

Abstract
Glioblastoma multiforme (GBM) remains an untreatable human brain malignancy. Despite promising preclinical studies using oncolytic herpes simplex virus (oHSV) vectors, efficacy in patients has been limited by inefficient virus replication in tumor cells. This disappointing outcome can be attributed in part to attenuating mutations engineered into these viruses to prevent replication in normal cells. Alternatively, retargeting of fully replication-competent HSV to tumor-associated receptors has the potential to achieve tumor specificity without impairment of oncolytic activity. Here, we report the establishment of an HSV retargeting system that relies on the combination of two engineered viral glycoproteins, gD and gB, to mediate highly efficient HSV infection exclusively through recognition of the abundantly expressed epidermal growth factor receptor (EGFR) on glioblastoma cells. We demonstrate efficacy in vitro and in a heterotopic tumor model in mice. Evidence for systemically administered virus homing to the tumor mass is presented. Treatment of orthotopic primary human GBM xenografts demonstrated prolonged survival with up to 73% of animals showing a complete response as confirmed by magnetic resonance imaging. Our study describes an approach to HSV retargeting that is effective in a glioma model and may be applicable to the treatment of a broad range of tumor types.
Introduction
Oncolytic viruses are lytic viruses engineered to replicate preferentially in tumor cells over normal cells.1,2,3,4 Oncolytic viruses based on herpes simplex virus (oHSV) have shown promise in the treatment of preclinical models of glioblastoma,5 but the mutations that achieve virus attenuation in normal cells, for example in genes that overcome innate immune mechanisms, also compromise efficient lytic replication in tumors.6 Moreover, it has been reported that some primary human glioblastomas are relatively resistant to HSV infection due to low expression of the main viral entry receptor on neuronal cells, nectin-1.7,8 Consistent with these observations, treatment efficacy in patient trials with attenuated oHSV has been disappointing although safety has been demonstrated.9,10,11,12 These results indicate a need for alternative approaches that will allow unimpaired virus replication in tumor cells without endangering normal cells. An attractive strategy is to retarget virus infection to tumor markers on the cell surface, such as overexpressed growth factor receptors. This strategy serves to limit infection to tumor cells while leaving intact viral functions that maximize virus replication and thus oncolytic activity.
HSV entry into cells requires the coordinated activities of four envelope glycoproteins, gB, gD, gH, and gL.13 After virion adsorption to the cells, mainly by the binding of gB and gC to nearly ubiquitous heparan sulfate moieties of cell surface proteoglycans, gD interacts with a specific receptor, such as herpes virus entry mediator (HVEM or HveA), nectin-1 (HveC), or 3-O sulfated heparan sulfate (3-OS HS).14,15,16 Receptor binding causes a conformational change in gD that triggers fusion of viral and cellular membranes by activation of gB and the gH/gL heterodimer.13 HSV retargeting requires elimination of the natural receptor-binding activities of gD and introduction of a recognition element for the target receptor in such a manner that the recombinant retargeted gD molecule retains the ability to efficiently activate the fusion mechanism upon binding to the novel receptor. Studies using IL-13 or an anti-HER2 single-chain antibody (scFv) as receptor recognition elements have indicated the feasibility of this general strategy.17,18,19
Here, we describe the establishment of novel retargeted HSV vectors through the introduction of scFv antibodies specific for either the human epidermal growth factor receptor (EGFR) or human carcinoembryonic antigen (CEA) into a detargeted mutant form of gD. Our analysis showed exclusive virus entry via either of these two targeted receptors without off-target infection. However, retargeted infectivity required high virus doses, suggesting that gD retargeting alone would not be adequate for use in oncolytic tumor therapy. To overcome this inefficiency, we combined gD retargeting with a pair of entry-accelerating mutations in gB (D285N/A549T, referred to here as NT) that lower the threshold for activation of the virus fusion mechanism.20 This modification increased the efficiency of retargeted virus entry as much as 100-fold, achieving infectivity comparable to that of wild-type (wt) virus via the standard entry receptors. Using the CEA-retargeted virus as a negative control, the EGFR-retargeted virus proved to be effective in an orthotopic mouse model of primary human glioma, engendering complete responses in up to 73% of treated animals. Our observations suggest that the retargeting strategy used in this study will prove useful for further clinical development of oHSV.
Results
Structure of retargeted gD
To detarget gD from its natural receptors, we deleted residues 2-24 in the HVEM-binding N-terminal region and introduced a single amino acid substitution, Y38C, to ablate responsiveness to nectin-1.21,22 For retargeting, we inserted an scFv directed against EGFR (scEGFR) or CEA (scCEA) at the position of the 2-24 deletion (Supplementary Figure S1). Retargeted gB:NT and wt gB (gB:wt) recombinant viruses were established by homologous recombination of gB:NT or gB:wt gD-null viruses with the scEGFR- or scCEA-containing gD constructs in gD-complementing VD60 cells. Recombinants were plaque-purified by multiple rounds of limiting dilution and finally passaged through non-complementing Vero cells to obtain virus preparations free of wt gD protein. Retargeted viruses containing the gB:NT allele were named KNE (retargeted to EGFR) and KNC (retargeted to CEA); their counterparts containing the wt gB gene are referred to as KE and KC. Absolute virus titers expressed as genome copies (gc)/ml were determined by real-time quantitative PCR (qPCR) for the viral immediate early gene ICP47, as described.23 The standardization of titers based on gc numbers allowed comparison of entry efficiencies between viruses that recognize different receptors.
Entry efficiency, specificity, and kinetics of retargeted recombinant viruses
Initial cell lines tested for virus entry included J1.1-2 cells that are resistant to HSV due to the absence of gD receptors,24 derivatives expressing human HVEM (J/A), nectin-1 (J/C) or EGFR (J/EGFR), and HSV-susceptible cells that endogenously express simian EGFR (Vero)25 or human CEA (MKN45).26 Virus entry was detected by immunostaining for the immediate early HSV protein ICP4. As shown in Figure 1a, entry of the EGFR-retargeted gB:NT recombinant virus (KNE) into J/EGFR cells was as efficient as entry of the parental wt HSV-1 strain KOS into J/A or J/C cells expressing typical HSV receptors. In contrast, the retargeted gB:wt virus (KE) entered J/EGFR cells approximately 100-fold less efficiently. Neither of the retargeted viruses detectably entered J1.1-2, J/A, or J/C cells even at high virus input (1,000 gc/cell), demonstrating that the detargeting mutations in the retargeted gD constructs were effective in abolishing virus entry via the natural HSV receptors. Consistent with these results, KNE entered EGFR-bearing Vero cells with an efficiency similar to wt KOS virus while KE showed very limited entry only at high virus input (Figure 1b). We confirmed that KNE entry into Vero cells was EGFR dependent by pretreating the cells with anti-EGFR monoclonal antibody, resulting in a dose-dependent inhibition of entry without effect on entry by wt KOS virus (Figure 1c). Together, these results indicated that the alternate receptor use of the gB:NT-enhanced EGFR-retargeted virus was highly efficient and selective.
Figure 1.
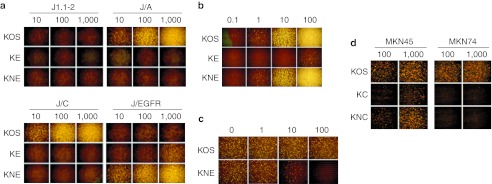
Entry efficiency and specificity of recombinant retargeted viruses. (a,d) Cells listed above the panels or (b) Vero cells were infected for 6 hours with the viruses shown to the left at the multiplicities in gc/cell shown above the columns and immunostained for ICP4. (c) Inhibition of retargeted virus entry by pretreatment of Vero cells with the amounts of 528 anti-EGFR monoclonal antibody (µg/ml) shown above the columns. Pretreated cells were incubated with KOS or KNE at 10 gc/cell for 2 hours, extracellular virus was inactivated, and the cells were stained for ICP4 at 6 hpi. EGFR, epidermal growth factor receptor; gc, genome copy.
We tested the gD-scCEA recombinant viruses (KC and KNC) on CEA-positive MKN45 and CEA-negative MKN74 human gastric carcinoma cells.26 As illustrated in Figure 1d, KOS entered both cell lines, consistent with previous observations,27 but entry of the scCEA viruses was limited to MKN45 cells. The gB:NT allele enhanced CEA-retargeted virus entry into MKN45 cells although less than observed with the EGFR-retargeted viruses on EGFR-bearing cells. These results with two different retargeting ligands, scEGFR and scCEA, along with similar results using EGF as a targeting ligand (data not shown), support the versatility of our gB:NT-enhanced retargeting system. In the remainder of this study, we limited our experiments to the EGFR-retargeted viruses and used KNC as a negative control in a glioblastoma treatment experiment.
We previously showed that the gB:NT allele accelerates normal HSV entry20 and thus we asked whether this mechanism also contributes to the enhancing effect of the gB:NT allele on retargeted virus entry. Vero-derived gD-complementing VD60 cells were infected for 0–60 minutes with KOS, KNE or KE, extracellular virus was inactivated, and plaques were counted 2 days later; we used VD60 cells to minimize differences in gD-dependent lateral virus spread and thus the rate of plaque formation following initial entry. Wt KOS virus entered VD60 cells gradually over 60 minutes while the gB:wt version of the retargeted virus entered at a highly reduced rate (Figure 2). However, the gB:NT version entered VD60 cells more rapidly not only than the gB:wt version, but also than KOS, reaching a plateau at ~20 minutes (Figure 2). These results demonstrated that the gB:NT allele dramatically increases the kinetics of virus entry via a novel receptor.
Figure 2.

Entry kinetics of retargeted viruses. VD60 cells were incubated at 4 °C for 30 minutes with 20,000 gc of the indicated viruses, washed thoroughly, incubated at 37 °C for 0–60 minutes, and treated with acidic buffer to inactivate extracellular virus. Media with methylcellulose was added and plaques were counted 2 days later. Values are the means ± SD of three replicates. gc, genome copy.
Infection and killing of human tumor lines
We compared the entry and cell-killing abilities of the two EGFR-retargeted viruses with those of wt KOS on a panel of HSV-permissive human tumor lines known to express EGFR, including lung carcinoma A549, colon carcinoma HT29, pancreatic carcinoma BxPC3, glioblastomas U87 and SNB19, and epidermoid carcinoma A431. As shown in Figure 3a, inoculation at 10 gc/cell yielded readily detectable entry of KNE, but only minimal entry of the gB:wt version, KE. KOS virus entered each of the cell lines at a similar level as KNE, as observed earlier with Vero cells (Figure 1b). We pretreated HT29 cells with anti-EGFR monoclonal antibody and observed dose-dependent inhibition of entry of KNE, but not of KOS (Figure 3b), confirming the receptor specificity of the retargeted virus on HSV-susceptible human cells.
Figure 3.

Retargeted virus entry and killing of established human tumor lines in culture. (a) Cells listed above the panels were infected for 6 hours at 10 gc/cell and immunostained for ICP4. (b) EGFR dependence of retargeted virus entry. HT29 cells were preincubated with the amounts of anti-EGFR monoclonal antibody 528 in µg/ml indicated above the panels before infection with KNE or KOS at 10 gc/cell for 2 hours. Extracellular virus was inactivated and the cells were stained for ICP4 at 16 hpi. (c) A549 or U87 cells were infected at 0.01–10 gc/cell for 3 days (upper panels) or 6 days (lower panels) and percent cell viability relative to uninfected cells was determined by MTT assay. Bars represent the means ± SD of six replicates. Significant differences between pairs of viruses under the same conditions (P < 0.05, unpaired Student's t-test) are indicated by brackets. EGFR, epidermal growth factor receptor; gc, genome copy; MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide.
To assess the oncolytic potential of these retargeted gB:wt and gB:NT viruses, A549 and U87 cells were infected with increasing amounts of the viruses and cell viability was determined by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay at 3 days (Figure 3c, top) and 6 days (Figure 3c, bottom) postinfection. At 10 gc/cell, KNE showed efficient cell killing comparable to that of KOS. However, at lower virus input, A549 cells were killed less efficiently by KNE than by KOS, an observation that remains to be further explored. At the highest multiplicity of infection, KE showed significantly less killing of both cell lines than KOS or KNE although at the lower multiplicity of infections the difference with KNE in particular was not uniformly pronounced. Based on the cumulative results to this point, we selected KNE for specificity and antitumor experiments in vivo.
Specificity and oncolytic potency of KNE in vivo
While wt HSV-1 KOS is neurotoxic in mouse strains such as BALB/c, the scFv used in our EGFR-retargeted viruses is specific for human EGFR.28 Thus, we performed neurotoxicity testing in mice as a stringent measure of the specificity of KNE. Groups of four nude mice were injected intracranially with 5 × 103 gc KOS or a 100,000-fold higher dose of KNE (5 × 108 gc). Of the animals injected with KOS virus, one died on day 6, two on day 7, and one on day 9. In contrast, all four mice injected with the retargeted virus remained alive and symptom-free throughout the 47-day observation period (Figure 4a). In a separate experiment, we analyzed brain sections of injected mice for the presence of virus by immunostaining for the viral ICP4 protein. The representative images of Figure 4b illustrate the abundant presence of virus in the brain of a mouse that had died on day 21 after receiving KOS at a dose of 1 × 103 gc, while little evidence of virus replication was detected in brain sections from a mouse that had been sacrificed on day 37 (no symptoms) after injection of 5 × 108 gc of retargeted virus. Virus stocks for these experiments were the same as those used in Figure 1a, arguing against the possibility that the observed differences in neurotoxicity between the two viruses were due to dosing differences. These results confirmed that KNE was effectively detargeted from the normal HSV entry receptors in mouse brain and was harmless in this complex in vivo environment lacking the targeted human receptor.
Figure 4.

Retargeted virus specificity and efficacy in vivo. (a) Toxicity in mouse brain. Nude mice were injected intracranially with 5 × 103 gc of KOS or 5 × 108 gc of KNE and animal survival was recorded. (b) Nude mice were injected intracranially with 1 × 103 gc of KOS (left) or 5 × 108 gc of KNE (right). Brain sections were prepared following death (day 21, left) or sacrifice (day 37, right) and stained with anti-ICP4 antibody and Cy3-labeled secondary antibody. gc, genome copy.
We also determined whether KNE would preferentially accumulate in EGFR-positive human tumors in nude mice. Following the establishment of subcutaneous U87 flank tumors (700–1,000 mm3), equal gc of KOS and KNE were administered by tail-vein injection. The animals were sacrificed 2 days later and the biodistribution of each virus in various organs was determined by qPCR for the viral ICP47 gene;23 10-fold serial dilutions of HSV viral DNA of known concentration were used to generate a standard curve (Figure 5, inset). As shown in Figure 5, the number of KOS genomes per 100 ng tissue DNA was comparable to or lower in the tumors than in most of the other tissues from the same animals. In contrast, the retargeted virus was detected at 100–1,000-fold higher levels in the tumors than in other tissues. These results indicated that the retargeted virus preferentially homed to the human tumor tissue.
Figure 5.

Vector biodistribution. U87 flank tumors in nude mice were allowed to grow to 700–1,000 mm3 in size, and 2 × 109 gc of KOS (white and light-gray bars for animals #1 and 2, respectively) or KNE (black and dark-gray bars) were administered by tail-vein injection (2 animals/virus). All animals were sacrificed 2 days later, and tumors and various organs were collected for DNA isolation and qPCR for the viral ICP47 gene. Virus load in the different tissues was calculated as gc/100 ng total DNA based on a standard curve (inset) generated with known amounts of HSV viral DNA (eight dilutions, duplicate determinations). The standard curve was generated by plotting threshold cycle values (CT) versus gc numbers (log10), showing reliable values for the slope (−3.1707x), intercept (39.64), and the correlation coefficient (R2 = 0.99656). The dotted horizontal line in the tissue plot represents the low limit of detection (10 gc/100 ng DNA). gc, genome copy; HSV, herpes simplex virus; qPCR, quantitative PCR.
In an initial experiment to explore the antitumor efficacy of KNE, we treated subcutaneous U87 tumors in nude mice (tumor size ~140 mm3) with a single intratumoral injection of phosphate-buffered saline (PBS) or 5 × 108 gc of virus. The results showed that PBS-injected tumors increased in size to 900–1,000 mm3 over a period of 29 days (first animal sacrificed), whereas the growth of tumors injected with the retargeted virus was suppressed during the first 20 days, resulting in only a limited increase in size at the end of the observation period (P < 0.0001; Supplementary Figure S2). While these data indicated that a single injection of the retargeted virus was not sufficient for complete tumor eradication, they provided compelling evidence of effective tumoricidal activity without virus-related adverse effects.
Treatment of intracranial tumors with KNE virus
Since glioblastoma therapy is the most sought-after application of oHSV, we were interested in testing the antitumor efficacy of KNE in an orthotopic xenograft model of GBM. A significant proportion of human glioblastomas overexpress EGFR or express a constitutively active mutant version called EGFRvIII.29,30 Using HSV entry receptor deficient hamster (CHO) cells engineered to express either version of the human receptor,25,31 we confirmed that KNE recognizes both for entry (data not shown). We examined eight primary human GBM lines cultured as tumor spheres for expression of wt and mutant EGFR by western blot (Figure 6). The majority of these lines vigorously expressed the mutant form of EGFR while GBM30 expressed substantially less of the mutant receptor and no wt EGFR; two other lines expressed wt EGFR only, but at very low levels. We chose GBM30 to study the therapeutic efficacy of KNE because this line might be particularly challenging for the virus as it expressed low levels of the target receptor, yet formed highly aggressive orthotopic tumors in nude mice.
Figure 6.

Immunoblot analysis of EGFR expression in GBM tumor spheres. Cells were grown as spheres (GBM spheroids) and lysates of equal numbers of cells were probed by immunoblotting for EGFR using an antibody recognizing a C-terminal epitope shared by EGFR and EGFRvIII (upper panel). The membranes were reprobed with an anti-α-tubulin antibody (lower panel). Control cell lines were CHO-K1 (CHO) or CHO-K1 stably expressing full-length EGFR (CHO-EGFR wt) or EGFRvIII (CHO-EGFR vIII). Size markers in kDa are indicated at the left. EGFR, epidermal growth factor receptor; GBM, glioblastoma multiforme.
We initially explored the ability of KNE to prolong the survival of nude mice carrying intracranial implants of GBM30 cells compared to treatment with KNC control virus or PBS. The results (Supplementary Figure S3a) showed that PBS- and control virus-treated animals died between 21 and 39 days after tumor cell implantation (15–33 days post-treatment) with no clear difference between these two groups. However, 50% of animals treated with KNE survived for 60 days when the experiment was terminated by humane sacrifice, suggesting that the EGFR-retargeted virus was effective and tumor specific in this aggressive GBM model.
We followed up with two larger studies carried out independently at the University of Pittsburgh and The Ohio State University (OSU), comparing the antitumor efficacy of KNE and PBS in the orthotopic GBM30 xenograft model. Figure 7a and Supplementary Figure S3b show the Kaplan-Meier survival plots obtained in the two studies. In both instances, the PBS-treated mice died within 35 days of tumor cell implantation (median survival: 23 days), while 63–73% of mice treated with KNE survived for the duration of the respective experiments (90 days; P < 0.001). The similarity between these results from the two independent laboratories provided confidence that KNE was highly effective in destroying an aggressive primary human glioblastoma.
Figure 7.

Retargeted HSV treatment of a nude mouse model of glioblastoma. (a) Kaplan-Meier survival plot. Triturated GBM30 cells were implanted intracranially and PBS (n = 8) or KNE virus (n = 11) were injected 5 days later at the same coordinates. Animals were observed daily and euthanized when showing signs of morbidity. *P < 0.001 (log rank and Breslow tests) compared to mice treated with PBS. (b) T2-weighted brain MRI images of one animal from each treatment group 1 day before (Pre) and on days 3, 10, and 15 after treatment. Arrows point to tumor growth. Study performed at The Ohio State University. HSV, herpes simplex virus; MRI, magnetic resonance imaging; PBS, phosphate-buffered saline.
As part of the OSU study, several randomly selected animals from each treatment group were assessed by magnetic resonance imaging (MRI) for changes in tumor size. Mice were imaged 1 day before treatment with PBS or KNE, and again on days 3, 10, and 15 post-treatment. Figure 7b shows the T2-weighted images of an example from each group. Consistent with the survival data, all of the imaged mice from the PBS group (n = 3) showed rapidly growing tumors while complete tumor regression was observed in three of the five imaged mice that had been treated with the retargeted virus (Figure 7b and Supplementary Figure S4). The remaining two virus-treated animals examined by MRI showed tumor progression (Supplementary Figure S4) and died on days 21 and 28, respectively (see Figure 7a). While it is not clear why a few animals in each experiment failed to respond to viral therapy, one possibility is that this was due to occasional inaccurate tumor cell or vector delivery (e.g., Supplementary Figure S4, virus animal #5).
Discussion
Oncolytic viruses derived from herpes simplex virus type 1 (oHSV) have shown promise for the treatment of GBM in animal model systems and have proven safe in patient trials.10,11 However, since the tumor selectivity of oHSV is typically based on defective genes,5 these viruses often fall far short of wt virus in replicative ability and thus have limited tumor killing efficacy.1 Virus retargeting offers a potential solution to this problem as selectivity for tumor cells does not depend on defective genes but rather is determined by the existence of targetable receptors that are preferentially or uniquely expressed on the tumor cells. EGFR is an attractive target as it is frequently overexpressed or mutated in GBM.30
Complete HSV retargeting was first achieved by Zhou and Roizman17 who overcame the key challenge of disrupting the binding of gD to its disparate cognate receptors without eliminating the protein's ability to respond to the binding of an inserted targeting ligand to a novel receptor. In addition, their virus contained an engineered version of gC retargeted by the same ligand to favor virion adsorption to tumor cells. However, virus yield and plaque size were reduced relative to wt virus and no tumor treatment experiments were reported. Based on new insights into the function of gD structural domains,32 the Campadelli-Fiume lab subsequently described a novel gD design incorporating an scFv for HER2 that mediated highly efficient HSV infection exclusively through recognition of the target receptor. This virus was shown to be effective in a subcutaneous xenograft model of ovarian carcinoma.19 Recently, the same group used an earlier, significantly different gD design to target the virus to HER2 in a platelet-derived growth factor receptor-induced, HER2-transduced orthotopic mouse glioma model. Although this design reduced virus growth and lateral spread on HER2- and HSV-receptor–positive cells compared with wt gD virus while showing residual infection through the canonical HSV receptor nectin-1,18 the glioma study demonstrated extended survival of tumor-bearing animals without virus-related central nervous system toxicity.33 Our lab previously described another fully retargeted gD construct mediating HSV entry exclusively by recognition of the human EGFR and we demonstrated that retargeted entry was enhanced by a pair of mutations in gB.20 In the present study, we incorporated a modified version of this construct into an unattenuated virus backbone and showed that in combination with the two mutations in gB, this design mediated highly efficient EGFR-dependent virus entry. The gB:NT-enhanced retargeted virus proved effective in the treatment of an aggressive orthotopic model of human GBM, showing cures in a majority of the animals. We have found that other targeting ligands can be effective in the same gD context, including the CEA-specific scFv described here as well as a natural receptor ligand, mature EGF (H. Uchida, J.B. Cohen, J.C. Glorioso, unpublished results). Thus our results support the broader utility of our targeting design.
In our study, high-dose inoculation of KNE in normal mouse brain had no apparent adverse effects and no evidence of virus spread was detected. However, the EGFR-targeting ligand of the recombinant virus is specific for human EGFR and thus in patients, low-level EGFR expression in nontumor cells, primarily neural precursor cells in the subventricular zone,34 may pose a safety risk. Likewise, HER2 is expressed in normal human brain35 and thus the reported safety in mice of HSV retargeted by a ligand specific for the human receptor33,36 does not exclude the possibility of unwanted side-effects in patients. These concerns may be addressed by further engineering of the vectors, for example by tagging essential genes with target sequences for microRNAs that are expressed in neural precursor cells, but minimally or not at all in GBM tumors.37 Such strategies may also permit the application of cocktails of viruses retargeted to different receptors to confront the intrinsic cellular heterogeneity of GBM tumors.30
Despite our results and those of others, the determinants of efficient HSV retargeting remain incompletely defined. For example, regardless of the gB allele, we have been unable to detect EGFR-retargeted virus entry using EGF or our anti-EGFR scFv in gD constructs of similar or identical design as those in the two HER2 studies referred to above (H. Uchida, J.B. Cohen, J.C. Glorioso, unpublished results). It is likely that the degree of gD activation in response to binding to a new receptor will vary with the position and type of targeting ligand, the affinity of the ligand for its receptor, and the abundance of the receptor on the surface of the target cell. In addition, the efficiency of retargeted virus infection may be influenced by the virus entry mechanism, fusion at the cell surface or with endosomal membranes,38 which may be determined by either or both the cell type39 and the natural response of the receptor to ligand binding. Insight into these variables will come from studies with additional ligands, receptors, and target cells, and will greatly benefit our ability to target unattenuated HSV to different types of cancer.
Materials and Methods
Cells. HSV-1–resistant baby hamster kidney J1.1-2 cells (provided by Gabriella Campadelli-Fiume, University of Bologna), HVEM-transduced J1.1-2 (J/A),40 nectin-1–transduced J1.1-2 (J/C)41 and EGFR-transduced J1.1-2 (J/EGFR),42 HSV-1–resistant Chinese hamster ovary CHO-K1 cells (ATCC CCL-61), EGFR-transduced CHO-K1 (CHO/EGFR)25 and EGFRvIII-transduced CHO-K131 (both from Stephen Russell, Mayo Clinic, Rochester, MN), African green monkey kidney Vero cells (ATCC CCL-81) and gD-complementing VD60 cells43 (provided by David Johnson, Oregon Health and Science University) were described previously. Human lung carcinoma A549 (ATCC CCL-185), colon carcinoma HT29 (ATCC HTB-38), pancreatic carcinoma BxPC3 (ATCC CRL-1687), glioblastoma U87 (ATCC HTB-14) and SNB19 (ATCC CRL-2219), epidermoid carcinoma A431 (ATCC CRL-1555), and gastric carcinoma MKN45 (JCRB 0254) and MKN74 (JCRB 0255) cells were cultured in Dulbecco's modified Eagle's medium (Life Technologies, Grand Island, NY) supplemented with 10% fetal bovine serum (Sigma, St. Louis, MO). GBM30 and other patient-derived GBM lines were isolated and maintained as tumor spheres in Neurobasal Medium supplemented with 2% B27 (both from Life Technologies), human EGF (50 ng/ml) and bFGF (50 ng/ml) (both from R&D Systems, Minneapolis, MN), and penicillin-streptomycin in low-attachment cell culture flasks (Corning, Corning, NY). GBM30, 34, 35, 88, 97 and OHG02 were obtained from freshly resected human glioma specimens from consenting patients as approved by the Institutional Review Board at The Ohio State University, essentially as described.44 GBM1123 was kindly provided by Ichiro Nakano (Ohio State University). X12 cells were isolated as a sphere-forming clone in culture from GBM12 passaged subcutaneously in the flanks of nude mice (from David James, Mayo Clinic).45 At the University of Pittsburgh, GBM30 cells were grown as tumor spheres in Neurobasal Medium (Invitrogen, Carlsbad, CA) supplemented with 2% B-27 without vitamin A (Invitrogen), 2 mg/ml amphotericin and 100 mg/ml gentamicin (Lonza, Allendale, NJ), 2 mmol/l L-glutamine (Mediatech-Cellgro, Manassas, VA), and 10 ng/ml each of EGF and bFGF (Shenandoah Biotechnology, Warwick, PA).
Viruses. The gD-null versions of HSV-1 KOS encoding the wt or gB:D285N/A549T mutant gB allele, designated K-gB:wtΔgD or K-gB:N/TΔgD, respectively, were described previously.20 Retargeted recombinant viruses KE and KC were established by co-transfection of VD60 cells with K-gB:wtΔgD viral DNA and plasmids pgD:Δ224/38C-scEGFR or pgD:Δ224/38C-scCEA, respectively, followed by plaque purification through three rounds of limiting dilution on VD60 cells. The gB:NT versions of these viruses, KNE and KNC, were created by recombination of the same plasmids with K-gB:N/TΔgD viral DNA. The recombinant viruses, along with wt KOS, were passaged through Vero cells and viral genome titers in gc/ml were determined by qPCR for the viral immediate early gene ICP47, as described previously.23 All recombinant viruses were confirmed by PCR and DNA sequencing through the relevant glycoprotein genes or deletions.
Plasmids. Plasmid pgD:Δ224/38C was derived from a previously described plasmid, pgD:A3C/Y38C,41 by deletion of gD codons 2-24 (codon numbers refer to the mature form of gD). Plasmids pgD:Δ224/38C-scEGFR and pgD:Δ224/38C-scCEA were created by inserting the coding sequences for the 528 anti-EGFR scFv (scEGFR, 248 amino acids)28 or the F39 anti-CEA scFv (scCEA, 238 amino acids)46 at the position of the 2-24 deletion in pgD:Δ224/38C. Plasmid constructs were confirmed by DNA sequencing.
Entry assay. Entry assays were performed as described.20 Briefly, cells were infected for 6 hours and immunostained using monoclonal mouse anti-ICP4 (1:300; Santa Cruz Biotechnology, Santa Cruz, CA) and Cy3-conjugated sheep anti-mouse IgG (1:400; Sigma). Images were collected under a Nikon Diaphot fluorescence microscope (Nikon, Melville, NY).
Entry-blocking assay. Cells were incubated with 0–100 µg/ml monoclonal mouse anti-EGFR antibody (528; Santa Cruz Biotechnology) for 1 hour at room temperature, infected with KNE or wt KOS at 10 gc/cell for 2 hours at 37 °C, washed with acidic buffer as described,20 and immunostained for ICP4 at 6 or 16 hpi.
Rate-of-entry assay. Rate-of-entry assays were performed as described previously.47 Briefly, VD60 cells in 48-well dishes were incubated with viruses at 20,000 gc/well at 4 °C for 30 minutes and washed three times with cold PBS. The cells were then shifted to 37 °C for various intervals followed by acidic wash. The cultures were overlaid with methylcellulose-containing media, incubated at 37 °C for 48 hours, and plaque numbers were counted. All infections were performed in triplicate.
Cell-killing assay. Cells were seeded in 96-well plates and infected at 0.01–10 gc/cell for 3 or 6 days. Cell viability was measured by MTT assay.48 Briefly, 0.5 mg/ml of MTT (Sigma) solution was added to the cells, and after 1 hour incubation at 37 °C, the MTT solution was removed, 100% ethanol was added, and the OD570 was recorded by a Wallac microplate reader (Perkin Elmer, Waltham, MA). Percent cell viability was calculated as 100% × OD (infected)/OD (uninfected).
Immunoblotting. GBM tumor spheres and CHO cells were dissociated with TripLE reagent (Life Technologies). The cells were counted, lysed with RIPA buffer (50 mmol/l Tris-Cl, pH 8.0, 150 mmol/l NaCl, 1% NP-40, 0.5% sodium deoxycholate, and 0.1% SDS) containing complete Protease Inhibitor Cocktail (Roche, Indianapolis, IN) and 2-mercaptoethanol (Sigma), and sonicated. After the addition of SDS sample buffer, the lysates were boiled and the equivalent of 200,000 cells of each sample electrophoresed on 4–20% SDS-PAGE gels. Proteins were transferred to polyvinylidene difluoride membranes (Millipore, Billerica, MA) and the membranes were sequentially reacted with polyclonal rabbit anti-EGFR sc-03 (Santa Cruz Biotechnology) and HRP-conjugated anti-rabbit IgG before development with ECL plus (GE Healthcare, Piscataway, NJ). The blots were then reprobed with T6793 mouse monoclonal anti-α-tubulin antibody (Sigma) and HRP-conjugated anti-mouse IgG.
Animals. Animals were 3–4 weeks old (University of Pittsburgh) or 6–8 weeks old (OSU) female BALB/c athymic nu/nu mice (Charles River, Frederick, MD) housed in a BSL2 facility. All animal procedures were performed in accordance with the requirements and recommendations in the Guide for the Care and the Use of Laboratory Animals (Institute for Laboratory Animal Research, 1985) approved by the University of Pittsburgh Institutional Animal Care and Use Committee and the Subcommittee on Research Animal Care of the Ohio State University.
Intracranial toxicity. Mice were anesthetized and inoculated with 5 × 103 gc of wt KOS or 5 × 108 gc of KNE virus per animal in a volume of 4 µl using a stereotactic head frame (David Kopf Instruments, Tujunga, CA). A small craniotomy was drilled 2 mm to the right of midline and 1 mm anterior to the coronal suture, and virus was injected stereotactically into the right frontal lobe 3 mm below the dura mater. The injection needle was removed, the craniotomy was sealed with bone wax, and the scalp was closed with staples. Animals were observed daily for appearance and behavioral changes and symptom-free animals were euthanized on day 47.
In a similar experiment, mice were injected with 1 × 103 gc KOS or 5 × 108 gc KNE. Upon death (KOS, 21 days) or humane sacrifice (retargeted virus, 37 days), 15 µm brain cryosections from one animal of each group were stained with anti-ICP4 antibody (1:400; Virusys, Taneytown, MD) and Cy3-conjugated secondary antibody (1:600; Sigma).
Treatment of subcutaneous glioblastomas. Anesthetized mice were injected subcutaneously in the right flanks with 1 × 107 U87 cells. Tumors were allowed to grow to ~140 mm3 and injected with 5 × 108 gc KNE (n = 6) or PBS (n = 5) in 50 µl volumes. The animals were monitored daily for signs of discomfort and tumor diameters measured every 4–5 days. Animals displaying loss of appetite, reduced activity or impaired movement were euthanized. Tumor volumes were calculated as (length × width)2/2.49 Statistical analysis was performed by analysis of variance with P < 0.05 considered significant.
Vector biodistribution. U87 flank tumors were generated as above and the animals observed daily until the tumor mass reached 700–1,000 mm3. Mice were placed into restraints under a heat lamp, their tails were warmed with warm-water moistened cotton balls, and 2 × 109 gc of KNE or KOS control in 100 µl of PBS were injected into the lateral tail vein. At 48 hpi, animals were sacrificed by cervical dislocation and tissues were removed using disposable sterile instruments. Isolated tissues were flash-frozen and stored at −80 °C until processing. Two pieces of 25 mg of each tissue were dissected using disposable scalpels and resuspended in 180 µl tissue lysis (ATL) buffer (Qiagen, Valencia, CA). The tissue samples were disrupted with sterile micropestles and a hand sonicator according to the manufacturer's instructions, and DNA was extracted using the DNeasy Blood & Tissue Kit (Qiagen). The DNA content of each sample was determined using a Thermo Genesys-6 spectrophotometer (Thermo-Fisher, Pittsburgh, PA). A sample of each tissue from an uninfected animal was spiked with HSV vector to examine whether DNA extraction and isolation resulted in loss of viral DNA. Recovery measured by both spectrophotometry and qPCR was very similar for all of the samples and showed no significant loss of input viral DNA (data not shown).
HSV gc content in the isolated DNAs was quantified by qPCR, essentially as described.23 Emission data were collected in real-time on an Applied Biosystems StepOnePlus sequence detector (Life Technologies) and analyzed by StepOne Software version 2.1 (Life Technologies). A standard curve over a range of tenfold serial dilutions (n = 8), each tested in duplicate, was created using a virus preparation of known concentration (3 × 106 gc/ml). The standard curve was used to convert experimental measurements to gc and the data was plotted as gc/100 ng total DNA in the sample.
Treatment of orthotopic human GBM30 tumors in nude mice. Anesthetized nude mice were fixed in a stereotactic apparatus, a burr hole was drilled 2 mm lateral and 0.5 mm anterior to the bregma to a depth of 3 mm, and 2 × 105 GBM30 cells in 4 µl Hank's buffered salt solution were implanted. At 5–6 days post-tumor cell implant, the mice were anesthetized again and stereotactically inoculated at the same coordinates with 2–5 µl PBS or virus (3.6 × 107 gc/2 µl (Pitt) or 8.6 × 108 gc/4.1 µl (OSU) of KNE or KNC). Animals were observed daily and euthanized at the indicated time points or when showing signs of morbidity.
MRI imaging. Several mice were randomly selected from each treatment group (PBS or KNE) of the OSU survival experiment for MRI imaging. Animals were imaged 1 day before treatment (4 days after GBM30 implantation) and on days 3, 10, and 15 post-treatment. Imaging was performed using a Bruker BioSpec 94/30 magnet (Bruker BioSpin, Karlsruhe, Germany), a 2.0 cm diameter receive-only mouse brain coil and a 70 mm diameter linear volume coil. Anesthetized mice were injected with 0.1 mmol/kg Magnevist (Bayer Health Care Pharmaceuticals, Wayne, NJ) intraperitoneally and T2-weighted images (repetition time = 3,500 ms, echo time = 12 ms, rare factor = 8, navgs = 4) were acquired coronally across the region of interest on a 400 MHz Bruker horizontal bore magnet running Paravision 4.0 (Bruker Biospin, Billerica, MA).
Survival statistics. Animal survival data were charted as Kaplan-Meier plots and compared using the log-rank and Breslow (generalized Wilcoxon) tests. Statistical analyses were performed with SPSS statistical software (version 19.0; SPSS, Chicago, IL).
SUPPLEMENTARY MATERIAL Figure S1. Structure of detargeted and retargeted gD constructs. Figure S2. KNE inhibition of flank tumor growth. Figure S3. Additional brain tumor survival plots. Figure S4. Brain MRI images of additional animals.
Acknowledgments
We thank David Johnson, Gabriella Campadelli-Fiume, Stephen Russell, Ichiro Nakano, and David James for reagents. We also thank David Clawson, Wendy Fellows, Mingdi Zhang, and George Huang for technical assistance (University of Pittsburgh), and Kimerly Powell and Anna Bratasz (Small Animal Imaging Shared Resources, The Ohio State University Wexner Medical Center) for help with magnetic resonance imaging experiments. This work was supported by National Institutes of Health grants CA119298, NS40923, and DK044935 to J.C.G.; CA069246 to E.A.C; and NS064607, CA150153, and NS045758 to B.K. The authors declared no conflict of interest.
Supplementary Material
Structure of detargeted and retargeted gD constructs.
KNE inhibition of flank tumor growth.
Additional brain tumor survival plots.
Brain MRI images of additional animals.
REFERENCES
- Markert JM, Gillespie GY, Weichselbaum RR, Roizman B., and, Whitley RJ. Genetically engineered HSV in the treatment of glioma: a review. Rev Med Virol. 2000;10:17–30. doi: 10.1002/(sici)1099-1654(200001/02)10:1<17::aid-rmv258>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- Cattaneo R, Miest T, Shashkova EV., and, Barry MA. Reprogrammed viruses as cancer therapeutics: targeted, armed and shielded. Nat Rev Microbiol. 2008;6:529–540. doi: 10.1038/nrmicro1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo ZS, Thorne SH., and, Bartlett DL. Oncolytic virotherapy: molecular targets in tumor-selective replication and carrier cell-mediated delivery of oncolytic viruses. Biochim Biophys Acta. 2008;1785:217–231. doi: 10.1016/j.bbcan.2008.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman GK, Cassady KA, Beierle EA, Markert JM., and, Gillespie GY. Targeting pediatric cancer stem cells with oncolytic virotherapy. Pediatr Res. 2012;71 4 Pt 2:500–510. doi: 10.1038/pr.2011.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manservigi R, Argnani R., and, Marconi P. HSV Recombinant Vectors for Gene Therapy. Open Virol J. 2010;4:123–156. doi: 10.2174/1874357901004010123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kambara H, Okano H, Chiocca EA., and, Saeki Y. An oncolytic HSV-1 mutant expressing ICP34.5 under control of a nestin promoter increases survival of animals even when symptomatic from a brain tumor. Cancer Res. 2005;65:2832–2839. doi: 10.1158/0008-5472.CAN-04-3227. [DOI] [PubMed] [Google Scholar]
- Friedman GK, Langford CP, Coleman JM, Cassady KA, Parker JN, Markert JM.et al. (2009Engineered herpes simplex viruses efficiently infect and kill CD133+ human glioma xenograft cells that express CD111 J Neurooncol 95199–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rueger MA, Winkeler A, Miletic H, Kaestle C, Richter R, Schneider G.et al. (2005Variability in infectivity of primary cell cultures of human brain tumors with HSV-1 amplicon vectors Gene Ther 12588–596. [DOI] [PubMed] [Google Scholar]
- Friedman GK, Pressey JG, Reddy AT, Markert JM., and, Gillespie GY. Herpes simplex virus oncolytic therapy for pediatric malignancies. Mol Ther. 2009;17:1125–1135. doi: 10.1038/mt.2009.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrow S, Papanastassiou V, Harland J, Mabbs R, Petty R, Fraser M.et al. (2004HSV1716 injection into the brain adjacent to tumour following surgical resection of high-grade glioma: safety data and long-term survival Gene Ther 111648–1658. [DOI] [PubMed] [Google Scholar]
- Markert JM, Liechty PG, Wang W, Gaston S, Braz E, Karrasch M.et al. (2009Phase Ib trial of mutant herpes simplex virus G207 inoculated pre-and post-tumor resection for recurrent GBM Mol Ther 17199–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markert JM, Medlock MD, Rabkin SD, Gillespie GY, Todo T, Hunter WD.et al. (2000Conditionally replicating herpes simplex virus mutant, G207 for the treatment of malignant glioma: results of a phase I trial Gene Ther 7867–874. [DOI] [PubMed] [Google Scholar]
- Connolly SA, Jackson JO, Jardetzky TS., and, Longnecker R. Fusing structure and function: a structural view of the herpesvirus entry machinery. Nat Rev Microbiol. 2011;9:369–381. doi: 10.1038/nrmicro2548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montgomery RI, Warner MS, Lum BJ., and, Spear PG. Herpes simplex virus-1 entry into cells mediated by a novel member of the TNF/NGF receptor family. Cell. 1996;87:427–436. doi: 10.1016/s0092-8674(00)81363-x. [DOI] [PubMed] [Google Scholar]
- Geraghty RJ, Krummenacher C, Cohen GH, Eisenberg RJ., and, Spear PG. Entry of alphaherpesviruses mediated by poliovirus receptor-related protein 1 and poliovirus receptor. Science. 1998;280:1618–1620. doi: 10.1126/science.280.5369.1618. [DOI] [PubMed] [Google Scholar]
- Shukla D, Liu J, Blaiklock P, Shworak NW, Bai X, Esko JD.et al. (1999A novel role for 3-O-sulfated heparan sulfate in herpes simplex virus 1 entry Cell 9913–22. [DOI] [PubMed] [Google Scholar]
- Zhou G., and, Roizman B. Construction and properties of a herpes simplex virus 1 designed to enter cells solely via the IL-13alpha2 receptor. Proc Natl Acad Sci USA. 2006;103:5508–5513. doi: 10.1073/pnas.0601258103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menotti L, Cerretani A, Hengel H., and, Campadelli-Fiume G. Construction of a fully retargeted herpes simplex virus 1 recombinant capable of entering cells solely via human epidermal growth factor receptor 2. J Virol. 2008;82:10153–10161. doi: 10.1128/JVI.01133-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menotti L, Nicoletti G, Gatta V, Croci S, Landuzzi L, De Giovanni C.et al. (2009Inhibition of human tumor growth in mice by an oncolytic herpes simplex virus designed to target solely HER-2-positive cells Proc Natl Acad Sci USA 1069039–9044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchida H, Chan J, Goins WF, Grandi P, Kumagai I, Cohen JB.et al. (2010A double mutation in glycoprotein gB compensates for ineffective gD-dependent initiation of herpes simplex virus type 1 infection J Virol 8412200–12209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon M, Zago A, Shukla D., and, Spear PG. Mutations in the N termini of herpes simplex virus type 1 and 2 gDs alter functional interactions with the entry/fusion receptors HVEM, nectin-2, and 3-O-sulfated heparan sulfate but not with nectin-1. J Virol. 2003;77:9221–9231. doi: 10.1128/JVI.77.17.9221-9231.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connolly SA, Landsburg DJ, Carfi A, Whitbeck JC, Zuo Y, Wiley DC.et al. (2005Potential nectin-1 binding site on herpes simplex virus glycoprotein d J Virol 791282–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsvitov M, Frampton AR, Jr, Shah WA, Wendell SK, Ozuer A, Kapacee Z.et al. (2007Characterization of soluble glycoprotein D-mediated herpes simplex virus type 1 infection Virology 360477–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cocchi F, Menotti L, Mirandola P, Lopez M., and, Campadelli-Fiume G. The ectodomain of a novel member of the immunoglobulin subfamily related to the poliovirus receptor has the attributes of a bona fide receptor for herpes simplex virus types 1 and 2 in human cells. J Virol. 1998;72:9992–10002. doi: 10.1128/jvi.72.12.9992-10002.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider U, Bullough F, Vongpunsawad S, Russell SJ., and, Cattaneo R. Recombinant measles viruses efficiently entering cells through targeted receptors. J Virol. 2000;74:9928–9936. doi: 10.1128/jvi.74.21.9928-9936.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka T, Huang J, Hirai S, Kuroki M, Kuroki M, Watanabe N.et al. (2006Carcinoembryonic antigen-targeted selective gene therapy for gastric cancer through FZ33 fiber-modified adenovirus vectors Clin Cancer Res 123803–3813. [DOI] [PubMed] [Google Scholar]
- Baek H, Uchida H, Jun K, Kim JH, Kuroki M, Cohen JB.et al. (2011Bispecific adapter-mediated retargeting of a receptor-restricted HSV-1 vector to CEA-bearing tumor cells Mol Ther 19507–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asano R, Sone Y, Makabe K, Tsumoto K, Hayashi H, Katayose Y.et al. (2006Humanization of the bispecific epidermal growth factor receptor x CD3 diabody and its efficacy as a potential clinical reagent Clin Cancer Res 124036–4042. [DOI] [PubMed] [Google Scholar]
- Gan HK, Kaye AH., and, Luwor RB. The EGFRvIII variant in glioblastoma multiforme. J Clin Neurosci. 2009;16:748–754. doi: 10.1016/j.jocn.2008.12.005. [DOI] [PubMed] [Google Scholar]
- Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD.et al. (2010Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1 Cancer Cell 1798–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura T, Peng KW, Harvey M, Greiner S, Lorimer IA, James CD.et al. (2005Rescue and propagation of fully retargeted oncolytic measles viruses Nat Biotechnol 23209–214. [DOI] [PubMed] [Google Scholar]
- Zhou G., and, Roizman B. Separation of receptor-binding and profusogenic domains of glycoprotein D of herpes simplex virus 1 into distinct interacting proteins. Proc Natl Acad Sci USA. 2007;104:4142–4146. doi: 10.1073/pnas.0611565104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gambini E, Reisoli E, Appolloni I, Gatta V, Campadelli-Fiume G, Menotti L.et al. (2012Replication-competent herpes simplex virus retargeted to HER2 as therapy for high-grade glioma Mol Ther 20994–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estrada C, Villalobo A.2006The Cell Cycle in the Central Nervous SystemJanigro, D (ed.). Humana Press: Totowa, NJ; pp. 265–280. [Google Scholar]
- Liu G, Ying H, Zeng G, Wheeler CJ, Black KL., and, Yu JS. HER-2, gp100, and MAGE-1 are expressed in human glioblastoma and recognized by cytotoxic T cells. Cancer Res. 2004;64:4980–4986. doi: 10.1158/0008-5472.CAN-03-3504. [DOI] [PubMed] [Google Scholar]
- Menotti L, Cerretani A., and, Campadelli-Fiume G. A herpes simplex virus recombinant that exhibits a single-chain antibody to HER2/neu enters cells through the mammary tumor receptor, independently of the gD receptors. J Virol. 2006;80:5531–5539. doi: 10.1128/JVI.02725-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavon I, Zrihan D, Granit A, Einstein O, Fainstein N, Cohen MA.et al. (2010Gliomas display a microRNA expression profile reminiscent of neural precursor cells Neuro-oncology 12422–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicola AV, McEvoy AM., and, Straus SE. Roles for endocytosis and low pH in herpes simplex virus entry into HeLa and Chinese hamster ovary cells. J Virol. 2003;77:5324–5332. doi: 10.1128/JVI.77.9.5324-5332.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gianni T, Campadelli-Fiume G., and, Menotti L. Entry of herpes simplex virus mediated by chimeric forms of nectin1 retargeted to endosomes or to lipid rafts occurs through acidic endosomes. J Virol. 2004;78:12268–12276. doi: 10.1128/JVI.78.22.12268-12276.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frampton AR, Jr, Stolz DB, Uchida H, Goins WF, Cohen JB., and, Glorioso JC. Equine herpesvirus 1 enters cells by two different pathways, and infection requires the activation of the cellular kinase ROCK1. J Virol. 2007;81:10879–10889. doi: 10.1128/JVI.00504-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchida H, Shah WA, Ozuer A, Frampton AR, Jr, Goins WF, Grandi P.et al. (2009Generation of herpesvirus entry mediator (HVEM)-restricted herpes simplex virus type 1 mutant viruses: resistance of HVEM-expressing cells and identification of mutations that rescue nectin-1 recognition J Virol 832951–2961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano K, Kobayashi M, Nakamura K, Nakanishi T, Asano R, Kumagai I.et al. (2011Mechanism of HSV infection through soluble adapter-mediated virus bridging to the EGF receptor Virology 41312–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ligas MW., and, Johnson DC. A herpes simplex virus mutant in which glycoprotein D sequences are replaced by beta-galactosidase sequences binds to but is unable to penetrate into cells. J Virol. 1988;62:1486–1494. doi: 10.1128/jvi.62.5.1486-1494.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otsuki A, Patel A, Kasai K, Suzuki M, Kurozumi K, Chiocca EA.et al. (2008Histone deacetylase inhibitors augment antitumor efficacy of herpes-based oncolytic viruses Mol Ther 161546–1555. [DOI] [PubMed] [Google Scholar]
- Pandita A, Aldape KD, Zadeh G, Guha A., and, James CD. Contrasting in vivo and in vitro fates of glioblastoma cell subpopulations with amplified EGFR. Genes Chromosomes Cancer. 2004;39:29–36. doi: 10.1002/gcc.10300. [DOI] [PubMed] [Google Scholar]
- Liao S, Khare PD, Arakawa F, Kuroki M, Hirose Y, Fujimura S.et al. (2001Targeting of LAK activity to CEA-expressing tumor cells with an anti-CEA scFv/IL-2 fusion protein Anticancer Res 213B1673–1680. [PubMed] [Google Scholar]
- Highlander SL, Dorney DJ, Gage PJ, Holland TC, Cai W, Person S.et al. (1989Identification of mar mutations in herpes simplex virus type 1 glycoprotein B which alter antigenic structure and function in virus penetration J Virol 63730–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods. 1983;65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- Tomayko MM., and, Reynolds CP. Determination of subcutaneous tumor size in athymic (nude) mice. Cancer Chemother Pharmacol. 1989;24:148–154. doi: 10.1007/BF00300234. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Structure of detargeted and retargeted gD constructs.
KNE inhibition of flank tumor growth.
Additional brain tumor survival plots.
Brain MRI images of additional animals.