

Abstract
Hepatocellular carcinoma (HCC) is one of the most prevalent malignancies resistant to current chemotherapies or radiotherapies, which makes it urgent to identify new therapeutic targets for HCC. In this study, we found that checkpoint kinase 1 (CHK1) was frequently overexpressed and correlated with poor clinical outcome in patients with HCC. We further showed that the CHK1 inhibitor GÖ6976 was capable of sensitizing HCC cells to cisplatin, indicating that CHK1 may have oncogenic function in HCC. We found that CHK1 phosphorylated the tumor suppressor spleen tyrosine kinase (L) (SYK[L]) and identified the phosphorylation site at Ser295. Furthermore, CHK1 phosphorylation of SYK(L) promoted its subsequent proteasomal degradation. Expression of a nonphosphorylated mutant of SYK(L) was more efficient at suppressing proliferation, colony formation, mobility, and tumor growth in HCC lines. Importantly, a strong inverse correlation between the expression levels of CHK1 and SYK(L) was observed in patients with HCC. Collectively, our data demonstrate that SYK(L) is a substrate of CHK1 in tumor cells and suggest that targeting the CHK1/SYK(L) pathway may be a promising strategy for treating HCC.
Introduction
Hepatocellular carcinoma (HCC) is one of the most prevalent malignancies worldwide (1, 2). Hepatic resection is still considered the gold standard for treating HCC patients who are healthy enough for such an operation. Unfortunately, even after curative surgical resection, the prognosis of patients with HCC is still poor due to the high incidence of tumor recurrence and distant metastasis (3). In addition, most patients with HCC do not respond to current chemotherapies or radiotherapies (4, 5), and only minimal effects are achieved by using sorafenib, a popular therapy that targets multiple kinases (6, 7). Therefore, there is an urgent need for further understanding of the molecular mechanisms in HCC tumorigenesis and for identifying new therapeutic targets for HCC.
Spleen tyrosine kinase (SYK) is a nonreceptor protein tyrosine kinase expressed in cells of either hematopoietic or epithelial origin. A significant drop in full-length SYK (the longer form, SYK[L]) levels was first observed in breast carcinoma (8). Recent evidence suggests that alterations in SYK expression are associated with malignant phenotypes, such as increased motility and invasion (9–12). Several clinical observations have indicated that a loss of SYK expression correlates with poor prognosis and metastasis (13–15). An alternatively spliced SYK transcript (the shorter form, SYK[S]) that lacks a 69-bp sequence has been reported to exist (16). This in-frame transcript variant creates a SYK isoform that lacks 23 residues in interdomain B (IDB). While its major structural domains are preserved (2 tandem SH2 domains and a kinase domain), SYK(S) is characterized by biologic functions that are different from those of SYK(L). Overexpression of SYK(L), but not SYK(S), has been shown to lead to reduced proliferation and invasiveness, indicating that SYK(L) may be a tumor suppressor (15, 17). Indeed, the loss of SYK(L) has been recently shown to be associated with tumorigenesis in multiple cancer types. For instance, we have demonstrated that decreased SYK(L) expression resulting from promoter methylation is an adverse prognostic factor in HCC patients (13). Interestingly, SYK(L) function is regulated by a protein phosphatase called T cell ubiquitin (TULA) (18, 19), indicating that the phosphorylation of SYK(L) plays a key role in its function. Recently, it has been reported that changing the splicing pattern of SYK impaired cell-cycle progression and anchorage-independent growth (20). Moreover, we found that the expression levels of SYK(L) and SYK(S) in tumor tissues have opposing indications for recurrence-free survival (RFS) and overall survival (OS) in patients with HCC (data not shown). Taken together, these results indicate that at least one of the extra 23 residues in the IDB in SYK(L) may be phosphorylated to regulate how SYK(L) functions. Nevertheless, the phosphorylation site or sites and corresponding kinase or kinases have yet to be identified.
Checkpoint kinase 1 (CHK1) is an evolutionarily conserved Ser/Thr kinase that becomes active after an event that damages DNA. In response to genotoxic damage, CHK1 is one of 2 key effector kinases activated either by ataxia telangiectasia mutated (ATM) or by ataxia telangiectasia and Rad3 related (ATR) (21, 22). Activated CHK1 is capable of phosphorylating a number of key regulators related to cell-cycle arrest, checkpoints, proliferation, apoptosis, DNA repair, and transcription (23). Thus, the functions of CHK1 extend beyond a single checkpoint; in fact, CHK1 has effects on DNA damage checkpoints, DNA replication checkpoints, and spindle checkpoints, and its substrates are still under investigation. This information might prove to be very important because CHK1 currently represents one of the most attractive targets for anticancer therapy in many types of cancer (24, 25). On the other hand, there have been no preclinical investigations on the effect of targeting CHK1 in HCC. Strikingly, the amino acid sequence 290ISRIK295SY localizes to 1 of 23 unique residues located in the IDB in SYK(L), and it combines with a potential CHK1 phosphorylation motif (ØxRxxS/TØ, where Ø represents any hydrophobic amino acid and x represents any amino acid) (26, 27). Consequently, we sought to determine whether SYK(L) is regulated by CHK1. We found that phosphorylation of Ser295 in SYK(L) by CHK1 is the key regulatory mechanism that modulates the various functions of SYK(L), including proliferation, migration, and invasion. Moreover, we demonstrated that the CHK1 inhibitor GÖ6976 sensitizes HCC cells to chemotherapeutic agents, in part through an increase in SYK(L) levels both in vitro and in vivo.
Results
SYK(L), but not SYK(S), is regulated by CHK1 in HCC cells.
The functional differences between SYK(L) and SYK(S) are obvious, as stated above, and phosphorylation of SYK(L) might be crucial for the regulation of SYK(L) function (18, 19). In addition, the potential CHK1 phosphorylation motif 290ISRIK295SY, which is a component in each of the 23 residues that SYK(S) lacks, is conserved among all SYK(L) mammalian orthologs (Figure 1A). Therefore, we considered this motif to be potentially of great importance and decided to perform several experiments to determine whether SYK(L) is actually regulated by CHK1. First, in each of the 6 HCC cell lines that we analyzed, there was an inverse correlation between the protein level of CHK1 and the protein level of SYK(L) (R2 = –0.732, P = 0.030) (Supplemental Figure 1; supplemental material available online with this article; doi: 10.1172/JCI61380DS1). Second, ectopic expression and siRNA of CHK1 were capable of decreasing and increasing the SYK(L) levels in both Huh7 and Hep-3B cells, respectively (Figure 1B), whereas ectopic expression or siRNA of CHK2, a close homolog that shares similar phosphor-targeting sequences, failed to alter the SYK(L) level in these cells (Supplemental Figure 2A). In contrast, the ectopic expression of CHK1 could not downregulate SYK(S), which lacks the potential CHK1 phosphorylation motif, 290ISRIK295SY (Supplemental Figure 2B). Interestingly, the alteration of SYK(L) by ectopic expression or siRNA of CHK1 was at the protein level, but not at the mRNA level, in these cells (Supplemental Figure 2C). Third, a complex containing both CHK1 and SYK(L) was detected using immunoprecipitation in the cells that were ectopically transfected with both Flag-CHK1 and SYK(L) (Figure 1C). More importantly, the interaction between CHK1 and SYK(L) was also reciprocally detected by the endogenous proteins (Figure 1D). In combination, our results strongly suggest that CHK1 physically interacts with and regulates SYK(L) in HCC cells.
Figure 1. SYK(L) is regulated by CHK1 in HCC cells.
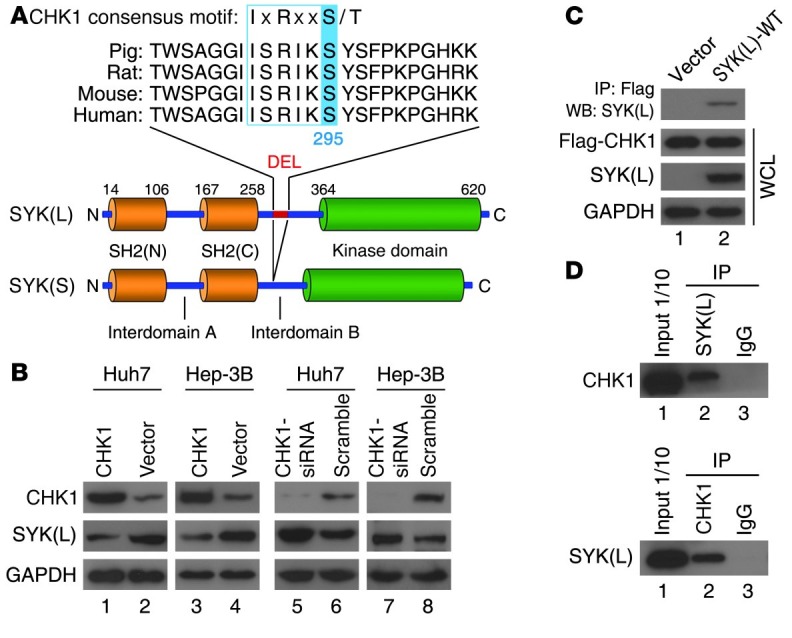
(A) Sequence alignment of the putative CHK1 phosphorylation motif in SYK(L) from different species. The domain structure of SYK(L) protein and its alternative splicing variant SYK(S) are shown. SYK(S) has 23 amino acid residues (DEL) missing in the IDB region, and the sequence homology of the 23-residue DEL across species is shown. (B) The indicated cells were transfected with the indicated plasmids for 24 hours (left) or the indicated siRNAs for 48 hours (right) and then were analyzed by Western blot. (C) SMMC7721 cells without detectable SYK(L) expression were transfected with the indicated plasmids for 24 hours, and the whole cell lysates (WCLs) were resolved directly by SDS-PAGE or were first incubated with Flag agarose and then analyzed by Western blot. (D) Cell lysates from Huh7 cells were immunoprecipitated with anti-CHK1, anti-SYK(L), or preimmune IgG as indicated and were subsequently immunoblotted with anti-SYK(L) or anti-CHK1 antibody. Note: CHK1 associates with endogenous SYK(L).
CHK1 is capable of phosphorylating SYK(L) on Ser295 in vitro and in vivo.
Next, we sought to determine whether SYK(L) is a substrate of CHK1 in vitro and in vivo; to do so, we generated a polyclonal substrate-directed phospho-specific antibody, p-SYK(L)-S295, based on the potential CHK1 phosphorylation motif in SYK(L), 290ISRIK295SY. Indeed, p-SYK(L)-S295 was clearly detected in the cells ectopically transfected with WT SYK(L), but p-SYK(L)-S295 was not detected in cells transfected with the S295A mutant in which the serine residue at position 295 was replaced with alanine (Supplemental Figure 3A). Furthermore, detection by the p-SYK(L)-S295 antibody was sensitive to phosphatase treatment, as the signal of p-SYK(L)-S295 completely disappeared after the cell lysates were incubated with λ-phosphatase (λ-PPase) (Supplemental Figure 3B). Our results demonstrate that in vivo phosphorylation on Ser295 of SYK(L) exists, and the anti–p-SYK(L)-S295 antibody is specific for phosphorylation on Ser295 in SYK(L). Using an in vitro CHK1 kinase assay, we demonstrated that CHK1 phosphorylated the WT SYK(L), but not the S295A mutant (Figure 2A). We also showed that the phosphorylation on Ser295 of SYK(L) was dependent upon CHK1 kinase activity, as the kinase-depleted CHK1 mutant (KD) lacked the ability to phosphorylate WT SYK(L) (Figure 2B). These results demonstrate that CHK1 directly phosphorylates SYK(L) in vitro.
Figure 2. SYK(L) is regulated by CHK1 through phosphorylation on Ser295.

(A) His-tagged SYK(L) WT or the S295A mutant that was translated in vitro were incubated with samples immunoprecipitated from cells with Flag-CHK1 in the presence or the absence of ATP (as indicated) for 2 hours. The samples were then analyzed by Western blot. (B) His-tagged SYK(L) WT protein translated in vitro was incubated with Flag-CHK1 WT or its kinase dead (KD) form, which was immunoprecipitated from cells as indicated for 2 hours. The samples were then analyzed by Western blot. (C) SMMC7721 cells transfected with the indicated plasmids for 24 hours were analyzed by Western blot. (D) SMMC7721 cells were stably transfected with the WT SYK(L) or the mutant SYK(L)-S295A as indicated, and the cells were treated with GÖ6976 (100 nM) or HU (2.5 mM) for 10 hours as indicated and subjected to Western blot. (E) Huh7 cells were transfected with scramble or 3 different CHK1 siRNA duplexes as indicated for 48 hours. The cell lysates were resolved directly by SDS-PAGE (WCL) or were first incubated with anti-SYK(L) antibodies (IP: SYK) and then analyzed by Western blot.
To determine whether CHK1 affects the protein levels of SYK(L) by phosphorylation on Ser295, SMMC7721 cells in which endogenous SYK(L) is undetectable were used (Supplemental Figure 4A). As shown in Figure 2C, WT CHK1, but not its KD form, was able to reduce the levels of WT SYK(L), but not the levels of the S295A mutant. However, the interaction between CHK1 and SYK(L) was not disrupted in cells that expressed the S295A mutant (Supplemental Figure 4B), whereas SYK(S) failed to form a complex with CHK1 (data not shown). GÖ6976 has been widely used to inhibit CHK1 (28–30), although it was initially used as a PKC inhibitor (31), while hydrourea (HU) is a common CHK1 activator. Strikingly, as shown in Figure 2D, the phosphorylation of SYK(L) on Ser295 was decreased and increased by treatment with GÖ6976 and HU, respectively, while the protein levels of WT SYK(L) were correspondingly increased and decreased by such treatments, respectively. However, the protein levels of the S295A mutant were only marginally altered in response to both treatments. These data further support a CHK1’s critical role in regulating the stability of SYK(L) through phosphorylation. Note that Ser345 of CHK1 was expected to increase after CHK1 inhibition due to a feedback mechanism (28–30). Most importantly, as shown in Figure 2E, the phosphorylation of endogenous SYK(L) on Ser295 was markedly reduced when the cells were treated with 3 different siRNA duplexes specifically targeting different CHK1-coding regions, whereas the protein levels of endogenous SYK(L) were correspondingly increased by these siRNA duplexes. These results demonstrate that the phosphorylation on Ser295 of SYK(L) by CHK1 may play a key role in regulating SYK(L) protein levels.
Regulation of SYK(L) by CHK1 occurs via the ubiquitin/proteasome pathway.
Because CHK1 may regulate SYK(L) protein levels by phosphorylating Ser295 and because the ubiquitin/proteasome pathway is the quickest known mechanism for irreversibly degrading proteins, we surmised that CHK1 may regulate SYK(L) by causing it to become ubiquitinated. To test this hypothesis, an in vivo ubiquitination assay was performed. As shown in Figure 3A, in the presence of ectopically expressed CHK1 and MG132 (a specific proteasome inhibitor), WT SYK(L) protein was more heavily ubiquitinated than the S295A mutant. Moreover, WT CHK1, but not its KD form, increased the polyubiquitination of SYK(L) (Figure 3B). As shown in Figure 3C, the half-life of the WT SYK(L) protein was consistently shorter than the half-life of the S295A mutant protein, and, as shown in Figure 3D, CHK1 knockdown by siRNA resulted in a longer half-life for the SYK(L) protein. Collectively, our results strongly indicate that CHK1 promotes SYK(L) protein degradation by phosphorylating Ser295, which elicits the polyubiquitination of SYK(L) and its subsequent degradation by the proteasome.
Figure 3. Regulation of SYK(L) by CHK1 occurs via the ubiquitin/proteasome pathway.
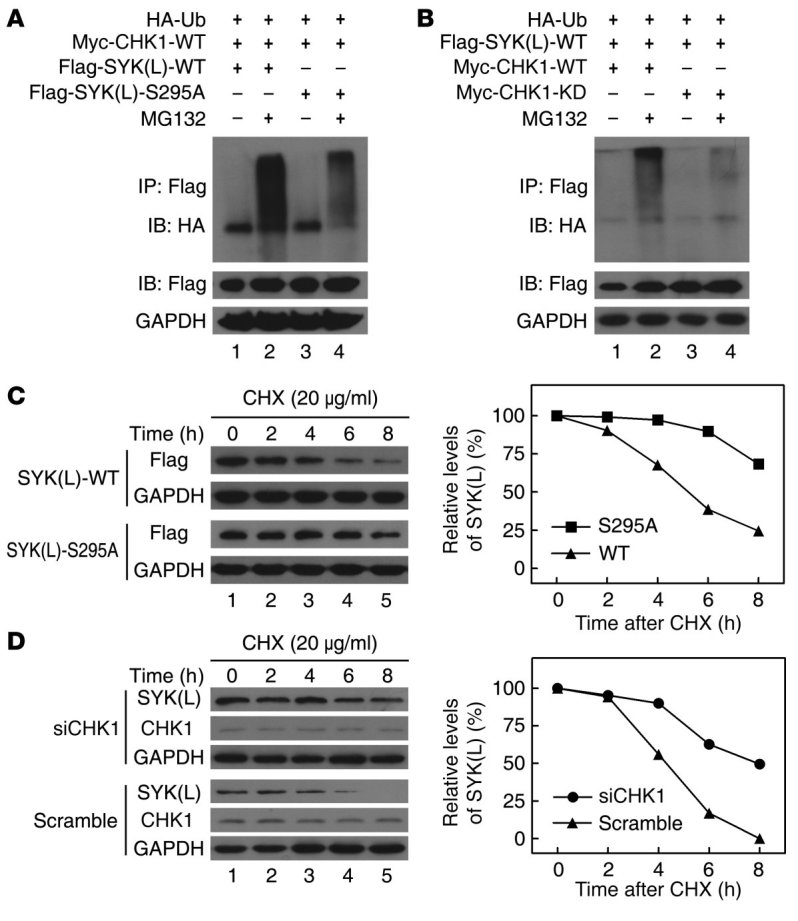
(A and B) Huh7 cells were cotransfected with the indicated plasmids for 20 hours and were treated with or without MG132 (10 μM) for 4 hours. The cell lysates were resolved directly by SDS-PAGE or were first incubated with Flag agarose and then analyzed by Western blot. (C) Huh7 cells stably expressing SYK(L) WT or the S295A mutant were incubated with 20 μg/ml of CHX for the indicated times. Cell lysates were then analyzed by Western blot (left panel), and the densities of the SYK(L) protein bands at each time point were normalized to GAPDH and were calculated into percentages using 100% as the value of the zero time point (right panel). (D) Huh7 cells transfected with scrambled siRNA or CHK1 siRNA were incubated with 20 μg/ml of CHX for the indicated times. Cell lysates were then analyzed as described in C.
The functions of SYK(L) are negatively regulated by CHK1-mediated phosphorylation of Ser295 in HCC.
To determine the biological functions of CHK1-mediated phosphorylation of Ser295 on SYK(L), we generated cells that were stably transfected with a vector control, WT SYK(L), or an S295A mutant. For these transfectants, we used SMMC7721, a HCC cell line with endogenous SYK(L) that is undetectable (Supplemental Figure 4A). As shown in Figure 4A, the cells that were stably transfected with SYK(L)-WT were characterized by an inhibition of cell proliferation in comparison with the cells that were transfected with the vector, which is consistent with the notion that SYK(L) is a tumor suppressor. Strikingly, the cells that were stably transfected with the SYK(L)-S295A mutant displayed less rapid cell proliferation than the cells that expressed the WT protein, indicating that phosphorylation on Ser295 of SYK(L) by CHK1 may negatively regulate the tumor suppressor function of SYK(L) in vitro. Using a colony formation assay with the same stable SMMC7721 transfectants, we further showed that the cells expressing the SYK(L)-S295A mutant grew the fewest number of colonies, whereas the cells expressing the WT protein grew fewer colonies than the vector-transfected cells (Figure 4B). This phenomenon was also observed in Huh7 cells, in which endogenous SYK(L) is detectable (Supplemental Figure 4A), which were stably transfected with these plasmids (Supplemental Figure 5). Since SYK(L) is involved in cancer cell mobility (9), we investigated whether expression of the S295A mutant of SYK(L) would affect cell migration or invasion. As shown in Figure 4C, the cells transfected with SYK(L)-WT displayed inhibited cell migration and inhibited invasion compared with the cells transfected with the vector. Likewise, the cells transfected with the SYK(L)-S295A mutant were more capable of suppressing both cell migration and invasion compared with the cells that expressed SYK(L)-WT. Using a xenograft nude mouse model with the same stable SMMC7721 transfectants, we found that the mice injected with SYK(L)-S295A–expressing cells had the smallest tumor weights, whereas smaller tumor weights were detected in mice injected with SYK(L)-WT–expressing cells compared with those injected with vector-expressing cells (Figure 4D). These results further supported the notion that CHK1 may negatively regulate the antitumor function of SYK(L) by phosphorylating it on Ser295. Mechanistically, as shown in Figure 4E, the Huh7 and SMMC7721 cells that were stably transfected with SYK(L)-S295A demonstrated the strongest inhibition in their expression of MMP2, MMP9, and cyclin D1, which are known downstream targets of SYK(L) (15, 32). These results demonstrate that the phosphorylation on Ser295 of SYK(L) may promote its degradation and modulate its tumor suppressor activity by increasing the expression of its downstream targets.
Figure 4. SYK(L) function is negatively regulated by CHK1-mediated phosphorylation of Ser295 in HCC in vitro and in vivo.

(A) SMMC7721 cells were stably transfected with the indicated plasmids, as shown by Western blot (insertion panel), and the proliferative capacity of these cells was measured by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay at the indicated times (n = 3). The dots represent the mean, while the bars indicate the SEM. *P < 0.05; **P < 0.001, using Student’s t test. (B) The stable cell lines used in A were cultured for 12 days, and the number of colonies was counted and graphed (n = 3). Bars indicate the SEM. P values were obtained by Student’s t test. (C) The stable cell lines used in A were subjected to migration or invasion assays as described in Methods, and the representative photographs are shown in the left panel. Scale bar, 80 μm. The quantification of the left panel was graphed (right panel; n = 3). Bars indicate the SEM. P values were obtained by Student’s t test. (D) The stable cell lines used in A were injected into nude mice (2 × 106 cells per mouse), and after 4 weeks, the xenografts were excised from the mice and weighed. Each dot represents a tumor weight, and the mean tumor weights in each group are indicated by solid lines (n = 8). P values were obtained by Student’s t test. (E) The stable cell lines made from both Huh7 and SMMC7721 cells were subjected to RT-PCR using primers specific for the indicated genes.V, empty vector as control.
Inhibition of CHK1 suppresses tumorigenicity through the CHK1/SYK(L) pathway in vitro and in vivo in HCC.
Because CHK1 regulates the stability of the SYK(L) protein (Figures 1–3) and the S295A mutant of SYK(L) increases its growth inhibitory effects in HCC (Figure 4), we reasoned that CHK1 inhibitors may be effective as potential therapeutic agents for HCC. Given that cisplatin is the standard therapy for chemoembolization for patients with HCC, we sought to determine the effects of the CHK1 inhibitor GÖ6976 alone or the combination of GÖ6976 with cisplatin on HCC in vitro and in vivo. Using an in vitro proliferation assay, as shown in Supplemental Figure 6, A and B, and Supplemental Figure 7, A and B, the CHK1 inhibition by GÖ6976 or siRNA alone suppressed cell proliferation, and GÖ6976 treatment was capable of sensitizing both Huh7 cells and MHCC-97H cells, which are positive for SYK(L), to cisplatin treatment. Furthermore, as shown in Supplemental Figure 8, the combination of GÖ6976 with cisplatin was synergistic. Moreover, as shown in Figure 5A, GÖ6976 further enhanced the inhibition of proliferation of the SMMC7721 cells that were stably transfected with SYK(L)-WT, but not with the S295A mutant, supporting the notion again that regulation of SYK(L) by CHK1 is mediated by the phosphorylation of Ser295.
Figure 5. Inhibition of CHK1 suppresses tumorigenicity through the CHK1/SYK(L) pathway in HCC in vitro and in vivo.
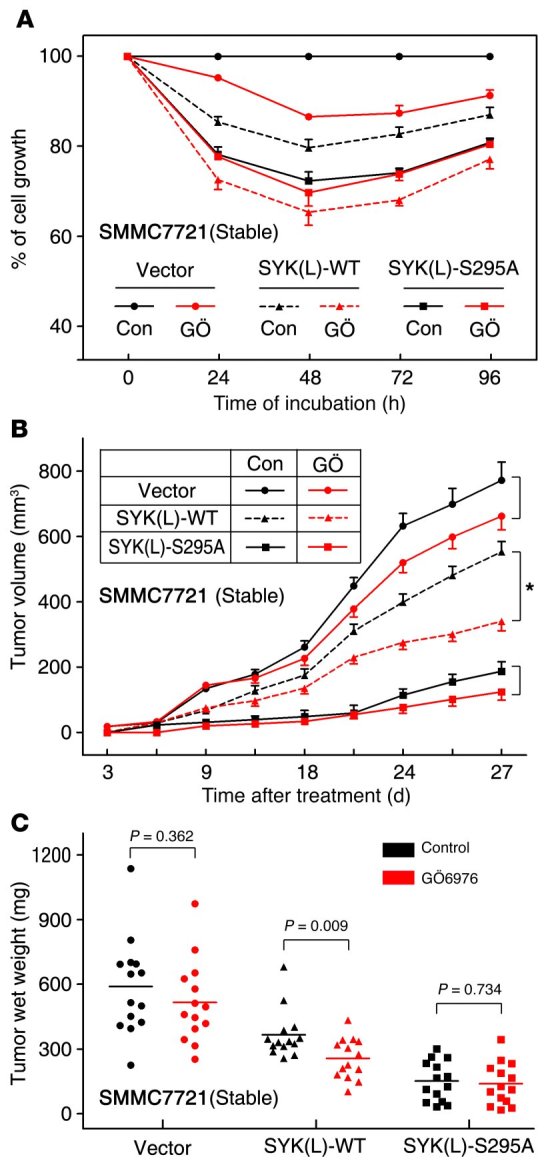
(A) SMMC7721 stable cell lines used in Figure 3A were treated with GÖ6976 (100 nM) or DMSO (Con) as indicated for different amounts of time and were subjected to an MTT assay (n = 3). The dots represent the mean, while the bars indicate the SEM. (B) SMMC7721 stable cell lines were injected into the flanks of nude mice and incubated for 6 days, and then the mice were treated with GÖ6976 (100 nM) or DMSO. The tumor volumes were measured and recorded every 3 days, and tumor growth curves were created for each group (n = 14). Dots represent the mean, while bars indicate the SEM. *P < 0.05, using Student’s t test. (C) On day 27, the xenografts were excised from mice and weighed, as shown in Supplemental Figure 9. Each dot represents a tumor weight; the mean tumor weights of each group were indicated by solid lines (left panel; n = 14), and P values were obtained using the Student’s t test.
To further assess whether CHK1 should be targeted as a potential HCC therapy via the CHK1/SYK(L) pathway, we evaluated tumorigenicity using a xenograft mouse model. After implanting cells into nude mice from HCC cell lines positive for SYK(L) (Huh7 and MHCC-97H cells), we found that tumor growth was significantly impaired by GÖ6976 treatment in each group of mice, and the combination treatment of GÖ6976 and cisplatin was more effective at inhibiting tumor growth than treatment with GÖ6976 or cisplatin alone (P < 0.05) (Supplemental Figure 6, C and D, and Supplemental Figure 7, C and D). Moreover, as shown in Figure 5, B and C, and Supplemental Figure 9, the results from the xenografts derived from the stable SMMC7721 transfectants showed that treatment with GÖ6976 alone had an obvious effect on the growth of the cells transfected with SYK(L)-WT, while GÖ6976 treatment did not affect the growth of cells transfected with the S295A mutant. Indeed, as shown in Supplemental Figure 10, GÖ6976 did decrease the p-SYK(L)-S295 levels and increase the SYK(L) protein levels in the xenografts from the cells transfected with SYK(L)-WT, whereas GÖ6976 had minimal effect on the SYK(L)-S295A protein levels from the corresponding xenografts. Notably, the results from the mice xenografted with the stable SMMC7721 transfectants also showed that the tumors derived from the cells transfected with the SYK(L)-S295A mutant were the smallest, while the tumors derived from cells transfected with the WT protein were smaller than those derived from the vector-transfected cells (Figure 5, B and C, and Supplemental Figure 9). These results reinforce the hypothesis that phosphorylation on Ser295 of SYK(L) by CHK1 may negatively regulate the antitumor function of SYK(L) in vivo. Collectively, these results indicate that the CHK1/SYK(L) pathway may play an important role in HCC and the combination treatment with both cisplatin and CHK1 inhibitors may prove to be an attractive strategy for treating HCC.
An inverse correlation between the protein levels of CHK1 and SYK(L) in HCC tissues.
CHK1 plays key roles in multiple pathways, and it functions as either a tumor suppressor or an oncogene, depending on the tumor type (23). Although there is a lack of information regarding the status of CHK1 in HCC, based on the results in Figure 5 and Supplemental Figures 6, 7, and 9, we speculated that CHK1 may have oncogenic function in HCC. To examine this possibility, we utilized the techniques of quantitative RT-PCR (qRT-PCR), immunoblotting, and immunohistochemistry (IHC) in HCC tissues. Using 20 fresh HCC tissues matched with adjacent nontumor tissues (NTs), our immunoblotting results showed that CHK1 was overexpressed in HCC tissues compared with adjacent NTs (Supplemental Figure 11A). For qRT-PCR and IHC, 162 consecutive HCC tissue samples were obtained from the curative liver resection, and among them, there were 80 cases suitable for qRT-PCR. As shown in Supplemental Figure 11B, the mRNA levels of CHK1 were elevated significantly in HCC tissues compared with NTs (P < 0.001). As shown in Supplemental Figure 11C, CHK1 protein levels were elevated in most of the HCC cases (135/162, 83.3%) by IHC, and CHK1 was mainly detected in the cytoplasm of tumor cells. Moreover, after resection of primary HCC samples, the patients with high CHK1 expression had a significantly shorter RFS and OS than those with low CHK1 expression in their tumors (Figure 6, A and B). It is well established that CHK1 expression is confined to proliferating cells, and as expected, CHK1 did have a correlation with Ki67 (r = 0.586, P < 0.001, χ2 tests), a marker of cell proliferation, in HCC (Supplemental Figure 13), indicating that CHK1 may be oncogenic in HCC and that CHK1 overexpression may be a valuable indicator of prognosis in the patients with HCC.
Figure 6. Overexpression of CHK1 indicates a poor prognosis, and there is an inverse correlation between the protein levels of SYK(L) and CHK1 in HCC.
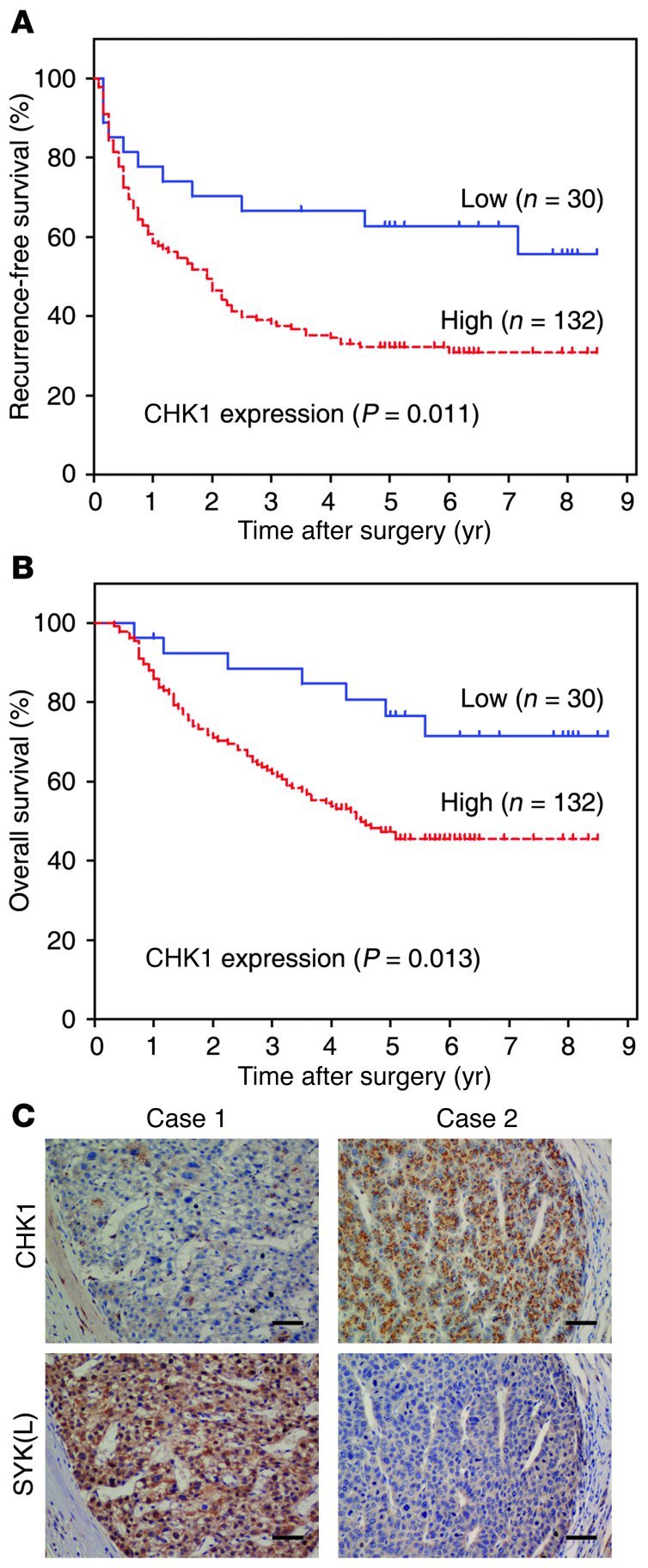
(A and B) RFS (A) and OS (B) curves were generated based on the CHK1 protein expression statuses from 162 HCC samples. Actuarial probabilities were calculated using the Kaplan-Meier method and were compared using the log-rank test. After resection of primary tumors, patients with low CHK1 expression in their primary tumors had better RFS and OS rates than those with high CHK1 expression (P = 0.011 and P = 0.013, respectively). (C) Immunohistochemical staining of CHK1 and SYK(L) was performed in tumor tissues of patients with HCC. Representative examples of CHK1 and SYK(L) staining in the serial sections from the same tumor tissues are shown. Scale bars: 50 μm. Data represent mean ± SEM.
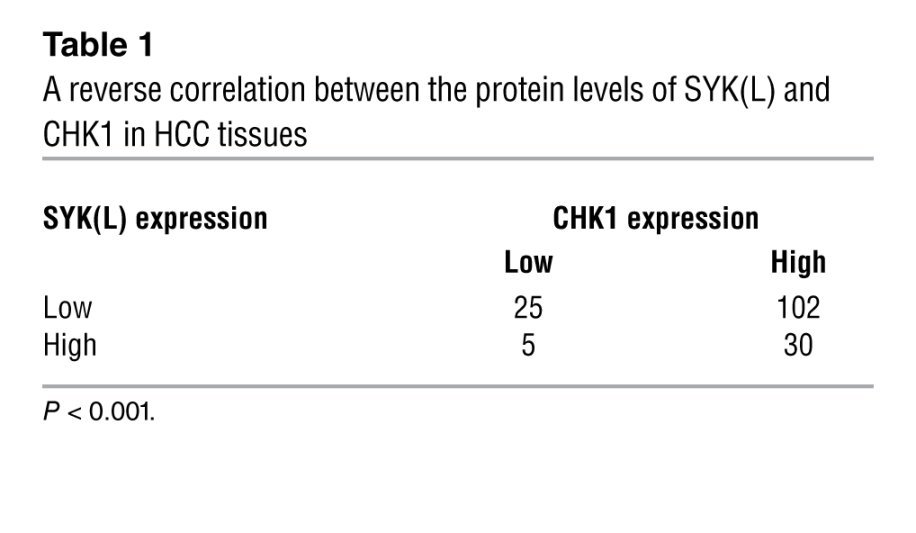
Previous studies have shown that SYK(L) is a tumor suppressor that is frequently silenced or downregulated in HCC (13), which is the opposite of CHK1 in HCC. Based on the negative correlation between the protein levels of CHK1 and SYK(L) (Figures 1 and 2), we were curious as to whether there was a correlation between CHK1 and SYK(L) levels in the 162 HCC tissues that we collected. The anti–p-SYK(L)-S295 antibody worked very well for Western blot, but not for IHC (data not shown). Therefore, we generated a SYK(L)-specific antibody called SYK-23 for IHC. As shown in Supplemental Figure 12, the anti–SYK-23 antibody detected only SYK(L), but not SYK(S). In the 162 HCC tissues, there was an inverse correlation between the protein levels of CHK1 and SYK(L) (P < 0.001, χ2 tests; Figure 6C and Table 1), whereas SYK(L) did not have a correlation with Ki67 in HCC (Supplemental Figure 13), demonstrating that CHK1 may negatively regulate SYK(L) protein in HCC tissues. Interestingly, there were 25 tumors that contained low levels of both SYK(L) and CHK1, indicating that other pathways, in addition to the CHK1 pathway, may be involved in the regulation of SYK(L) protein levels in HCC; for example, methylation on the SYK(L) promoter reduces its expression, as we showed previously (13).
Table 1.
A reverse correlation between the protein levels of SYK(L) and CHK1 in HCC tissues
Discussion
In this report, we demonstrate for what we believe is the first time that phosphorylation on Ser295 of SYK(L) by CHK1 regulates the stability of the protein and its antitumor function; in addition, we show, using in vitro and in vivo models, that the CHK1 inhibitor GÖ6976 sensitizes HCC cells to cisplatin. Importantly, CHK1 protein levels inversely correlated with SYK(L) protein levels in HCC clinical samples. Furthermore, CHK1 may have oncogenic function in HCC, and CHK1 is frequently overexpressed in HCC samples and correlates with poor clinical outcome.
Loss of SYK(L), albeit not exclusively, by hypermethylation of its promoter occurs in many types of cancer (13). In fact, SYK(L) tyrosine promoter methylation, with an associated loss of SYK(L) protein and tumor suppressor activity, has been detected in HCC (13). However, the posttranslational regulation of SYK(L) protein has not yet been explored. In this report, we demonstrate that SYK(L) protein is regulated by the CHK1-mediated phosphorylation on Ser295, which induces polyubiquitination and degradation through the ubiquitin/proteasome pathway. The mutant SYK(L)-S295A was more stable than the WT, and it more efficiently impeded the proliferation of HCC cells than the WT protein in vitro and in vivo. As a tumor suppressor, the function of SYK(L) was obviously enhanced by treatment with the CHK1 inhibitor GÖ6976 in vitro and in vivo. These results strongly indicate that the phosphorylation of SYK(L) on Ser295 is mainly mediated by CHK1. Additionally, the mutant SYK(L)-S295A protein was able to more efficiently impede the proliferation of HCC cells than the WT protein after treatment with GÖ6976 in vitro and in vivo, suggesting that other unknown factors may also be involved in the phosphorylation of SYK(L). We believe this is the first evidence suggesting that the loss or low levels of SYK(L) protein might be partially due to high CHK1 expression in cancers such as HCC, and our results propose a regulatory mechanism for SYK(L) at the posttranslational level. We have previously reported that 27% of clinical HCC samples (33 out of 124) were methylated at the promoter of SYK(L) (13), indicating that the SYK(L) promoter methylation may only account for a smaller percentage of low expression of SYK(L) in HCC. On the other hand, we show here that most of the HCC samples had low expression of SYK(L) (80%, 127 out of 162) and high expression of CHK1 (83%, 132 out of 162), and that there was a strong inverse correlation between the expression levels of CHK1 and SYK(L) in these HCC samples. Therefore, the high expression of CHK1 may account for a larger percentage of low expression of SYK(L) in HCC. For instance, the expression of SYK(L) is regulated by CHK1 in both Huh7 and Hep-3B cells (Figure 1B and Figure 2, C–E), whereas SYK(L) is silenced by its promoter methylation in SMMC7721 (Supplemental Figure 14). Of course, both the SYK(L) promoter methylation and the high expression of CHK1 may simultaneously exist in the same HCC cases. Collectively, we speculate that the low expression of SYK(L) may be due to the high expression of CHK1 and/or its promoter methylation in HCC.
CHK1, a key component of the pathways that preserve genomic integrity during the cell cycle in response to endogenously and exogenously generated DNA lesions, is commonly considered a tumor suppressor in multiple cancer types (33, 34). Conversely, CHK1 has been found to be overexpressed in various tumors compared with the adjacent normal tissues (35–37), and expression of CHK1 protein has been shown to positively correlate with tumor grade and cancer cell proliferation of breast tumors (38). Here, we provide evidence that CHK1 is upregulated in HCC tumor tissues compared with their matched nontumor species and that higher expression of CHK1 is associated with worse OS and RFS in HCC patients, indicating that CHK1 may have oncogenic function in HCC and that CHK1 overexpression may be a valuable indicator of prognosis in the patients with HCC. Interestingly, CHK1 (11q24.2) is located in the chromosomal arm 11q, which was shown to be one of the areas of frequent gain or amplification in HCC (39, 40). Therefore, we speculate that the amplification of chromosomal arm 11q may be one of the mechanisms of increased CHK1 in HCC. Moreover, CHK1 expression levels inversely correlated with SYK(L) protein levels in HCC, which is consistent with the notion that SYK(L) is a tumor suppressor, whereas CHK1 might be oncogenic in HCC. In the setting of the cancer types in which CHK1 is an oncogene (35–37), CHK1 is a potential therapeutic target (41, 42). Indeed, CHK1 inhibitors, such as UCN-01 and GÖ6976, have been utilized as sensitizers in preclinical and clinical trials (28, 43–45). The p53 pathway is crucial, but not required, for the targeting of CHK1 in cancer (46). Cellular responses, including apoptosis, mitotic catastrophe, and senescence, may contribute to the positive outcome (47), and combining CHK1 inhibition with DNA-damaging agents does not lead to preferential killing of p53-deficient cells over p53-proficient cells (48). In this report, we examined the effects of treatment with GÖ6976 alone or in combination with cisplatin, a relatively effective chemotherapeutic drug used in transarterial chemoembolization for advanced-stage HCC patients. We demonstrated that GÖ6976 was much more effective in treating HCC cells expressing SYK(L) than those without SYK(L) in vitro and in vivo, indicating that the combination of CHK1 inhibitors with DNA-damaging agents might be beneficial for HCC patients, depending on the status of the CHK1/SYK(L) pathway in their tumor tissues. These results also suggest that SYK(L), as a downstream target of CHK1, may play a critical role in the function of CHK1 in HCC. We believe this is the first direct evidence that other targets of CHK1, besides key cell-cycle regulators, may also be crucial for mediating the effects of CHK1 inhibitors for the treatment of cancer. This might also partially explain why combining CHK1 inhibition with DNA-damaging agents does not lead to preferential killing of p53-deficient cells over p53-proficient cells, as was shown recently (48).
In summary, we propose a model for the regulation of SYK(L) by CHK1 in HCC (Figure 7): highly expressed CHK1 phosphorylates SYK(L) on Ser295, which triggers the degradation of SYK(L) by the ubiquitin/proteasome pathway and consequently results in the loss of SYK(L)’s inhibitory effects on tumorigenesis. On the other hand, when CHK1 is inhibited by small molecule inhibitors, such as GÖ6976, SYK(L) protein is stabilized and suppresses HCC tumor progression.
Figure 7. A proposed model for the regulation of SYK(L) by CHK1 in HCC.
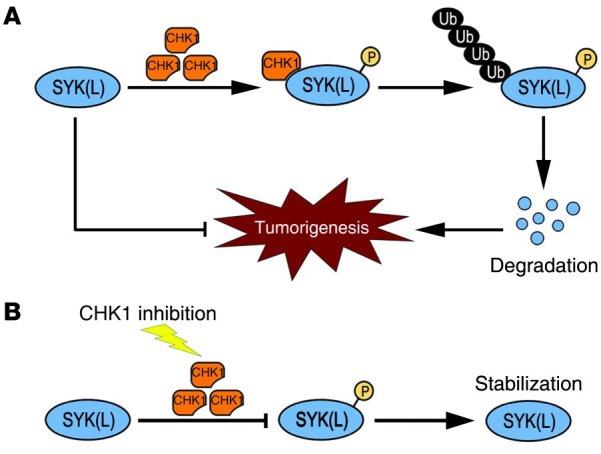
(A) High levels of CHK1 protein may phosphorylate SYK(L) on Ser295, which triggers the degradation of SYK(L) by the ubiquitin/proteasome pathway and consequently favors the tumorigenesis of HCC. (B) When CHK1 is inhibited using small molecule inhibitors, such as GÖ6976, the phosphorylation of SYK(L) is diminished to stabilize the SYK(L) protein, which in turn suppresses tumor progression in HCC.
Methods
Cell lines, stable cell lines, and plasmids.
Human HCC cell lines (MHCC-LM3, MHCC-97H, Huh7, BEL-7402, and SMMC7721) were obtained from the Liver Cancer Institute of Fudan University (Shanghai, China). Human PLC/PRF/5 and Hep-3B HCC cell lines were purchased from ATCC. All cell lines were cultured in high-glucose DMEM (Invitrogen) supplemented with 10% FBS. In some experiments, cells were treated for 5 days with a DNA methyltransferase inhibitor, 5-Aza-2′-deoxycytidine (Sigma-Aldrich), at a final concentration of 2.0 μM, as described previously (13).
Three HCC cell lines, SMMC7721, Huh7, and MHCC-97H, were selected to generate stable cell lines in this study. The retroviral packaging system was purchased from Clontech. Recombinant retroviruses expressing either vector pLNCX2 or pLNCX2 inserted with SYK(L)-WT or SYK(L)-S295A were generated according to the procedure described in the manufacturer’s instructions. These retroviruses were used to infect SMMC7721 and Huh7 cells with 8 μg/ml Polybrene (Sigma-Aldrich) and then were selected with 750 μg/ml of G418 (Calbiochem).
Expression plasmids containing pcDNA3.1-Flag-SYK(L), pcDNA3.1-Flag-CHK1, pcDNA3.1-Flag-CHK2, and HA-tagged ubiquitin were constructed as previously described (32, 49). The pcDNA3.1-myc-CHK1 plasmid was obtained by subcloning from pcDNA3.1-Flag-CHK1. Mutations were introduced using the QuikChange Site-Directed Mutagenesis Kit (Stratagene), and all mutations were verified by DNA sequencing. His-SYK(L) and the mutant His-SYK(L)-295A were inserted into the pET-22b(+) vector by standard cloning methods.
Antibodies and reagents.
Anti-CHK1 was obtained from Novus Biologicals. Antibodies against SYK (N-19), Ki67, actin, and GAPDH were from Santa Cruz Biotechnology Inc. Antibodies against phosphorylated-CHK1 (Ser 345), phosphorylated-cdc2 (Tyr 15), CHK2, His, and HA were obtained from Cell Signaling Technology. Anti-Flag and cisplatin were purchased from Sigma-Aldrich. The CHK1 inhibitor GÖ6976 was purchased from Calbiochem. Anti–SYK-23, an antibody specific for SYK(L), was generated by immunizing rabbits with the coupled peptide 283TWSAGGIISRIKSYSFPKPGHRK305C. p-SYK(L)-S295, an antibody specific for phosphorylation on Ser295 of SYK(L), was generated by immunizing rabbits with the coupled peptide SRIKS295YSFPKPGHRKC.
Transfection experiments.
Transfections were performed using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions. Briefly, HCC cells seeded at 2.5 × 105 cells per well in a 6-well tissue culture dish or at 1 × 106 cells per 10-cm tissue culture dish were transfected with 2 μg or 12 μg plasmid DNA for 24 hours, respectively, and then the cells were treated with the indicated chemicals.
RNAi treatment and cycloheximide chase assay.
This procedure was performed as previously described (49). Briefly, Huh7 cells were transfected with siRNA oligonucleotides using the Lipofectamine RNAiMAX transfection reagent (Invitrogen) for 48 hours. The oligos corresponding to CHK1 were termed siRNA-CHK1-1 (5′-GCGUGCCGUAGACUGUCCA-3′), siRNA-CHK1-2 (5′-GAAGCAGTCGCAGTGAAGA-3′), and siRNA-CHK1-3 (5′-ACAGUAUUUCGGUAUAAUA-3′). For the cycloheximide (CHX) chase assay, cells were treated with 20 μg/ml CHX for 0, 2, 4, 6, and 8 hours and then were lysed and analyzed by Western blot.
Immunoblotting and immunoprecipitation.
The immunoblotting and immunoprecipitation procedures were performed as previously described (49). Sixty micrograms of total protein from each sample was resolved by 10% SDS-PAGE and transferred to polyvinylidene difluoride membrane (Millipore). The blots were then incubated with various antibodies. For immunoprecipitation, cell lysates from 5 × 106 cells were incubated with 1 μg antibody at 4°C overnight, followed by the addition of 30 μl of protein A/G-conjugated agarose beads (Santa Cruz Biotechnology Inc.), which were prewashed with lysis buffer 4 times. The precipitates were washed 4 times with ice-cold PBS, resuspended in 6× Laemmli buffer, and resolved by SDS-PAGE, followed by immunoblotting.
In vitro kinase assays.
His-SYK(L) and His-SYK(L)-295A were synthesized in vitro using the S30 T7 High-Yield Protein Expression System (Promega). The in vitro kinase assay has been described previously (49). Briefly, Flag-CHK1-WT or Flag-CHK1-KD was obtained by immunoprecipitation using anti–Flag agarose from the lysates of cells overexpressing Flag-CHK1-WT or Flag-CHK1-KD. The samples were then incubated with 3 μg of His-SYK(L) or His-SYK(L)-295A in 50 μl of kinase buffer (Cell Signaling Technology) containing 10 μM ATP for 30 minutes at 30°C. Subsequently, the samples were dissolved in SDS-PAGE sample buffer and subjected to Western blotting.
λ-PPase treatment.
Flag-SYK(L) was immunoprecipitated with anti–Flag agarose from the lysates of cells overexpressing Flag-SYK(L), resuspended in bacteriophage λ-PPase buffer containing 200 U of enzyme (New England BioLabs), and incubated for 1 hour at 30°C. The reaction was stopped by heat inactivation at 65°C for 1 hour.
In vivo ubiquitination assay.
This procedure was performed as previously described (49). Briefly, Huh7 cells were transfected with the indicated plasmids for 24 hours and were treated with 10 μM MG132 for 6 hours prior to harvesting. The cells were lysed in RIPA buffer with protease inhibitor cocktail (Sigma-Aldrich) and phosphatase inhibitor cocktail (Calbiochem). SYK(L)-WT and SYK(L)-295A were immunoprecipitated using anti–Flag agarose for 4 hours at 4°C. Polyubiquitinated SYK(L) was detected using an anti-HA antibody.
Colony formation assays and tumorigenicity assays.
Colony formation assays were performed as previously described (50). Five-week-old male nude mice were purchased from Shanghai SLAC Laboratory Animal Co. and maintained in microisolator cages. A total of 2 × 106 cells (SMMC7721 stable cell lines overexpressing vector, SYK[L]-WT or SYK[L]-S295A) were suspended in 200 μl serum-free DMEM culture with 25% Matrigel (BD Biosciences) and injected into the flanks of nude mice for 4 weeks. The tumors were then excised from the mice and harvested, weighed, and photographed.
Cell migration and invasion assays.
In vitro cell migration assays were performed in Transwell chambers (8-μm pore size; Costar) according to the manufacturer’s instructions. 2 × 104 cells were placed into the top chamber of each insert (BD Biosciences) and incubated at 37°C for 48 hours. Similar inserts coated with Matrigel were used to determine invasive potential for cell invasion assays.
Cell viability assays.
SMMC7721, Huh7 and MHCC-97H cells were seeded at 2,500, 6,000, and 5,500 cells per well, respectively, in 96-well microplates. These cells were then treated with different concentrations of cisplatin (6.7 μM), GÖ6976 (100 nM), or DMSO control for the indicated hours, and cell viability was measured by the Cell Counting Kit-8 (CCK-8) assay kit (Dojindo Corp.) according to the manufacturer’s instructions. Three independent experiments were performed.
MTS assay.
An MTS assay (CellTiter 96 Aqueous One Solution Reagent; Promega Corp.) was used to determine the number of viable cells in proliferation, as described previously (51). Cells were plated in quadruplicate onto the 96-well plates according to the manufacturer’s instructions. After attachment (12 hours later), cells were treated with cisplatin and GÖ6976 in a serial fixed-ratio concentration for 72 hours. After treatment with drugs for 68 hours, 20 μl MTS was added to each well and incubated for 4 hours. Absorbance was read at a wavelength of 490 nm. Values for untreated groups were considered as 100% viability. The values of treatment groups were compared with the control groups. The effects of cotreatment with these 2 drugs were determined by using CalcuSyn software, according to the manufacturer’s instruction. The combination index (CI) was characterized as follows: CI < 1, synergy; CI = 1, antagonism; CI > 1, additive.
RT-PCR.
RT-PCR was performed as previously described (13). Briefly, total RNA was treated with DNase I (TaKaRa), and 2 μg aliquots were used for cDNA synthesis using random hexamers with Superscript III (Invitrogen). The cDNA templates were subjected to PCR amplification. The primers are shown in Supplemental Table 1.
Patients and specimens.
HCC samples and their adjacent nontumor samples were obtained from 162 consecutive patients who had undergone curative liver resection from January 2003 to April 2004 at the Sun Yat-sen University Cancer Center. The diagnoses were confirmed by histological reviews. None of the patients received anticancer treatment before hepatectomy. The last follow-up was on May 31, 2011. The end point for RFS was defined as the date when evidence of metastasis or recurrence was clear or as the date of last follow-up when death without recurrence ended the observation. OS was computed from the day of surgery to the day of death or to the last follow-up.
IHC staining.
The IHC procedure we used has been described in previous studies (13, 49). Briefly, formalin-fixed, paraffin-embedded tissue samples from consenting patients were cut in 4-μm sections and placed on polylysine-coated slides; then the samples were deparaffinized in xylene and rehydrated using a series of graded alcohols. The tissue slides were then treated with 3% hydrogen peroxide in methanol for 10 minutes to exhaust endogenous peroxidase activity, and the antigens were retrieved in 0.01 M sodium citrate buffer (pH 6.0) using a microwave oven. After 30 minutes of preincubation in 10% normal goat serum to prevent nonspecific staining, the samples were incubated overnight using a primary antibody, either anti-Ki67 (1:100), anti-CHK1 (1:200), or anti-SYK-23 (1:100), in a humidified container at 4°C. The tissue slides were treated with a nonbiotin horseradish peroxidase detection system according to the manufacturer’s instructions (Gene Tech). The results of IHC were evaluated by 2 different pathologists who specialize in liver cancer. The intensity of staining for Ki67, CHK1 and SYK(L) on the scale of 0 to 3 according to the percentage of positive tumor (0, <5% positive cells; 1, 5%–20%; 2, 20%–50%; and 3, >50%) was recorded. Ki67, CHK1, and SYK(L) expression were classified as high level when the score reached 2; if the score was 1 or less, the case was classified as low expressing.
Antitumor assay using a nude mouse model in vivo.
A total of 2 × 106 cells were suspended in 200 μl serum-free DMEM with 25% Matrigel and injected into the flanks of mice. Intraperitoneal injections of cisplatin (3 mg/kg per week for 4 weeks), GÖ6976 (2 mg/kg), or a mixture of cisplatin (2 mg/kg per week for 4 weeks) and GÖ6976 (2 mg/kg) were administered every 3 days; the control group received 200 μl of 0.1% DMSO. Once palpable tumors were observed, tumor volume measurements were taken every 3 days using calipers. The tumor volume was calculated using the following formula: V = (width2 × length)/2. Body weights were also recorded. The tumor samples from mice were collected and analyzed for Western blots or IHC.
Statistics.
Data represent mean ± SEM. A 2-tailed, unpaired Student’s t test or the Mann-Whitney U test was employed to compare the values between subgroups. An association between CHK1 and SYK(L) abundance was analyzed using χ2 tests. Survival curves were constructed using the Kaplan-Meier method and were compared using the log-rank test. Statistical analyses were performed using SPSS 16.0 software. We considered P < 0.05 as significant and P < 0.001 as strongly significant.
Study approval.
Animal experiments were approved by the Animal Research Committee of Sun Yat-sen University Cancer Center and were performed in accordance with established guidelines. The use of human HCC tissues was reviewed and approved by the ethical committee of Sun Yat-sen University Cancer Center, and informed consent was obtained. The samples were retrospectively acquired from the surgical pathology archives of Sun Yat-sen University Cancer Center.
Supplementary Material
Acknowledgments
We would like to thank the members of the laboratory for their helpful comments on the manuscript. This work was supported by grants from the 973 project (2010CB912201 and 2012CB967000 to T. Kang) and from the National Nature Science Foundation in China (NSFC) (30872489, 30972916, and 81172344 to Y. Yuan, 30930045 and 81125015 to T. Kang).
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Citation for this article: J Clin Invest. 2012;122(6):2165–2175. doi:10.1172/JCI61380.
References
- 1.El-Serag HB, Rudolph KL. Hepatocellular carcinoma: epidemiology and molecular carcinogenesis. Gastroenterology. 2007;132(7):2557–2576. doi: 10.1053/j.gastro.2007.04.061. [DOI] [PubMed] [Google Scholar]
- 2. Caldwell S, Park SH. The epidemiology of hepatocellular cancer: from the perspectives of public health problem to tumor biology. J Gastroenterol. 2009. 44 suppl 19: 96 101 10.1007/s00535-008-2258-6 [DOI] [PubMed] [Google Scholar]
- 3.Portolani N, et al. Early and late recurrence after liver resection for hepatocellular carcinoma: prognostic and therapeutic implications. Ann Surg. 2006;243(2):229–235. doi: 10.1097/01.sla.0000197706.21803.a1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lopez PM, Villanueva A, Llovet JM. Systematic review: evidence-based management of hepatocellular carcinoma--an updated analysis of randomized controlled trials. Aliment Pharmacol Ther. 2006;23(11):1535–1547. doi: 10.1111/j.1365-2036.2006.02932.x. [DOI] [PubMed] [Google Scholar]
- 5.Yeo W, et al. A randomized phase III study of doxorubicin versus cisplatin/interferon alpha–2b/doxorubicin/fluorouracil (PIAF) combination chemotherapy for unresectable hepatocellular carcinoma. J Natl Cancer Inst. 2005;97(20):1532–1538. doi: 10.1093/jnci/dji315. [DOI] [PubMed] [Google Scholar]
- 6.Llovet JM, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359(4):378–390. doi: 10.1056/NEJMoa0708857. [DOI] [PubMed] [Google Scholar]
- 7.Cheng AL, et al. Efficacy and safety of sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma: a phase III randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2009;10(1):25–34. doi: 10.1016/S1470-2045(08)70285-7. [DOI] [PubMed] [Google Scholar]
- 8.Coopman PJ, et al. The Syk tyrosine kinase suppresses malignant growth of human breast cancer cells. Nature. 2000;406(6797):742–747. doi: 10.1038/35021086. [DOI] [PubMed] [Google Scholar]
- 9.Coopman PJ, Mueller SC. The Syk tyrosine kinase: a new negative regulator in tumor growth and progression. Cancer Lett. 2006;241(2):159–173. doi: 10.1016/j.canlet.2005.11.004. [DOI] [PubMed] [Google Scholar]
- 10.Luangdilok S, et al. Syk tyrosine kinase is linked to cell motility and progression in squamous cell carcinomas of the head and neck. Cancer Res. 2007;67(16):7907–7916. doi: 10.1158/0008-5472.CAN-07-0331. [DOI] [PubMed] [Google Scholar]
- 11.Sung YM, et al. Tumor suppressor function of Syk in human MCF10A in vitro and normal mouse mammary epithelium in vivo. PLoS One. 2009;4(10):e7445. doi: 10.1371/journal.pone.0007445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang X, Shrikhande U, Alicie BM, Zhou Q, Geahlen RL. Role of the protein tyrosine kinase Syk in regulating cell–cell adhesion and motility in breast cancer cells. Mol Cancer Res. 2009;7(5):634–644. doi: 10.1158/1541-7786.MCR-08-0371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yuan Y, et al. Frequent epigenetic inactivation of spleen tyrosine kinase gene in human hepatocellular carcinoma. Clin Cancer Res. 2006;12(22):6687–6695. doi: 10.1158/1078-0432.CCR-06-0921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nakashima H, et al. Clinical significance of nuclear expression of spleen tyrosine kinase (Syk) in gastric cancer. Cancer Lett. 2006;236(1):89–94. doi: 10.1016/j.canlet.2005.05.022. [DOI] [PubMed] [Google Scholar]
- 15.Layton T, Stalens C, Gunderson F, Goodison S, Silletti S. Syk tyrosine kinase acts as a pancreatic adenocarcinoma tumor suppressor by regulating cellular growth and invasion. Am J Pathol. 2009;175(6):2625–2636. doi: 10.2353/ajpath.2009.090543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang L, et al. Alternative splicing disrupts a nuclear localization signal in spleen tyrosine kinase that is required for invasion suppression in breast cancer. Cancer Res. 2003;63(15):4724–4730. [PubMed] [Google Scholar]
- 17.Ogane S, Onda T, Takano N, Yajima T, Uchiyama T, Shibahara T. Spleen tyrosine kinase as a novel candidate tumor suppressor gene for human oral squamous cell carcinoma. Int J Cancer. 2009;124(11):2651–2657. doi: 10.1002/ijc.24237. [DOI] [PubMed] [Google Scholar]
- 18.Agrawal R, Carpino N, Tsygankov A. TULA proteins regulate activity of the protein tyrosine kinase Syk. J Cell Biochem. 2008;104(3):953–964. doi: 10.1002/jcb.21678. [DOI] [PubMed] [Google Scholar]
- 19.Chen X, et al. Determination of the substrate specificity of protein–tyrosine phosphatase TULA–2 and identification of Syk as a TULA–2 substrate. J Biol Chem. 2010;285(41):31268–31276. doi: 10.1074/jbc.M110.114181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Prinos P, et al. Alternative splicing of SYK regulates mitosis and cell survival. Nat Struct Mol Biol. 2011;18(6):673–679. doi: 10.1038/nsmb.2040. [DOI] [PubMed] [Google Scholar]
- 21.Zhao H, Piwnica–Worms H. ATR–mediated checkpoint pathways regulate phosphorylation and activation of human Chk1. Mol Cell Biol. 2001;21(13):4129–4139. doi: 10.1128/MCB.21.13.4129-4139.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jazayeri A, et al. ATM– and cell cycle–dependent regulation of ATR in response to DNA double–strand breaks. Nat Cell Biol. 2006;8(1):37–45. doi: 10.1038/ncb1337. [DOI] [PubMed] [Google Scholar]
- 23.Dai Y, Grant S. New insights into checkpoint kinase 1 in the DNA damage response signaling network. Clin Cancer Res. 2010;16(2):376–383. doi: 10.1158/1078-0432.CCR-09-1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Merry C, Fu K, Wang J, Yeh IJ, Zhang Y. Targeting the checkpoint kinase Chk1 in cancer therapy. Cell Cycle. 2010;9(2):279–283. doi: 10.4161/cc.9.2.10445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dent P, Tang Y, Yacoub A, Dai Y, Fisher PB, Grant S. CHK1 inhibitors in combination chemotherapy: thinking beyond the cell cycle. Mol Interv. 2011;11(2):133–140. doi: 10.1124/mi.11.2.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hutchins JR, Hughes M, Clarke PR. Substrate specificity determinants of the checkpoint protein kinase Chk1. FEBS Lett. 2000;466(1):91–95. doi: 10.1016/S0014-5793(99)01763-9. [DOI] [PubMed] [Google Scholar]
- 27.O’Neill T, et al. Determination of substrate motifs for human Chk1 and hCds1/Chk2 by the oriented peptide library approach. J Biol Chem. 2002;277(18):16102–16115. doi: 10.1074/jbc.M111705200. [DOI] [PubMed] [Google Scholar]
- 28.Feng Z, Xu S, Liu M, Zeng YX, Kang T. Chk1 inhibitor Go6976 enhances the sensitivity of nasopharyngeal carcinoma cells to radiotherapy and chemotherapy in vitro and in vivo. Cancer Lett. 2010;297(2):190–197. doi: 10.1016/j.canlet.2010.05.011. [DOI] [PubMed] [Google Scholar]
- 29.Beckerman R, et al. A role for Chk1 in blocking transcriptional elongation of p21 RNA during the S–phase checkpoint. Genes Dev. 2009;23(11):1364–1377. doi: 10.1101/gad.1795709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Leung–Pineda V, Ryan CE, Piwnica–Worms H. Phosphorylation of Chk1 by ATR is antagonized by a Chk1–regulated protein phosphatase 2A circuit. Mol Cell Biol. 2006;26(20):7529–7538. doi: 10.1128/MCB.00447-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Martiny–Baron G, et al. Selective inhibition of protein kinase C isozymes by the indolocarbazole Go 6976. J Biol Chem. 1993;268(13):9194–9197. [PubMed] [Google Scholar]
- 32.Wang L, Devarajan E, He J, Reddy SP, Dai JL. Transcription repressor activity of spleen tyrosine kinase mediates breast tumor suppression. Cancer Res. 2005;65(22):10289–10297. doi: 10.1158/0008-5472.CAN-05-2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Niida H, et al. Cooperative functions of Chk1 and Chk2 reduce tumour susceptibility in vivo. EMBO J. 2010;29(20):3558–3570. doi: 10.1038/emboj.2010.218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tort F, et al. Checkpoint kinase 1 (CHK1) protein and mRNA expression is downregulated in aggressive variants of human lymphoid neoplasms. Leukemia. 2005;19(1):112–117. doi: 10.1038/sj.leu.2403571. [DOI] [PubMed] [Google Scholar]
- 35.Madoz-Gúrpide J, Cañamero M, Sanchez L, Solano J, Alfonso P, Casal JI. A proteomics analysis of cell signaling alterations in colorectal cancer. Mol Cell Proteomics. 2007;6(12):2150–2164. doi: 10.1074/mcp.M700006-MCP200. [DOI] [PubMed] [Google Scholar]
- 36.Verlinden L, et al. The E2F–regulated gene Chk1 is highly expressed in triple–negative estrogen receptor /progesterone receptor /HER–2 breast carcinomas. Cancer Res. 2007;67(14):6574–6581. doi: 10.1158/0008-5472.CAN-06-3545. [DOI] [PubMed] [Google Scholar]
- 37.Cho SH, Toouli CD, Fujii GH, Crain C, Parry D. Chk1 is essential for tumor cell viability following activation of the replication checkpoint. Cell Cycle. 2005;4(1):131–139. doi: 10.4161/cc.4.1.1299. [DOI] [PubMed] [Google Scholar]
- 38.Lundgren K, Holm K, Nordenskjold B, Borg A, Landberg G. Gene products of chromosome 11q and their association with CCND1 gene amplification and tamoxifen resistance in premenopausal breast cancer. Breast Cancer Res. 2008;10(5):R81. doi: 10.1186/bcr2150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wilkens L, Bredt M, Flemming P, Kubicka S, Klempnauer J, Kreipe H. Cytogenetic aberrations in primary and recurrent fibrolamellar hepatocellular carcinoma detected by comparative genomic hybridization. Am J Clin Pathol. 2000;114(6):867–874. doi: 10.1309/BMTT-JBPD-D13H-1UVD. [DOI] [PubMed] [Google Scholar]
- 40.Chen YJ, Chen PJ, Lee MC, Yeh SH, Hsu MT, Lin CH. Chromosomal analysis of hepatic adenoma and focal nodular hyperplasia by comparative genomic hybridization. Genes Chromosomes Cancer. 2002;35(2):138–143. doi: 10.1002/gcc.10103. [DOI] [PubMed] [Google Scholar]
- 41.Bolderson E, Richard DJ, Zhou BB, Khanna KK. Recent advances in cancer therapy targeting proteins involved in DNA double–strand break repair. Clin Cancer Res. 2009;15(20):6314–6320. doi: 10.1158/1078-0432.CCR-09-0096. [DOI] [PubMed] [Google Scholar]
- 42.Ma CX, Janetka JW, Piwnica-Worms H. Death by releasing the breaks: CHK1 inhibitors as cancer therapeutics. Trends Mol Med. 2011;17(2):88–96. doi: 10.1016/j.molmed.2010.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Welch S, et al. UCN-01 in combination with topotecan in patients with advanced recurrent ovarian cancer: a study of the Princess Margaret Hospital Phase II consortium. Gynecol Oncol. 2007;106(2):305–310. doi: 10.1016/j.ygyno.2007.02.018. [DOI] [PubMed] [Google Scholar]
- 44.Perez RP, et al. Modulation of cell cycle progression in human tumors: a pharmacokinetic and tumor molecular pharmacodynamic study of cisplatin plus the Chk1 inhibitor UCN-01 (NSC 638850). Clin Cancer Res. 2006;12(23):7079–7085. doi: 10.1158/1078-0432.CCR-06-0197. [DOI] [PubMed] [Google Scholar]
- 45.Fracasso PM, et al. A Phase 1 study of UCN–01 in combination with irinotecan in patients with resistant solid tumor malignancies. Cancer Chemother Pharmacol. 2011;67(6):1225–1237. doi: 10.1007/s00280-010-1410-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen Z, et al. Selective Chk1 inhibitors differentially sensitize p53–deficient cancer cells to cancer therapeutics. Int J Cancer. 2006;119(12):2784–2794. doi: 10.1002/ijc.22198. [DOI] [PubMed] [Google Scholar]
- 47.Al-Ejeh F, Kumar R, Wiegmans A, Lakhani SR, Brown MP, Khanna KK. Harnessing the complexity of DNA–damage response pathways to improve cancer treatment outcomes. Oncogene. 2010;29(46):6085–6098. doi: 10.1038/onc.2010.407. [DOI] [PubMed] [Google Scholar]
- 48.Zenvirt S, Kravchenko–Balasha N, Levitzki A. Status of p53 in human cancer cells does not predict efficacy of CHK1 kinase inhibitors combined with chemotherapeutic agents. Oncogene. 2010;29(46):6149–6159. doi: 10.1038/onc.2010.343. [DOI] [PubMed] [Google Scholar]
- 49.Kang T, et al. GSK–3 beta targets Cdc25A for ubiquitin–mediated proteolysis, and GSK–3 beta inactivation correlates with Cdc25A overproduction in human cancers. Cancer Cell. 2008;13(1):36–47. doi: 10.1016/j.ccr.2007.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liang Y, et al. Stem–like cancer cells are inducible by increasing genomic instability in cancer cells. J Biol Chem. 2010;285(7):4931–4940. doi: 10.1074/jbc.M109.048397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jin Y, et al. Antineoplastic mechanisms of niclosamide in acute myelogenous leukemia stem cells: inactivation of the NF-kappaB pathway and generation of reactive oxygen species. Cancer Res. 2010;70(6):2516–2527. doi: 10.1158/0008-5472.CAN-09-3950. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.