardoll DM. Targeting the PD-1/B7-H1(PD-L1) pathway to activate anti-tumor immunity. Curr Opin Immunol. 2012;24:207–212. doi: 10.1016/j.coi.2011.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmadzadeh M, Johnson LA, Heemskerk B, Wunderlich JR, Dudley ME, White DE.et al. (2009Tumor antigen-specific CD8 T cells infiltrating the tumor express high levels of PD-1 and are functionally impaired Blood 1141537–1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwai Y, Ishida M, Tanaka Y, Okazaki T, Honjo T., and, Minato N. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc Natl Acad Sci USA. 2002;99:12293–12297. doi: 10.1073/pnas.192461099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strome SE, Dong H, Tamura H, Voss SG, Flies DB, Tamada K.et al. (2003B7-H1 blockade augments adoptive T-cell immunotherapy for squamous cell carcinoma Cancer Res 636501–6505. [PubMed] [Google Scholar]
- Nomi T, Sho M, Akahori T, Hamada K, Kubo A, Kanehiro H.et al. (2007Clinical significance and therapeutic potential of the programmed death-1 ligand/programmed death-1 pathway in human pancreatic cancer Clin Cancer Res 132151–2157. [DOI] [PubMed] [Google Scholar]
- Brahmer JR, Drake CG, Wollner I, Powderly JD, Picus J, Sharfman WH.et al. (2010Phase I study of single-agent anti-programmed death-1 (MDX-1106) in refractory solid tumors: safety, clinical activity, pharmacodynamics, and immunologic correlates J Clin Oncol 283167–3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attia P, Phan GQ, Maker AV, Robinson MR, Quezado MM, Yang JC.et al. (2005Autoimmunity correlates with tumor regression in patients with metastatic melanoma treated with anti-cytotoxic T-lymphocyte antigen-4 J Clin Oncol 236043–6053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck KE, Blansfield JA, Tran KQ, Feldman AL, Hughes MS, Royal RE.et al. (2006Enterocolitis in patients with cancer after antibody blockade of cytotoxic T-lymphocyte-associated antigen 4 J Clin Oncol 242283–2289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ban G, Song MS., and, Lee SW. Cancer cell targeting with mouse TERT-specific group I intron of Tetrahymena thermophila. J Microbiol Biotechnol. 2009;19:1070–1076. doi: 10.4014/jmb.0812.692. [DOI] [PubMed] [Google Scholar]
- Gamrekelashvili J, Krüger C, von Wasielewski R, Hoffmann M, Huster KM, Busch DH.et al. (2007Necrotic tumor cell death in vivo impairs tumor-specific immune responses J Immunol 1781573–1580. [DOI] [PubMed] [Google Scholar]
- Iida N, Nakamoto Y, Baba T, Kakinoki K, Li YY, Wu Y.et al. (2008Tumor cell apoptosis induces tumor-specific immunity in a CC chemokine receptor 1- and 5-dependent manner in mice J Leukoc Biol 841001–1010. [DOI] [PubMed] [Google Scholar]
- Lee SJ.et al. (2012Over-expression of miR-145 enhances the effectiveness of HSVtk gene therapy for malignant glioma Cancer Lett 32072–80. [DOI] [PubMed] [Google Scholar]
- McCormick F. Cancer gene therapy: fringe or cutting edge. Nat Rev Cancer. 2001;1:130–141. doi: 10.1038/35101008. [DOI] [PubMed] [Google Scholar]
- Nishimura H, Nose M, Hiai H, Minato N., and, Honjo T. Development of lupus-like autoimmune diseases by disruption of the PD-1 gene encoding an ITIM motif-carrying immunoreceptor. Immunity. 1999;11:141–151. doi: 10.1016/s1074-7613(00)80089-8. [DOI] [PubMed] [Google Scholar]
- Okazaki T, Tanaka Y, Nishio R, Mitsuiye T, Mizoguchi A, Wang J.et al. (2003Autoantibodies against cardiac troponin I are responsible for dilated cardiomyopathy in PD-1-deficient mice Nat Med 91477–1483. [DOI] [PubMed] [Google Scholar]
- He YF, Zhang GM, Wang XH, Zhang H, Yuan Y, Li D.et al. (2004Blocking programmed death-1 ligand-PD-1 interactions by local gene therapy results in enhancement of antitumor effect of secondary lymphoid tissue chemokine J Immunol 1734919–4928. [DOI] [PubMed] [Google Scholar]
- Song MY, Park SH, Nam HJ, Choi DH., and, Sung YC. Enhancement of vaccine-induced primary and memory CD8(+) T-cell responses by soluble PD-1. J Immunother. 2011;34:297–306. doi: 10.1097/CJI.0b013e318210ed0e. [DOI] [PubMed] [Google Scholar]
- Fridlender ZG, Sun J, Singhal S, Kapoor V, Cheng G, Suzuki E.et al. (2010Chemotherapy delivered after viral immunogene therapy augments antitumor efficacy via multiple immune-mediated mechanisms Mol Ther 181947–1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miles BJ, Shalev M, Aguilar-Cordova E, Timme TL, Lee HM, Yang G.et al. (2001Prostate-specific antigen response and systemic T cell activation after in situ gene therapy in prostate cancer patients failing radiotherapy Hum Gene Ther 121955–1967. [DOI] [PubMed] [Google Scholar]
- Benson DM, Jr, Bakan CE, Mishra A, Hofmeister CC, Efebera Y, Becknell B.et al. (2010The PD-1/PD-L1 axis modulates the natural killer cell versus multiple myeloma effect: a therapeutic target for CT-011, a novel monoclonal anti-PD-1 antibody Blood 1162286–2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiner LM, Surana R., and, Wang S. Monoclonal antibodies: versatile platforms for cancer immunotherapy. Nat Rev Immunol. 2010;10:317–327. doi: 10.1038/nri2744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguilar LK, Guzik BW., and, Aguilar-Cordova E. Cytotoxic immunotherapy strategies for cancer: mechanisms and clinical development. J Cell Biochem. 2011;112:1969–1977. doi: 10.1002/jcb.23126. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.