

Abstract
Outcomes for patients with glioblastoma (GBM) remain poor despite aggressive multimodal therapy. Immunotherapy with genetically modified T cells expressing chimeric antigen receptors (CARs) targeting interleukin (IL)-13Rα2, epidermal growth factor receptor variant III (EGFRvIII), or human epidermal growth factor receptor 2 (HER2) has shown promise for the treatment of gliomas in preclinical models and in a clinical study (IL-13Rα2). However, targeting IL-13Rα2 and EGFRvIII is associated with the development of antigen loss variants, and there are safety concerns with targeting HER2. Erythropoietin-producing hepatocellular carcinoma A2 (EphA2) has emerged as an attractive target for the immunotherapy of GBM as it is overexpressed in glioma and promotes its malignant phenotype. To generate EphA2-specific T cells, we constructed an EphA2-specific CAR with a CD28-ζ endodomain. EphA2-specific T cells recognized EphA2-positive glioma cells as judged by interferon-γ (IFN-γ) and IL-2 production and tumor cell killing. In addition, EphA2-specific T cells had potent activity against human glioma-initiating cells preventing neurosphere formation and destroying intact neurospheres in coculture assays. Adoptive transfer of EphA2-specific T cells resulted in the regression of glioma xenografts in severe combined immunodeficiency (SCID) mice and a significant survival advantage in comparison to untreated mice and mice treated with nontransduced T cells. Thus, EphA2-specific T-cell immunotherapy may be a promising approach for the treatment of EphA2-positive GBM.
Introduction
Glioblastoma (GBM) is the most aggressive primary brain tumor in adults.1 The current standard of care consists of surgical resection, radiation, and chemotherapy with temozolomide but results in 5-year overall survival rates of <10%.2,3 Immunotherapy is an attractive strategy to improve outcomes for patients with GBM as it does not rely on the cytotoxic mechanisms employed by chemotherapy or radiation. Indeed, dendritic cell vaccines have shown encouraging results, producing clinical responses and increased progression-free survival in patients with recurrent and newly diagnosed GBM.4,5,6 Although these results await confirmation in randomized clinical trials, published studies have also shown that it is difficult to reliably induce GBM-specific T cells in vivo.
One strategy to overcome this limitation is to generate tumor-specific T cells in vitro by genetically modifying T cells to express chimeric antigen receptors (CARs), which consist of a single chain variable fragment, a transmembrane domain, and signaling domains derived from the T-cell receptor complex and costimulatory molecules.7 The clinical experience with CAR T cells in patients with GBM is limited, but given the recent encouraging clinical results using CAR T cells to treat GD2-positive neuroblastoma and CD19-positive leukemia, further exploration is warranted.8,9
The success of CAR T-cell immunotherapies for GBM will require preventing immune escape by targeting antigens that are important for sustaining the malignant GBM phenotype. The erythropoietin-producing hepatocellular carcinoma A2 (EphA2) receptor, a member of the Eph family of receptor tyrosine kinases, has emerged as a target antigen as such. EphA2 is overexpressed in GBM10,11 and is associated with poor outcomes.12,13 EphA2 overexpression induces pro-oncogenic effects including enhanced tumorigenesis,14 tumor cell migration and invasion,15 angiogenesis, and metastasis.16,17,18,19
Here, we report the development of an EphA2-specific CAR to redirect T cells to EphA2-positive GBMs. We show that these T cells are able to recognize and kill EphA2-positive glioma cells and glioma-initiating cells in vitro and induce tumor regression in an orthotopic xenograft severe combined immunodeficiency (SCID) mouse model of GBM.
Results
EphA2 is expressed in glioma cell lines and primary GBM
We confirmed the expression of EphA2 in GBMs by western blot analysis. EphA2 was expressed in the glioma cell lines U87 and U373 but not in normal whole brain or frontal lobe tissue, T cells, or the leukemia cell line K562 (Figure 1a). To determine the expression of EphA2 in primary GBM, protein was extracted from cell lines established after short-term culture of five different GBM tumor biopsies.20 EphA2 was detected in 5/5 primary GBM cell lines, although the level of expression varied between patients (Figure 1b). These results confirm that EphA2 is expressed in GBM in contrast to normal brain.
Figure 1.

Erythropoietin-producing hepatocellular carcinoma A2 (EphA2) is expressed in glioma but not in normal brain. (a) Western blot showed high expression of EphA2 in the glioma cell lines U87 and U373. EphA2 was not detectable in normal brain tissue (whole brain or frontal lobe), K562, or normal T cells. (b) EphA2 was detected in 5/5 primary cell lines established from tumors of patients with GBM. GAPDH, glyceraldehyde 3-phosphate dehydrogenase; GBM, glioblastoma.
Generation of EphA2-specific CAR-modified T cells
To redirect T cells to the EphA2 receptor, a second-generation EphA2-specific CAR was designed based on the humanized EphA2 monoclonal antibody (MAb) 4H5.21,22 A codon-optimized synthetic gene encoding 4H5 in single chain variable fragment format was cloned into a SFG retroviral vector upstream of an IgG1-CH2CH3 domain, a CD28 transmembrane domain, and costimulatory domains derived from CD28 and CD3-ζ (Figure 2a). Gibbon ape leukemia virus -pseudotyped retroviral particles encoding the EphA2-specific CAR were used to transduce CD3/CD28-activated T cells from normal healthy donors. Following T-cell transduction, fluorescence activated cell sorting (FACS) analysis was used to determine the cell surface expression of the EphA2-specific CAR. The percentage of CAR-expressing T cells ranged from 49.9 to 95.0% with a median of 73.2% (n = 7; Figure 2b). The resultant T-cell lines consisted of a mixed population of CD4- and CD8-positive cells, with both subsets expressing EphA2-specific CARs (Figure 2b).
Figure 2.

Generation of erythropoietin-producing hepatocellular carcinoma A2 (EphA2)-specific T cells. (a) The EphA2-specific chimeric antigen receptors (CAR) was generated by cloning a single chain variable fragment derived from the EphA2 monoclonal antibody 4H5 upstream of an IgG1-CH2CH3 domain, a CD28 TM domain, and costimulatory domains derived from CD28 and CD3-ζ into an SFG retroviral vector. (b) EphA2-CAR expression was detected by staining T cells with a CH2CH3 antibody. Fluorescence activated cell sorting analysis revealed expression of EphA2-specific CARs on the cell surface of transduced T cells as compared with controls (median = 73.2%, n = 7, range = 49.9–95.0%, representative plot shown). Transduced T cells consisted of CD4- and CD8-positive cells with both subsets expressing EphA2-specific CARs. LTR, long terminal repeats; NT, nontransduced; TM, transmembrane.
EphA2-specific T cells recognize and kill EphA2-positive gliomas
We used ELISAs and cytotoxicity assays to test the functionality of EphA2-specific T cells in vitro. EphA2-specific T cells were cocultured with EphA2-positive and negative targets, and after 24 hours, we determined the concentration of interferon-γ (IFN-γ) and interleukin (IL)-2 in the cell culture supernatants by ELISA. EphA2-specific T cells recognized EphA2-positive glioma cell lines U373 and U87 as evidenced by the production of significantly higher levels of IFN-γ and IL-2 in comparison to the EphA2-negative T cells and K562 (Figure 3a). Nontransduced-T cells produced little to no IFN-γ or IL-2 in response to all targets (Figure 3a). In standard 4-hour 51Cr release assays, EphA2-specific T cells had significant cytotoxic activity against U373 and U87 whereas nontransduced T cells did not (P < 0.0004 and P < 0.002; Figure 3b). Although low levels of killing of the EphA2-negative targets was observed in the presence of EphA2-specific T cells and nontransduced T cells, there was no difference between both effector T-cell populations (P > 0.15 for T cells and P > 0.27 for K562; Figure 3b). Recognition of EphA2-positive tumor cells by EphA2-specific T cells was confirmed for three other EphA2-positive tumor cell lines (Supplementary Figure S1). In addition, all five primary EphA2-positive GBM cell lines were killed in cytotoxicity assays whereas nontransduced T cells produced only background levels of killing (Figure 3c). To provide further evidence that the recognition of EphA2-specific T cells depends on the expression of EphA2, we genetically modified K562 cells to express EphA2 (K562-EphA2). EphA2-specific T cells killed K562-EphA2 as efficiently as U87 cells, but had no effect on parental K562 cells. By contrast, nontransduced T cells had no cytotoxic activity on any of these target cells. (Supplementary Figure S2a–c). In addition, U87 cells, genetically modified to express an EphA2-specific shRNA, expressed lower levels of EphA2 and were less sensitive to EphA2-specific T-cell killing (Supplementary Figure S2d,e). These results indicate that T-cell activation and killing depend on the expression of EphA2-specific CARs and the presence of EphA2 on target cells.
Figure 3.

Erythropoietin-producing hepatocellular carcinoma A2 (EphA2)-specific T cells recognize and kill EphA2-positive gliomas. (a) Nontransduced (NT) or EphA2-specific T cells were cocultured with target cells at a 1:5 ratio, and after 24 hours, the production of interferon-γ (IFN-γ) and interleukin- 2 (IL-2) by T cells was determined by ELISA. EphA2-specific T cells produced significantly higher levels of IFN-γ (U373 versus K562 P = 0.006; U373 versus T cells P = 0.007; U87 versus K562 P = 0.049; U87 versus T cells P = 0.034); and IL-2 (U373 versus K562 P = 0.011; U373 versus T cells P = 0.014; U87 versus K562 P = 0.018; U87 versus T cells P = 0.029) in response to EphA2-positive targets U373 and U87 than to EphA2-negative targets K562 and normal T cells (mean + SD; n = 5). (b) EphA2-specific T cells were tested in 4-hour chromium release assays. EphA2-specific T cells had significant cytotoxic activity against U373 and U87 whereas NT-T cells did not at effector to target (E:T) ratios of 10:1 (left panel) or 20:1 (right panel; U373: P < 0.0004; U87 P < 0.002 at both E:T ratios). There was no difference between both effector T-cell populations for EphA2-negative targets (T cells: P > 0.15; K562: P > 0.27 at both E:T ratios; mean + SD; n = 5). (c) Cytotoxicity assays with EphA2-specific T cells and NT-T cells as effectors and five primary EphA2-positive glioblastoma cell lines as targets.
Neurospheres express EphA2 and are killed by EphA2-specific T cells
Glioma-initiating cells are resistant to traditional chemotherapy and radiation therapy.23,24 To determine whether EphA2-specific T cells have activity against this cell population, we generated neurospheres from a U87 cell line that was genetically modified with a retrovirus encoding an eGFP.FFLuc fusion gene. Secondary neurospheres expressed EphA2 at similar levels to U87 cells cultured in monolayer (Figure 4a). EphA2-specific T cells killed U87 cells isolated from secondary neurospheres as equally well as U87 cells cultured in monolayer in standard cytotoxicity assays (Figure 4b).
Figure 4.
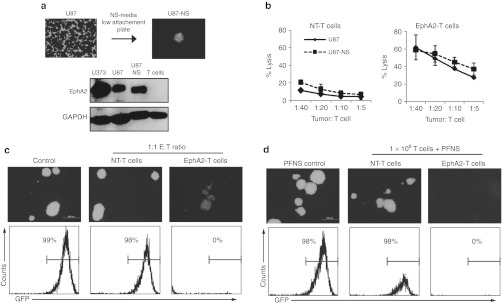
Erythropoietin-producing hepatocellular carcinoma A2 (EphA2)-specific T cells inhibit neurosphere formation and destroy established neurospheres. (a) Neurospheres were formed by culturing U87.eGFP.FFLuc in neurosphere (NS) media on low-attachment plates. Western blot showed that these U87-neurospheres expressed an equivalent amount of EphA2 as compared with U87 cells cultured in monolayer. (b) EphA2-specific T cells killed U87-NS in a 4-hour chromium release assay (mean + SD; n = 2). (c) Nontransduced (NT) or EphA2-specific T cells were plated with U87-NS cells at a 1:1 ratio in neurosphere media on low-attachment plates. Although NT-T cells had no effect on the ability of U87-NS cells to form neurospheres, EphA2-specific T cells prevented neurosphere formation as shown by fluorescence microscopy and fluorescence activated cell sorting (FACS) analysis for GFP-positive U87-NS. (d) 1 × 106 NT or EphA2-specific T cells were plated with PFNS. Only EphA2-specific T cells were able to destroy preformed neurospheres as judged by fluorescence microscopy and FACS analysis for GFP-positive U87-NS (data shown are representative of two independent experiments). GAPDH, glyceraldehyde 3-phosphate dehydrogenase; GFP, green fluorescent protein; PFNS, preformed U87-NS.
To provide further evidence that EphA2-specific T cells have activity against glioma-initiating cells, we determined if EphA2-specific T cells prevent the formation of neurospheres. U87 cells derived from neurospheres were plated at a ratio of 1:1 with nontransduced or EphA2-specific T cells under neurosphere culture conditions. EphA2-specific T cells inhibited the formation of neurospheres as shown by fluorescence microscopy and FACS analysis whereas nontransduced T cells did not (Figure 4c). In addition, EphA2-specific T cells, but not nontransduced T cells, were able to destroy intact neurospheres (Figure 4d). These results indicate that EphA2-specific T cells have potent antineurosphere activity.
EphA2-specific T cells induce GBM regression in vivo
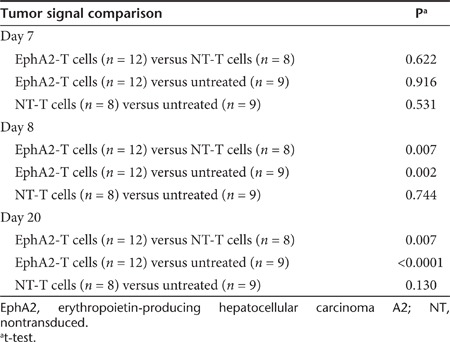
To evaluate the anti-GBM activity of EphA2-specific T cells in vivo, we used an orthotopic xenograft mouse model described earlier.20 U373 glioma cells were modified to express an eGFP.FFLuc fusion protein (U373.eGFP.FFLuc) allowing us to track tumor growth using serial noninvasive in vivo bioluminescence imaging. 1 × 105 U373.eGFP.FFLuc cells were injected into the brains of SCID mice on day 0. Tumors engrafted and grew exponentially in the week post-tumor cell injection. On day 7, the mice were injected intracranially with 2 × 106 EphA2-specific T cells or nontransduced T cells into the previous stereotactic tumor coordinates. Untreated mice served as controls. Although control mice and mice treated with nontransduced T cells showed continuous tumor growth, mice treated with EphA2-specific T cells did not (Figure 5a,b). Comparison of bioluminescence imaging results revealed no significant difference between all three groups on the day of T-cell injection; however, mice treated with EphA2-specific T cells had significantly lower tumor signals as early as one day post-treatment in comparison to untreated mice or mice treated with nontransduced T cells (P = 0.002 and P = 0.007; Table 1). Treatment with EphA2-specific T cells significantly prolonged survival of mice as compared with no treatment (P = 0.009) or treatment with nontransduced T cells (P = 0.010). By contrast, nontransduced T cells did not prolong survival in the untreated mice (P = 0.852; Figure 5c).
Figure 5.

Erythropoietin-producing hepatocellular carcinoma A2 (EphA2)-specific T cells induce regression of established glioblastoma in vivo. 1 × 105 U373.eGFP.FFLuc were injected intracranially into severe combined immunodeficiency mice on day 0 and treated with a single injection of 2 × 106 EphA2-specific T cells into the same tumor coordinates on day 7. Untreated mice and mice treated with nontransduced (NT) T cells served as controls. (a) Bioluminescence imaging was used to follow tumor progression. All mice had detectable tumors just prior to treatment (day 7). By day 40, all mice in the control groups had progressive tumors (9/9 for untreated, 8/8 for NT-T cells). The tumor signal decreased in all mice treated with EphA2-specific T cells, and 50% (6/12) were long-term survivors. (b) The bioluminescence signal from the tumors (radiance = photons/sec/cm2/sr) over time. Dotted lines represent each individual mouse while the solid black line represents the median radiance for the group at the given time. (c) Mice treated with EphA2-specific T cells had a significantly prolonged survival compared with untreated (P = 0.009) and NT-T cell (P = 0.010) treated mice. There was no significant difference between the survival of untreated versus NT-T cell treated mice (P = 0.852).
Table 1. Tumor signal comparison.
To evaluate the potency of EphA2-specific T cells, we modified the studies and injected fourfold fewer T cells into day 7 tumors or the same number of T cells into day 14 tumors (Figure 6a,b). All mice (n = 4) injected on day 7 with a lower number of T cells had regression of their tumors with one mouse having a complete response (CR), and two of four mice with day 14 tumors had a response, including one CR (Figure 6d). To discover if the systemic administration of human EphA2-specific T cells have antitumor activity in our xenograft model, mice with day 7 tumors received 1 × 107 EphA2-specific T cells by tail vein injection (Figure 6c,d). No antitumor effects were observed.
Figure 6.

Antitumor activity of erythropoietin-producing hepatocellular carcinoma A2 (EphA2)-specific T cells in vivo depends on T-cell number and tumor size. (a) 5 × 105 EphA2-specific T cells were injected intratumorally on day 7 or (b) 2 × 106 EphA2-specific T cells on day 14. All mice (n = 4) injected on day 7 had a regression of their tumors with one mouse having a complete response (CR). Two of four mice treated on day 14 had a response, including one CR. (c) Mice with day 7 tumors received 1 × 107 EphA2-specific T cells intravenously through their tail vein (n = 4). No antitumor effects were observed. (d) Kaplan–Meier survival curve of all treated groups of mice.
Discussion
In this study, we describe the development and characterization of a novel CAR specific for the EphA2 receptor and show that T cells expressing this CAR can effectively target and kill EphA2-positive GBM. We show that EphA2-specific T cells produce the immunostimulatory cytokines IFN-γ and IL-2 when cocultured with EphA2-positive targets and cause tumor cell lysis in cytotoxicity assays in vitro. EphA2-specific T cells were also able to target glioma-initiating cells as shown in neurosphere inhibition and killing assays. Finally, we demonstrated that EphA2-specific T cells have potent antitumor activity in vivo.
Over the last decade, several tumor-associated antigens have been identified in GBM, including the membrane-bound tumor antigens, epidermal growth factor receptor variant III (EGFRvIII), IL-13Rα2, human epidermal growth factor receptor 2 (HER2), transmembrane glycoprotein NMB, and EphA2.10,11,25,26,27,28,29 T cells expressing EGFRvIII-, IL-13Rα2-, and HER2-specific CARs have been evaluated in preclinical animal models with encouraging results;20,30,31 the clinical experience with these CAR T cells, however, is limited. Nevertheless, limitations of individual CARs are starting to emerge. Three patients with GBM have received intratumoral injection of IL-13Rα2-specific CAR T cells. T-cell administration was safe, and tumor responses were observed; however, tumors recurred, and these malignant cells were negative for IL-13Rα2.32 The risk of immune escape is also high for EGFRvIII targeted T-cell therapy given the emergence of GBM antigen loss variants in patients who received EGFRvIII peptide vaccines.5 In addition, there are safety concerns with regards to HER2-targeted T-cell therapies due to the death of a patient who received a high dose of HER2-CAR T cells.33
Given these limitations of currently available CARs for GBM, new targets need to be explored. Moreover, a panel of GBM-specific CARs may facilitate clinical protocols in which patients are infused with T-cell products that target multiple GBM antigens. EphA2 has emerged as an attractive target for GBM immunotherapy owing to recent findings that EphA2 signaling is involved in glioma cell proliferation, migration, and invasion.12,15
We detected the expression of EphA2 by western blot analysis in all GBM samples tested. Western blot was used, as commercially available EphA2 antibodies failed to reliably detect EphA2 by FACS analysis. Coculture of EphA2-specific T cells with EphA2-positive target cells resulted in secretion of IFN-γ and IL-2 in an antigen-dependent manner. EphA2-specific T cells also prevented neurosphere formation and destroyed intact neurospheres. Although CAR T cells as well as conventional antigen-specific T cells are able to kill CD133-positive glioma-initiating cells,20,34,35 recent studies showed that a subset of CD133-negative glioma cells also have glioma-initiating cell characteristics.36 Thus, our finding that EphA2-specific T cells destroy neurospheres and prevent their formation provides further evidence that glioma-initiating cells are sensitive to immune-mediated killing.
In vivo, EphA2-specific T cells induced the regression of GBMs grown in the brains of SCID mice. Mice were injected with 1 × 105 tumor cells and at the time of T-cell injection (day 7) the tumor signal had increased 4.8-fold (median). All mice had a decrease in their tumor signal, and 6 of 12 mice treated with 2 × 106 and one of four mice treated with 5 × 105 EphA2-specific T cells had a CR. There was no significant difference in tumor size at the time of EphA2-specific T-cell injection between long-term survivors and mice which died. These data imply that this model requires the tumor cell:T-cell ratio to be approximately equivalent to induce complete tumor regression. In support of this approximate assessment is our observation that of four mice treated on day 14, when tumor had a 16.8-fold (median) increase in signal over baseline, 2 × 106 EphA2-specific T cells induced a response in two animals in one of which there was a CR. Hence, we suggest that the overall CR rate of 40% is likely a reflection of the limited persistence and expansion of human T cells in SCID mice in vivo. Although this limitation may be overcome by modifying the tumor cells to secrete T-cell growth factors such as IL-2,31 such manipulations remain highly artificial and may not provide any better prediction of future clinical potency.
Is targeting EphA2 with EphA2-specific T cells safe? The EphA2 epitope recognized by the CAR is preferentially accessible in dividing malignant cells and not in quiescent normal epithelial cells, though these may also express EphA2 at low levels.22,37 As shown in Figure 1a and as reported by others, normal adult brain does not express EphA2.10 In addition, we observed no acute toxicities following local and systemic injection of EphA2-specific T cells. Histological examination of the brains of two tumor-free mice sacrificed at 118 days post T-cell injection showed normal brain morphology (Supplementary Figure S3). These results are particularly reassuring since our EphA2-specific CAR recognizes both human and murine EphA2 as judged by the ability of EphA2-specific T cells to secrete IFN-γ in response to recombinant murine EphA2 protein, and to kill murine target cells in an EphA2-specific manner (Supplementary Figure S4). This favorable safety profile is supported by findings of other investigators who found that intravenous administration of a bispecific EphA2-specific antibody targeting the same EphA2 epitope and human CD3 (bscEphA2xCD3) in combination with human CD3-positive T cells resulted in no systemic side effects.37 In addition, patients with GBM receiving a dendritic cell vaccine loaded with a cocktail of HLA-A2 restricted peptides derived from IL-13Rα2, EphA2, YKL-40, and gp100 had no adverse immune events.4
In conclusion, T cells redirected to EphA2 by an EphA2-specific CAR have potent antitumor activity against glioma and glioma-initiating cells in vitro, and induce the regression of established GBM xenografts in vivo, indicating that they may be of value in the treatment of EphA2-positive GBM.
Materials and Methods
Blood donors, primary tumor cells, and cell lines. Blood samples from healthy donors and primary tumor cells from patients with GBM were obtained in accordance to protocols approved by the Institutional Review Board of Baylor College of Medicine. The GBM cell lines U373 and U87 and the leukemia cell line K562 were purchased from the American Type Culture Collection (ATCC; Manassas, VA). U373 and U87 cells expressing enhanced green fluorescent protein and firefly luciferase (U373.eGFP.FFLuc and U87.eGFP.FFLuc) were generated by retroviral transduction. T cells and cell lines were grown in Roswell Park Memorial Institute or Dulbecco's Modified Eagle's Medium (Thermo Scientific HyClone, Waltham, MA; Lonza, Basel, Switzerland) with 10% fetal calf serum (HyClone, Logan, UT) and 2 mmol/l GlutaMAX-I (Invitrogen, Carlsbad, CA). The “Characterized Cell Line Core Facility” at MD Anderson Cancer Center, Houston, TX, performed cell line validation. Primary GBM cell lines were established as previously described20 and were grown in Dulbecco's Modified Eagle's Medium with 10% fetal calf serum, 2 mmol/l GlutaMAX-I, 1.5 g/l sodium bicarbonate, 0.1 mmol/l nonessential amino acids, and 1.0 mmol/l sodium pyruvate (all media supplements from Invitrogen).
Western blot. Cells were dissociated with phosphate buffered saline + 3 mmol/l EDTA and lysed in a buffer containing 50 mmol/l Tris, 150 mmol/l NaCl, 5 mmol/l EDTA, 1% Triton X-100 (all from Sigma, St. Louis, MO), and protease inhibitors (Thermo Scientific, Waltham, MA). Protein concentrations were determined using a Bio-Rad protein assay (Bio-Rad, Hercules, CA) with bovine serum albumin as the standard. Samples were denatured in Laemmli buffer (Bio-Rad) at 95 °C for 5 minutes. Electrophoresis-ready protein lysates from normal human brain tissue (whole brain and frontal lobes) were obtained from Clontech (Mountain View, CA). Protein of 10 µg were loaded per well and run on an 8% polyacrylamide gel. Proteins were transferred to Immobilon-P polyvinylidene difluoride membranes (Millipore, Billerica, MA). Using the SNAP i.d. protein detection system (Millipore), membanes were blocked with 0.25% milk powder in tris-buffered saline + 0.1% Tween-20 (Sigma) and then probed with EphA2 (clone D7; Sigma) or glyceraldehyde 3-phosphate dehydrogenase (sc-47724; Santa Cruz Biotechnology, Santa Cruz, CA) mouse monoclonal antibodies followed by a horseradish peroxidase conjugated goat mouse IgG antibody (sc-2005; Santa Cruz Biotechnology). Blots were developed using SuperSignal West Dura Extended Duration Substrate (Thermo Scientific) and exposed to GeneMate Blue Basic Autoradiography Film (BioExpress, Kaysville, UT).
Generation of an EphA2-specific CAR. The EphA2-specific single chain variable fragment was derived from the EphA2 MAb 4H5, a humanized version of the EphA2 MAb EA2.21,22 A codon-optimized gene was synthesized by GeneArt (Invitrogen) containing the immunoglobulin heavy-chain leader peptide,38 the 4H5 heavy-chain, a glycine (G) serine (S) linker [(G4S)3], and the 4H5 light chain flanked by 5' NcoI and 3' BamHI sites. This mini gene was subcloned into an SFG retroviral vector containing the human IgG1-CH2CH3 domain, a CD28 transmembrane domain, and costimulatory domains derived from CD28 and the CD3 ζ-chain.39,40 The cloning of the EphA2-specific CAR was verified by sequencing (Seqwright, Houston, TX).
Retrovirus production and transduction of T cells. Retroviral particles were generated by transient transfection of 293T cells with the EphA2-specific CAR encoding SFG retroviral vector, Peg-Pam-e plasmid containing the sequence for MoMLV gag-pol, and pMEVSVg plasmid containing the sequence for VSV-G, using GeneJuice transfection reagent (EMD Biosciences, San Diego, CA). Supernatants containing the retrovirus were collected after 48 and 72 hours. The VSV-G pseudotyped viral particles were used to transduce the PG-13 producer cell line for the production of retroviral particles for T-cell transduction. To generate EphA2-specific CAR T cells, peripheral blood mononuclear cells were isolated by Lymphoprep (Greiner Bio-One, Monroe, NC) gradient centrifugation and then stimulated on nontissue culture treated 24-well plates, which were precoated with OKT3 (Ortho Biotech, Bridgewater, NJ) and CD28 (Becton Dickinson, Mountain View, CA) antibodies. Recombinant human interleukin-2 (IL-2; Proleukin; Chiron, Emeryville, CA) of 100 U/ml was added to cultures on day 2. On day 3, OKT3/CD28-stimulated T cells (2.5 × 105 cells/well) were transduced on RetroNectin (Clontech, Mountainview, CA) coated plates in the presence of IL-2. On day 5 or 6, T cells were transferred into new wells and subsequently expanded with 50–100 U/ml IL-2. Nontransduced T cells, used as controls, were activated with OKT3/CD28 and expanded in parallel with 50–100 U/ml IL-2. EphA2-specific CAR expression was determined 3 to 4 days post-transduction. In all experiments, the functionality of matched (from the same donor) transduced and nontransduced T cells was compared.
Flow cytometry. A FACSCalibur instrument (BD, Becton Dickinson, Mountain View, CA) was used to acquire immunofluorescence data which were analyzed with CellQuest (BD) or FCS Express software (De Novo Software, Los Angeles, CA). Isotype controls were immunoglobulin G1–fluorescein isothiocyanate (IgG1-FITC; BD), IgG1–phycoerythrin (IgG1-PE; BD), IgG1–peridinin chlorophyll protein (IgG1-PerCP; BD), and isotype Cy5 (Jackson ImmunoResearch Laboratories, West Grove, PA). T cells were analyzed for EphA2-CAR expression using a CH2CH3 Cy5 antibody (Jackson ImmunoResearch Laboratories) along with anti-CD8 FITC, -CD4 PE, and -CD3 PerCP. U87 cells were analyzed for CD133 expression using a CD133 PE antibody (Miltenyi Biotec, Bergisch Gladbach, Germany). Forward and side scatter gating were used to discriminate live cells from dead cells. Cells were collected and washed once with phosphate buffered saline (Sigma) containing 1% fetal bovine serum (Hyclone; FACS buffer) prior to the addition of antibodies. Cell were incubated for 30 minutes on ice in the dark, washed once, and fixed in 0.5% paraformaldehyde/FACS buffer prior to analysis.
Analysis of cytokine production. EphA2-specific or nontransduced T cells from healthy donors were cocultured with normal T cells, K562, U373, or U87 cells at a 1:5 effector to target (E:T) ratio in a 48-well plate. After 24 hours of incubation, culture supernatants were harvested, and the presence of IFN-γ and IL-2 was determined by ELISA as per the manufacturer's instructions (R&D Systems, Minneapolis, MN).
Cytotoxicity assay. Standard chromium (51Cr) release assays were performed as previously described.41 Briefly, 1 × 106 target cells were labeled with 0.1 mCi (3.7 MBq) 51Cr and mixed with decreasing numbers of effector cells to give effector to target ratios of 40:1, 20:1, 10:1, and 5:1. Target cells incubated in complete medium alone or in 1% Triton X-100 were used to determine spontaneous and maximum 51Cr release, respectively. After 4 hours, supernatants were collected and radioactivity was measured in a gamma counter (Cobra Quantum; PerkinElmer; Wellesley, MA). The mean percentage of specific lysis of triplicate wells was calculated according to the following formula: [test release − spontaneous release]/[maximal release − spontaneous release] × 100.
Neurosphere assay. Primary U87-neurospheres were formed by seeding 1 × 105 U87 or U87.eGFP.FFLuc glioma cells in serum-free neurosphere media (1:1 Dulbecco's Modified Eagle's Medium:F-12 supplemented with 2 mmol/l GlutaMAX-I, 1x B27, 50 ng/ml epidermal growth factor, and 20 ng/ml fibroblast growth factor) on ultra-low attachment 6-well plates (Corning, Lowell, MA). These primary neurospheres were then dissociated with trypsin and reseeded to form secondary neurospheres. To determine if EphA2-specific T cells could inhibit neurosphere formation, secondary neurospheres were dissociated and cocultured with T cells in a 1:1 ratio in neurosphere media on ultra-low attachment 24-well plates (Corning). To determine if the T cells could destroy established neurospheres, secondary spheres were collected without dissociating and cocultured with 1 × 106 T cells. Cultures were imaged by fluorescent microscopy on day 4, and analyzed by FACS analysis on day 6.
Orthotopic xenograft SCID mouse model. All animal experiments followed a protocol approved by the Baylor College of Medicine Institutional Animal Care and Use Committee. Experiments were performed as described previously20 with a few modifications. ICR-SCID mice were purchased from Taconic (IcrTac:ICR-Prkdcscid; Fox Chase C.B-17 SCID ICR; Taconic, Hudson, NY). Male 8- to 12-week-old mice were anesthetized with rapid sequence inhalation isofluorane (Abbot Laboratories, England, UK) followed by an intraperitoneal injection of 225–240 mg/kg Avertin solution and then maintained on isofluorane by inhalation throughout the procedure. The head was shaved and the mice were immobilized in a CunninghamMouse/Neonatal Rat Adaptor (Stoelting, Wood Dale, IL) stereotactic apparatus fitted into an E15600 Lab Standard Stereotaxic Instrument (Stoelting), then scrubbed with 1% povidone-iodine. A 10 mm skin incision was made along the midline. The tip of a 30G ½ inch needle mounted on a Hamilton syringe (Hamilton, Reno, NV) served as the reference point. A 1 mm burr-hole was drilled into the skull 1 mm anterior and 2 mm to the right of the bregma. Firefly-luciferase expressing U373 cells (U373.eGFP.FFLuc; 1 × 105 in 2.0 µl) were injected 3 mm deep to the bregma, corresponding to the center of the right caudate nucleus over 5 minutes. The needle was left in place for 3 minutes, to avoid tumor cell extrusion, and then withdrawn over 5 minutes. Seven days after tumor cell injection, animals were treated with 2 × 106 nontransduced or EphA2-specific T cells from the same donor in 2 µl to the same tumor coordinates. The incision was closed with 2–3 interrupted 7.0 Ethilon sutures (Ethicon, Somerville, NJ). A subcutaneous injection of 0.03–0.1 mg/kg buprenorphine (Buprenex RBH, Hull, England) was given for pain control.
Bioluminescence imaging. Isofluorane anesthetized animals were imaged using the IVIS system (IVIS, Xenogen, Alameda, CA) 10–15 minutes after 150 mg/kg D-luciferin (Xenogen) was injected per mouse intraperitoneally. The photons emitted from the luciferase-expressing tumor cells were quantified using Living Image software (Caliper Life Sciences, Hopkinton, MA). A pseudo-color image representing light intensity (blue least intense and red most intense) was generated and superimposed over the grayscale reference image. Animals were imaged every other day for one week after injections, then twice weekly thereafter. Mice were euthanized when the tumor radiance was >1 × 109 on two occasions or when they met euthanasia criteria (neurological deficits, weight loss, signs of distress) in accordance with the Center for Comparative Medicine at Baylor College of Medicine.
Statistical analysis. For the mouse experiments, tumor radiance data were log-transformed and summarized using mean ± SD at baseline and multiple subsequent time points for each group of mice. Changes in tumor radiance from baseline at each time point were calculated and compared between groups using t-test. Survival determined from the time of tumor cell injection was analyzed by the Kaplan–Meier method and by the log-rank test.
SUPPLEMENTARY MATERIAL Figure S1. EphA2-specific T cells recognize EphA2-positive lung cancer cell lines. Figure S2. EphA2-specific target cell killing depends on the presence of EphA2. Figure S3. Normal brain morphology in mice treated with EphA2-specific CAR T cells. Figure S4. EphA2-specific T cells recognize murine EphA2.
Acknowledgments
We thank Cliona M. Rooney and Malcolm K. Brenner (Center for Cell and Gene Therapy) for helpful discussion and advice. The authors declare no conflict of interest. K.K.H.C. is a Melnick scholar and was supported by NIH grants 5T32HL092332 and 5T32GM007330, S.N. by CPRIT grant RP101499, S.G. by the Clayton Foundation for Research, the James McDonald Foundation and CPRIT grant RP110553, and S.G. and N.A.. by the Dana Foundation. The Characterized Cell Line Core Facility at MD Anderson Cancer Center is funded by NCI no. CA16672.
Supplementary Material
EphA2-specific T cells recognize EphA2-positive lung cancer cell lines.
EphA2-specific target cell killing depends on the presence of EphA2.
Normal brain morphology in mice treated with EphA2-specific CAR T cells.
EphA2-specific T cells recognize murine EphA2.
REFERENCES
- DeAngelis LM. Brain tumors. N Engl J Med. 2001;344:114–123. doi: 10.1056/NEJM200101113440207. [DOI] [PubMed] [Google Scholar]
- Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ.et al. (2005Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma N Engl J Med 352987–996. [DOI] [PubMed] [Google Scholar]
- Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJ, Janzer RC.et al. (2009Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial Lancet Oncol 10459–466. [DOI] [PubMed] [Google Scholar]
- Okada H, Kalinski P, Ueda R, Hoji A, Kohanbash G, Donegan TE.et al. (2011Induction of CD8+ T-cell responses against novel glioma-associated antigen peptides and clinical activity by vaccinations with {alpha}-type 1 polarized dendritic cells and polyinosinic-polycytidylic acid stabilized by lysine and carboxymethylcellulose in patients with recurrent malignant glioma J Clin Oncol 29330–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampson JH, Heimberger AB, Archer GE, Aldape KD, Friedman AH, Friedman HS.et al. (2010Immunologic escape after prolonged progression-free survival with epidermal growth factor receptor variant III peptide vaccination in patients with newly diagnosed glioblastoma J Clin Oncol 284722–4729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liau LM, Prins RM, Kiertscher SM, Odesa SK, Kremen TJ, Giovannone AJ.et al. (2005Dendritic cell vaccination in glioblastoma patients induces systemic and intracranial T-cell responses modulated by the local central nervous system tumor microenvironment Clin Cancer Res 115515–5525. [DOI] [PubMed] [Google Scholar]
- Eshhar Z, Waks T, Gross G., and, Schindler DG. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proc Natl Acad Sci USA. 1993;90:720–724. doi: 10.1073/pnas.90.2.720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pule MA, Savoldo B, Myers GD, Rossig C, Russell HV, Dotti G.et al. (2008Virus-specific T cells engineered to coexpress tumor-specific receptors: persistence and antitumor activity in individuals with neuroblastoma Nat Med 141264–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter DL, Levine BL, Kalos M, Bagg A., and, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med. 2011;365:725–733. doi: 10.1056/NEJMoa1103849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wykosky J, Gibo DM, Stanton C., and, Debinski W. EphA2 as a novel molecular marker and target in glioblastoma multiforme. Mol Cancer Res. 2005;3:541–551. doi: 10.1158/1541-7786.MCR-05-0056. [DOI] [PubMed] [Google Scholar]
- Hatano M, Eguchi J, Tatsumi T, Kuwashima N, Dusak JE, Kinch MS.et al. (2005EphA2 as a glioma-associated antigen: a novel target for glioma vaccines Neoplasia 7717–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, Park PJ, Lai W, Maher E, Chakravarti A, Durso L.et al. (2006A genome-wide screen reveals functional gene clusters in the cancer genome and identifies EphA2 as a mitogen in glioblastoma Cancer Res 6610815–10823. [DOI] [PubMed] [Google Scholar]
- Wang LF, Fokas E, Bieker M, Rose F, Rexin P, Zhu Y.et al. (2008Increased expression of EphA2 correlates with adverse outcome in primary and recurrent glioblastoma multiforme patients Oncol Rep 19151–156. [PubMed] [Google Scholar]
- Zelinski DP, Zantek ND, Stewart JC, Irizarry AR., and, Kinch MS. EphA2 overexpression causes tumorigenesis of mammary epithelial cells. Cancer Res. 2001;61:2301–2306. [PubMed] [Google Scholar]
- Miao H, Li DQ, Mukherjee A, Guo H, Petty A, Cutter J.et al. (2009EphA2 mediates ligand-dependent inhibition and ligand-independent promotion of cell migration and invasion via a reciprocal regulatory loop with Akt Cancer Cell 169–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa K, Pasqualini R, Lindberg RA, Kain R, Freeman AL., and, Pasquale EB. The ephrin-A1 ligand and its receptor, EphA2, are expressed during tumor neovascularization. Oncogene. 2000;19:6043–6052. doi: 10.1038/sj.onc.1204004. [DOI] [PubMed] [Google Scholar]
- Cheng N, Brantley DM, Liu H, Lin Q, Enriquez M, Gale N.et al. (2002Blockade of EphA receptor tyrosine kinase activation inhibits vascular endothelial cell growth factor-induced angiogenesis Mol Cancer Res 12–11. [PubMed] [Google Scholar]
- Dobrzanski P, Hunter K, Jones-Bolin S, Chang H, Robinson C, Pritchard S.et al. (2004Antiangiogenic and antitumor efficacy of EphA2 receptor antagonist Cancer Res 64910–919. [DOI] [PubMed] [Google Scholar]
- Brantley-Sieders DM, Fang WB, Hwang Y, Hicks D., and, Chen J. Ephrin-A1 facilitates mammary tumor metastasis through an angiogenesis-dependent mechanism mediated by EphA receptor and vascular endothelial growth factor in mice. Cancer Res. 2006;66:10315–10324. doi: 10.1158/0008-5472.CAN-06-1560. [DOI] [PubMed] [Google Scholar]
- Ahmed N, Salsman VS, Kew Y, Shaffer D, Powell S, Zhang YJ.et al. (2010HER2-specific T cells target primary glioblastoma stem cells and induce regression of autologous experimental tumors Clin Cancer Res 16474–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damschroder MM, Widjaja L, Gill PS, Krasnoperov V, Jiang W, Dall'Acqua WF.et al. (2007Framework shuffling of antibodies to reduce immunogenicity and manipulate functional and biophysical properties Mol Immunol 443049–3060. [DOI] [PubMed] [Google Scholar]
- Coffman KT, Hu M, Carles-Kinch K, Tice D, Donacki N, Munyon K.et al. (2003Differential EphA2 epitope display on normal versus malignant cells Cancer Res 637907–7912. [PubMed] [Google Scholar]
- Murat A, Migliavacca E, Gorlia T, Lambiv WL, Shay T, Hamou MF.et al. (2008Stem cell-related “self-renewal” signature and high epidermal growth factor receptor expression associated with resistance to concomitant chemoradiotherapy in glioblastoma J Clin Oncol 263015–3024. [DOI] [PubMed] [Google Scholar]
- Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB.et al. (2006Glioma stem cells promote radioresistance by preferential activation of the DNA damage response Nature 444756–760. [DOI] [PubMed] [Google Scholar]
- Sampson JH, Archer GE, Mitchell DA, Heimberger AB., and, Bigner DD. Tumor-specific immunotherapy targeting the EGFRvIII mutation in patients with malignant glioma. Semin Immunol. 2008;20:267–275. doi: 10.1016/j.smim.2008.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debinski W, Obiri NI, Powers SK, Pastan I., and, Puri RK. Human glioma cells overexpress receptors for interleukin 13 and are extremely sensitive to a novel chimeric protein composed of interleukin 13 and pseudomonas exotoxin. Clin Cancer Res. 1995;1:1253–1258. [PubMed] [Google Scholar]
- Zhang JG, Eguchi J, Kruse CA, Gomez GG, Fakhrai H, Schroter S.et al. (2007Antigenic profiling of glioma cells to generate allogeneic vaccines or dendritic cell-based therapeutics Clin Cancer Res 132 Pt 1566–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuan CT, Wakiya K, Dowell JM, Herndon JE 2nd, Reardon DA, Graner MW.et al. (2006Glycoprotein nonmetastatic melanoma protein B, a potential molecular therapeutic target in patients with glioblastoma multiforme Clin Cancer Res 127 Pt 11970–1982. [DOI] [PubMed] [Google Scholar]
- Kurpad SN, Zhao XG, Wikstrand CJ, Batra SK, McLendon RE., and, Bigner DD. Tumor antigens in astrocytic gliomas. Glia. 1995;15:244–256. doi: 10.1002/glia.440150306. [DOI] [PubMed] [Google Scholar]
- Bullain SS, Sahin A, Szentirmai O, Sanchez C, Lin N, Baratta E.et al. (2009Genetically engineered T cells to target EGFRvIII expressing glioblastoma J Neurooncol 94373–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahlon KS, Brown C, Cooper LJ, Raubitschek A, Forman SJ., and, Jensen MC. Specific recognition and killing of glioblastoma multiforme by interleukin 13-zetakine redirected cytolytic T cells. Cancer Res. 2004;64:9160–9166. doi: 10.1158/0008-5472.CAN-04-0454. [DOI] [PubMed] [Google Scholar]
- Brown CE, Starr R, Naranjo A, Wright C, Bading J, Ressler J, A.et al. (2011Adoptive transfer of autologous IL13-zetakine+ engineered T cell clones for the treatment of recurrent glioblastoma: lessons from the Clinic Mol Ther 19suppl 1): S136–S137. [Google Scholar]
- Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case Report of a Serious Adverse Event Following the Administration of T Cells Transduced With a Chimeric Antigen Receptor Recognizing ERBB2. Mol Ther. 2010;18:843–851.. doi: 10.1038/mt.2010.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown CE, Starr R, Martinez C, Aguilar B, D'Apuzzo M, Todorov I.et al. (2009Recognition and killing of brain tumor stem-like initiating cells by CD8+ cytolytic T cells Cancer Res 698886–8893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua W, Yao Y, Chu Y, Zhong P, Sheng X, Xiao B.et al. (2011The CD133+ tumor stem-like cell-associated antigen may elicit highly intense immune responses against human malignant glioma J Neurooncol 105149–157. [DOI] [PubMed] [Google Scholar]
- Sun Y, Kong W, Falk A, Hu J, Zhou L, Pollard S.et al. (2009CD133 (Prominin) negative human neural stem cells are clonogenic and tripotent PLoS ONE 4e5498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond SA, Lutterbuese R, Roff S, Lutterbuese P, Schlereth B, Bruckheimer E.et al. (2007Selective targeting and potent control of tumor growth using an EphA2/CD3-Bispecific single-chain antibody construct Cancer Res 673927–3935. [DOI] [PubMed] [Google Scholar]
- Wels W, Harwerth IM, Zwickl M, Hardman N, Groner B., and, Hynes NE. Construction, bacterial expression and characterization of a bifunctional single-chain antibody-phosphatase fusion protein targeted to the human erbB-2 receptor. Biotechnology (NY) 1992;10:1128–1132. doi: 10.1038/nbt1092-1128. [DOI] [PubMed] [Google Scholar]
- Vera J, Savoldo B, Vigouroux S, Biagi E, Pule M, Rossig C.et al. (2006T lymphocytes redirected against the kappa light chain of human immunoglobulin efficiently kill mature B lymphocyte-derived malignant cells Blood 1083890–3897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulè MA, Straathof KC, Dotti G, Heslop HE, Rooney CM., and, Brenner MK. A chimeric T cell antigen receptor that augments cytokine release and supports clonal expansion of primary human T cells. Mol Ther. 2005;12:933–941. doi: 10.1016/j.ymthe.2005.04.016. [DOI] [PubMed] [Google Scholar]
- Gottschalk S, Edwards OL, Sili U, Huls MH, Goltsova T, Davis AR.et al. (2003Generating CTL against the subdominant Epstein-Barr virus LMP1 antigen for the adoptive Immunotherapy of EBV-associated malignancies Blood 1011905–1912. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
EphA2-specific T cells recognize EphA2-positive lung cancer cell lines.
EphA2-specific target cell killing depends on the presence of EphA2.
Normal brain morphology in mice treated with EphA2-specific CAR T cells.
EphA2-specific T cells recognize murine EphA2.