

Abstract
Objective:
To assess the frequency of mutations in C19orf12 in the greater neurodegeneration with brain iron accumulation (NBIA) population and further characterize the associated phenotype.
Methods:
Samples from 161 individuals with idiopathic NBIA were screened, and C19orf12 mutations were identified in 23 subjects. Direct examinations were completed on 8 of these individuals, and medical records were reviewed on all 23. Histochemical and immunohistochemical studies were performed on brain tissue from one deceased subject.
Results:
A variety of mutations were detected in this cohort, in addition to the Eastern European founder mutation described previously. The characteristic clinical features of mitochondrial membrane protein-associated neurodegeneration (MPAN) across all age groups include cognitive decline progressing to dementia, prominent neuropsychiatric abnormalities, and a motor neuronopathy. A distinctive pattern of brain iron accumulation is universal. Neuropathologic studies revealed neuronal loss, widespread iron deposits, and eosinophilic spheroidal structures in the basal ganglia. Lewy neurites were present in the globus pallidus, and Lewy bodies and neurites were widespread in other areas of the corpus striatum and midbrain structures.
Conclusions:
MPAN is caused by mutations in C19orf12 leading to NBIA and prominent, widespread Lewy body pathology. The clinical phenotype is recognizable and distinctive, and joins pantothenate kinase–associated neurodegeneration and PLA2G6-associated neurodegeneration as one of the major forms of NBIA.
Since the discovery of the first major gene causing neurodegeneration with brain iron accumulation (NBIA) in 2000, significant progress has been made in delineating the genetic and clinical features of various forms. This first gene, PANK2, accounts for one-third of all NBIA cases, and disease in these cases has been denoted pantothenate kinase–associated neurodegeneration (PKAN).1 Subsequently identified genes each account for a small proportion of idiopathic cases, but a significant population of patients with clinical and radiographic features of NBIA still have no identified underlying genetic cause.
A new NBIA gene, C19orf12, was recently reported with a description of the associated phenotype called MPAN (mitochondrial membrane protein-associated neurodegeneration).2 Because the cohort described is ethnically homogeneous, and the majority of subjects are homozygous for a founder mutation, the relevance of this gene to the greater NBIA population remains unclear. We screened all idiopathic NBIA cases in our research registry for mutations in C19orf12. Established 21 years ago, this registry contains a heterogeneous mix of subjects from the United States and 37 other countries. This approach has enabled us to identify novel pathogenic mutations in C19orf12 and to expand our understanding of the phenotype and natural history of MPAN beyond that of the original cohort described.
METHODS
Subjects
We recruited subjects into an NBIA research repository that includes clinical data and associated DNA samples. Subjects were recruited through a research listing on GeneTests, the Oregon Health & Science University Web site, and at NBIA Disorders Association international family conferences. Samples from 161 individuals with idiopathic NBIA were screened for mutations in C19orf12 as described below. The cohort of subjects screened for C19orf12 mutations were all determined to have NBIA based on MRI evidence of high brain iron in the basal ganglia, most often in the globus pallidus and without an eye-of-the-tiger sign, as well as accompanying neurologic signs and symptoms consistent with NBIA (dystonia, spasticity, or parkinsonism). Most were previously screened for PANK2 and PLA2G6 mutations. The group was ethnically diverse, originating from 5 continents, and clinically heterogeneous with regard to age at onset and symptom progression. In 23 subjects, mutations were identified in C19orf12. Direct examinations were completed on 8 of these subjects and medical records were reviewed on all 23. Only partial clinical data were available on some subjects.
Standard protocol approvals, registrations, and patient consents
DNA and clinical information were collected and used after participants gave written informed consent according to the protocol approved by the institutional review board at Oregon Health & Science University.
Analysis of mutations
We used primer sets (table e-1 on the Neurology® Web site at www.neurology.org) to amplify all C19orf12 exons and adjacent intronic sequences, including splice signals, and sequenced DNA from at least one affected individual in each family. We did not routinely confirm sequence variants in parental samples. We analyzed sequence variants that did not cause a frameshift or stop codon using SIFT, PolyPhen-2, PMut, and PhD-SNP algorithms to predict pathogenicity.3–6
Neuropathologic studies
We detected 2 C19orf12 mutations in DNA from a deceased individual who was previously coded as having idiopathic NBIA in our repository. Brain tissue was already in our possession, obtained from the NICHD Brain and Tissue Bank for Developmental Disorders (http://medschool.umaryland.edu/BTBank/). On this specimen, we performed histochemical and immunohistochemical studies as previously described7 using tau-2 anti-tau antibody (Sigma, St. Louis, MO), anti-α-synuclein (Thermo Scientific, Rockford, IL), anti-ubiquitin (Dako, Carpinteria, CA), and anti-TDP-43 (Proteintech Group, Chicago, IL). In brief, paraffin sections were stained with Perls Prussian blue iron stain, Luxol fast blue, and hematoxylin & eosin using standard methods. Immunohistochemistry was performed on deparaffinized sections after antigen retrieval and results developed with appropriate secondary antibodies and diaminobenzidine or Vector red chromagens.
RESULTS
Genetic findings
All subjects were either sporadic within a family or had affected siblings, consistent with an autosomal recessive pattern of inheritance. We found mutations in C19orf12 in 23 patients with idiopathic NBIA. Table e-2 summarizes the mutations identified in this cohort, including several not previously reported. The common deletion c.204_214del11, reported originally in a Polish cohort,2 was found in a homozygous state in 3 unrelated subjects. One originated from Bosnia, the second from Ukraine, and the third had German, Hungarian, and Scottish ancestry. Four additional individuals, including one sibpair, were compound heterozygotes for c.204_214del11 and a second missense mutation (table e-2). These individuals also reported Polish or other Eastern European ancestry.
In 4 families, we identified only single mutations (table e-2). However, based on the predicted deleterious nature of these mutations and clinical findings similar to those from 2-mutation cases, we categorized them as having MPAN. In 6 additional cases, we identified heterozygous variations and determined them most likely to be single nucleotide polymorphisms (SNPs) without clinical consequence. We based these conclusions on algorithm predictions of neutrality and, in 2 cases, lack of homozygosity in cases of known consanguinity. We also noted that the SNP c.424A>G was always found in association with the c.205G>A mutation.
Clinical findings
Table 1 summarizes the main clinical features of patients with MPAN in our cohort. The reported age at onset ranged from 4 to 30 years with a mean of 11 years. Among sibships, the age at onset was within the same year for 2 pairs and within 2 years for a sibship of 3. Of the 23 subjects in our cohort, 19 are still living and are in their teens to late 30s with varying degrees of disability.
Table 1.
Clinical features of mitochondrial membrane protein-associated neurodegenerationa
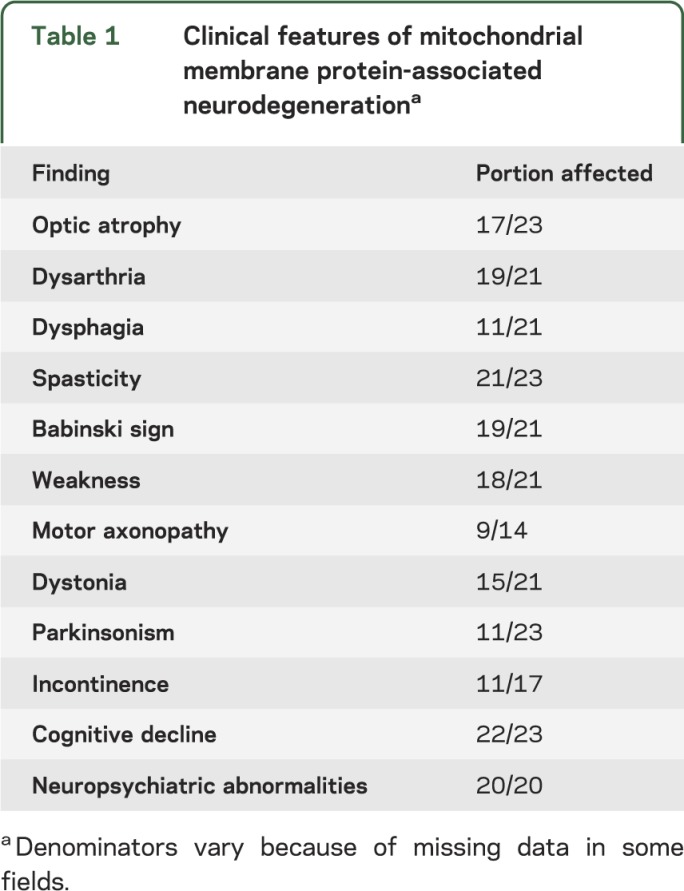
Individuals with MPAN most often presented with gait abnormalities (17 of 23). In 4 cases, visual impairment was one of the initial symptoms, associated with optic atrophy. One subject had sudden onset of optic atrophy with rapid progression over a few months following severe varicella infection. Optic atrophy was a consistent feature in juvenile-onset subjects. Overall, 74% of subjects in our cohort had optic atrophy, including all 5 subjects with homozygous null mutations, similar to previously reported cases.2 Among those with optic atrophy, there were also occasional reports of color vision loss, end-gaze nystagmus, abnormal pursuits, and hypometric saccades.
Early, nonspecific gait abnormalities were followed in the majority of subjects by the appearance of weakness (18 of 21) and prominent upper motor neuron signs, including pathologically brisk deep tendon reflexes and spasticity (21 of 23), usually affecting the lower limbs earlier and more significantly than the upper extremities. Extensor plantar responses were present in 19 of 21 subjects evaluated and were bilateral in all but 2 cases. Later in disease, evidence of lower motor neuron dysfunction emerged, with a distal-to-proximal pattern of deep tendon reflex loss in all subjects who had multiple assessments over time periods ranging from 2 to 12 years, often accompanied by muscle atrophy. Sensory symptoms were not prominent. Lower extremity proprioceptive abnormalities were noted in 4 subjects. Curiously, this appeared to be dissociated in severity from vibratory loss, which was mild when present.
Dysarthria was reported in all individuals with homozygous null mutations and in 19 of 21 subjects in total. Dysphagia was reported in 11 of 21 subjects.
Dystonia was common (15 of 21) and frequently affected the hands and feet, although it was generalized in some subjects. Eleven of 23 subjects had parkinsonism, with varying combinations of bradykinesia, rigidity, tremor, postural instability, and, in one subject, REM sleep behavior disorder. Parkinsonism was more common in subjects with later-onset disease, though it was also seen late in the course of some juvenile-onset subjects. Response to dopaminergic drugs was variable in treated subjects, and levodopa-induced dyskinesias were not observed.
Eleven of 17 subjects were reported to have incontinence, and in 4 subjects, both bowel and bladder function were affected. While we have anecdotally noted incontinence late in the course of other forms of NBIA, several subjects with MPAN had this complication earlier in their disease, including many while still ambulatory and prior to severe cognitive decline. Detailed urodynamic studies were not available on any subject in our cohort, so further characterization of the presumed neurogenic bladder dysfunction was not possible.
Unlike in PKAN, cognitive decline progressing to severe dementia was nearly universal in this group (22 of 23). The youngest subject, assessed only once at age 10 years, was the only one without reported cognitive impairment. Neuropsychiatric changes were also prominent (20 of 20) and were more varied than those reported in PKAN.1 Inattention, hyperactivity, emotional lability, depression, anxiety, impulsivity, compulsions, hallucinations, and perseveration were all reported in more than one subject. In several individuals, 2 or more of these occurred comorbidly. Of note, one previously normal subject (196) with unusually late onset at 30 years presented with isolated memory impairment that progressed to frank dementia over 2 years before signs of parkinsonism developed and brain iron accumulation was recognized on MRI.
Four subjects with advanced disease were noted to have stereotypic hand or head movements with alteration in consciousness, prompting evaluation for seizure activity. Many subjects in our cohort underwent EEG evaluation at some point in their disease course, though none was found to have EEG evidence of seizures. One 35-year-old subject for whom we do not have EEG records has been treated for seizures for the past 7 years.
Special studies
EMG and nerve conduction studies of varying quality and completeness confirmed the presence of a motor axonopathy in 9 subjects; studies in 4 other subjects did not. A pattern of distal denervation was seen, with variable motor unit amplitudes, decreased recruitment, fibrillations, and positive sharp waves. Two subjects undergoing skin or nerve biopsies were found to have axonal spheroids, previously thought to be limited to the PLA2G6-associated neurodegeneration (PLAN8) form of NBIA.
Radiographic findings
The subjects in this cohort were referred to our repository due to high brain iron. All subjects had increased iron deposition in the globus pallidus (n = 23), and the vast majority also had iron deposition in the substantia nigra (21 of 23). None had an eye-of-the-tiger sign.9 However, 5 subjects had hyperintense streaking of the medial medullary lamina between the globus pallidus interna and externa on T2-weighted images that might be mistaken for this (figure 1). Seven subjects also had generalized cortical atrophy, and 3 had cerebellar atrophy.
Figure 1. MRI findings in mitochondrial membrane protein-associated neurodegeneration.

T2 fluid-attenuated inversion recovery images from 3 individuals (248, 437, 438, images A–C, respectively) with hyperintense streaking of the medial medullary lamina. Substantia nigra from the same individuals (D–F).
Neuropathologic findings
Brain tissue was available from a single case, 196, described briefly above. This subject had late disease onset with dementia as the primary clinical finding and 2 C19orf12 mutations, including the common deletion c.204_214del11 and c.294G>C. A left parietal biopsy was performed approximately 2 years after onset of dementia but before the appearance of parkinsonism or other neurologic signs. This revealed abundant cortical Lewy bodies and axonal spheroids. The subject died 9 years later following a course marked by progressive dementia and parkinsonism, and brain tissue was donated for further studies.
Examination of the postmortem tissue revealed findings in the basal ganglia that were very similar to those previously reported in PKAN; namely, neuronal loss, gliosis, widespread iron deposits, and eosinophilic spheroidal structures in the globus pallidus (figure 2). As in PKAN, larger spheroidal structures appeared to be derived from degenerating neurons. Also as in PKAN, these displayed uniformly strong immunoreactivity for ubiquitin but variable staining with anti-tau antibody and no detectable TDP-43 or α-synuclein staining (figure 2). However, in marked contrast to PKAN, immunostaining for α-synuclein revealed widespread Lewy neurites in the globus pallidus, especially in more peripheral areas away from the rarefied zone of maximal tissue destruction. Lewy bodies and neurites were also widespread in other areas of the corpus striatum and in midbrain substantia nigra and neocortex (figure 3). These were associated with near complete neuronal loss in the substantia nigra. Lewy neurites were also identified in the pons, and Lewy bodies and neurites were abundant in the hippocampus with relative sparing of the CA1 region, which contained occasional tau-positive pretangles. Minimal iron deposition was identified in the substantia nigra, and no significant iron was observed in the cortex. Of note, the burden of neocortical Lewy pathology was substantially greater than in typical cases of sporadic Lewy body disease. We determined an average of 12.4 Lewy bodies per 40× magnification field in frontal cortex in MPAN, vs an average of 0.3 Lewy bodies per similar field in frontal cortex in 9 patients with Parkinson disease (PD) with dementia and an average of 1.9 Lewy bodies per field in 2 patients with PLA2G6 mutations. Similar analysis of limbic (anterior insular) cortex yielded values of 4.0, 0.7, and 1.4 for MPAN, PD dementia, and PLAN, respectively. The cerebellum appeared normal with the exception of rare Lewy bodies and neurites in the dentate nucleus.
Figure 2. Histopathologic features of mitochondrial membrane protein-associated neurodegeneration.
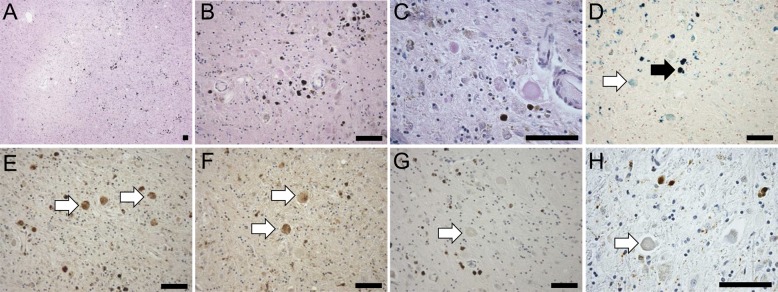
(A) Low-magnification (4×) view of hematoxylin & eosin stain of the globus pallidus, showing central pallor and widespread dark hemosiderin deposits. (B) At higher magnification (20×), the predominantly perivascular distribution of hemosiderin and the near complete loss of neurons in the globus pallidus are apparent; residual small nuclei are glial-derived. (C) Eosinophilic spheroidal structures as described in pantothenate kinase–associated neurodegeneration (PKAN) are readily apparent; some of these harbor residual nuclear outlines and lipofuscin pigment, indicating an origin in degenerating neurons (40×). (D) Perls stain (blue) for iron highlights densely stained hemosiderin deposits (white arrowhead) as well as more diffuse iron in the globus pallidus and increased iron associated with eosinophilic spheroids (black arrowhead, 20×). (E) Ubiquitin immunohistochemistry shows uniform, strong staining of spheroids (white arrowheads) as in PKAN (20×). (F) An immunostain for τ labels a subpopulation of spheroids (white arrowheads); occasional τ-positive neurites are in the background (20×). (G) TDP-43 immunostaining does not label any structures in the globus pallidus (white arrowhead); the brown signal is derived from hemosiderin deposits (20×). (H) Spheroids in the globus pallidus are not labeled with an antibody to α-synuclein (white arrowhead, 40×). Scale bars: 100 μm.
Figure 3. Histopathologic features of mitochondrial membrane protein-associated neurodegeneration.
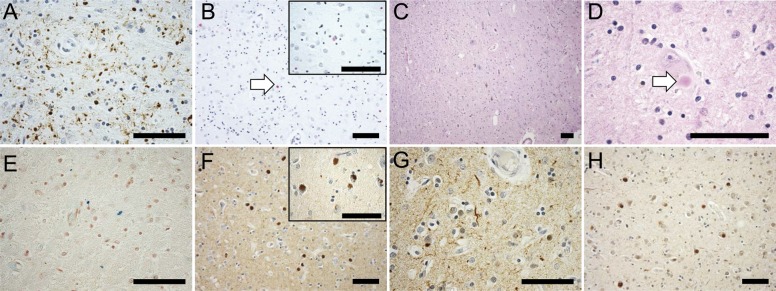
(A) Lewy neurites are abundant in relatively intact areas at the periphery of the globus pallidus (40×). (B) α-Synuclein immunohistochemistry of the putamen developed with Vector Red identifies Lewy bodies (white arrowhead) and neurites (20×), inset at higher magnification (60×). (C) Hematoxylin & eosin stain of the midbrain demonstrates sclerosis of the substantia nigra with only isolated residual neurons (10×); (D) many of these contain Lewy bodies (white arrowhead, 60×). (E) Perls stain (blue) of the midbrain shows only occasional areas of minute parenchymal iron deposits, predominantly in a perivascular distribution (40×). (F) α-Synuclein immunohistochemistry revealed abundant Lewy bodies in the neocortex (20×), inset at higher magnification (60×); (G) α-synuclein immunohistochemistry also highlighted Lewy neurites in a widespread distribution, including the cerebral cortex (depicted, 40×), basal ganglia, pons, and midbrain. (H) Neocortical Lewy bodies were ubiquitin positive, as in the case of sporadic Lewy body disease (20×). Scale bars: 100 μm.
DISCUSSION
MPAN is caused by mutations in C19orf12 leading to NBIA and prominent, widespread Lewy body pathology. The clinical phenotype, while sharing features with other forms of NBIA, is recognizable and distinctive (table 2). This newly recognized autosomal recessive subtype joins PKAN and PLAN as one of the major forms of NBIA, accounting for approximately 5% of cases. In our registry of patients ascertained for the presence of high basal ganglia iron by MRI or postmortem examination, the distribution of NBIA subtypes is depicted in figure e-1 (MPAN frequency is based on the number of mutation-positive unrelated families in our cohort).
Table 2.
Distinguishing features of the most common neurodegeneration with brain iron accumulation subtypes
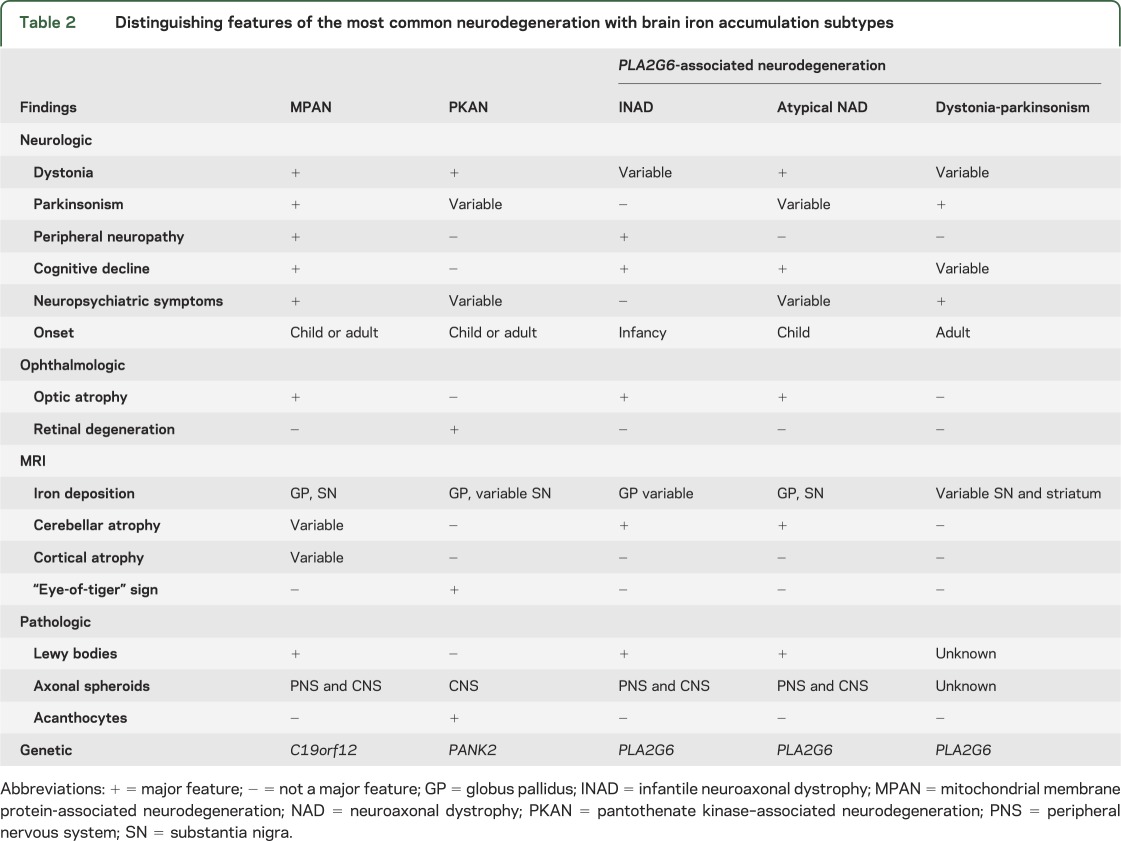
Our cohort offers an expanded view of the MPAN phenotype compared to the more ethnically homogenous population reported previously.2 The characteristic clinical features of MPAN across all age groups include cognitive decline progressing to dementia, prominent neuropsychiatric abnormalities, and a motor neuronopathy, with early upper motor neuron findings followed later by signs of lower motor neuron dysfunction and spheroid pathology. In juvenile-onset MPAN, these findings are joined by early optic atrophy, and in adult-onset disease by parkinsonism. Features that are variably present include dysarthria, dysphagia, dystonia-parkinsonism, bowel and bladder dysfunction, stereotypic hand and head movements, and proprioceptive sensory abnormalities dissociated from vibratory loss. Universally present in our cohort ascertained for this feature was a distinctive pattern of brain iron accumulation, characterized by T2/gradient echo hypointensities in the substantia nigra and globus pallidus, often with unique T2-hyperintense streaking between the hypointense internal globus pallidus and external globus pallidus. Like other forms of NBIA, MPAN spans a phenotypic spectrum that is likely to continue to broaden as more cases are described. Phenotypes outside of this recognized spectrum but associated with mutations in C19orf12 are likely to be discovered as more individuals with undiagnosed neurologic and neuropsychiatric disorders undergo whole genome sequencing.
A variety of mutations account for disease in our ethnically diverse cohort. While a substantial number harbor an Eastern European founder mutation, most patients have private mutations. Nonsense, frameshift, and missense mutations were distributed throughout the small coding region of C19orf12. Based on our results in 3 patients in whom we found only a single heterozygous mutant allele, occult deleterious mutations are likely to be present in noncoding sequence. Therefore, we recommend that intronic and regulatory sequences be screened in order to increase detection of disease-associated mutations. Multiplex ligation-dependent probe amplification may increase detection rates; however, we found no evidence for large duplications or deletions in our cohort. Additionally, we observed the co-occurrence of sequence variations c.205G>A and c.424A>G in 4 families (table e-1), 3 of which also had a third variation predicted to be pathogenic. Based on prediction algorithms described in Methods and the presence of the third variant in 3 of the families, we postulated that c.424A>G is an innocuous SNP that travels in cis with the pathogenic c.205G>A mutation. Clinical testing for MPAN is now available (www.genetests.org).
Though many lines of evidence support an autosomal recessive pattern in most cases of MPAN, one of our single-mutation patients (437) had a family history consistent with dominant inheritance. His father died at age 47 following a long course of progressive dementia with parkinsonism. Postmortem examination of the father's brain demonstrated neuropathologic changes nearly identical to those reported herein from an unrelated patient, including widespread abundant Lewy bodies, and both axonal spheroids and increased iron in globus pallidus and substantia nigra. Interestingly, the single unique frameshift mutation identified in this family leads to a series of 32 amino acid substitutions followed by a premature stop codon only 2 amino acids before the normal termination. The aberrant protein product is likely to escape nonsense-mediated mRNA decay and may exert a dominant negative effect on normal protein. Further studies in this family are underway to examine this possibility.
Lewy bodies are common in nearly all forms of NBIA, including MPAN, with PKAN being the only major form not associated with α-synuclein pathology. They appear much more abundant in non-PKAN hereditary NBIA, however, than in sporadic α-synucleinopathies, especially in neocortical areas in which Lewy bodies are typically relatively sparse in sporadic α-synucleinopathies such as dementia associated with PD, over which they were increased approximately 40× in MPAN (vs 6× in PLAN). Lewy bodies in sporadic illness are hypothesized to arise through increased α-synuclein expression or decreased degradation through the ubiquitin-proteasome and autophagy-lysosomal pathways.10 In MPAN, the markedly increased burden of Lewy bodies throughout the neocortex, deep gray matter, and midbrain suggest that alterations in the protein encoded by C19orf12 have profound effects on these pathways. Although amyloid-β abnormalities strongly drive α-synuclein accumulation in the cortex in sporadic neurodegenerative disease, there was no evidence in MPAN brain for amyloid-β by immunohistochemistry. There is a large body of literature that implicates mitochondrial dysfunction in the pathogenesis of PD, and the major toxicologic models of PD (MPTP, rotenone, and paraquat) uniformly affect complex I of the respiratory chain. However, the precise target of these toxins remains unknown. Given the massive α-synucleinopathy observed in MPAN, we speculate that C19orf12 may be a key susceptibility factor in PD and other α-synucleinopathies, as well as in models of these diseases.
Although little is known about the function of the protein encoded by C19orf12, its profile fits with proteins that are defective in other forms of NBIA. Mitochondrial membrane association and coregulation with proteins of fatty acid biogenesis and branched chain amino acid degradation expression profiles suggest similarities to other NBIA proteins, including PANK2, PLA2G6, and FA2H. Insight into protein function and disease pathogenesis will be accelerated by the creation of animal models of disease, which are currently in development.
Supplementary Material
ACKNOWLEDGMENT
The authors thank the patients and their families who participate in their research repository, the NBIA Disorders Association, Hoffnungsbaum e.V., the Associazione Italiana Sindromi Neurodegenerative Da Accumulo Di Ferro, Dr. Sue Richards, Dr. Stephen Moore, Dr. Jau-Shin Lou, the American Academy of Neurology Foundation, the American Philosophical Society, and the NICHD Brain and Tissue Bank for Developmental Disorders.
GLOSSARY
- MPAN
mitochondrial membrane protein-associated neurodegeneration
- NBIA
neurodegeneration with brain iron accumulation
- PD
Parkinson disease
- PKAN
pantothenate kinase–associated neurodegeneration
- PLAN
PLA2G6-associated neurodegeneration
- SNP
single nucleotide polymorphism
Footnotes
Supplemental data at www.neurology.org
AUTHOR CONTRIBUTIONS
P. Hogarth: design of study, interpretation of data, drafting and revising of manuscript. A. Gregory: design of study, analysis of data, drafting and revising of manuscript. M.C. Kruer: analysis of data, revising of manuscript. L. Sanford: analysis of data, revising of manuscript. W. Wagoner: analysis of data, revising of manuscript. M.R. Natowicz: analysis of data, revising of manuscript. R.T. Egel: analysis of data, revising of manuscript. S.H. Subramony: analysis of data, revising of manuscript. J.G. Goldman: analysis of data, revising of manuscript. E. Berry-Kravis: analysis of data, revising of manuscript. N.C. Foulds: analysis of data, revising of manuscript. S.R. Hammans: analysis of data, revising of manuscript. I. Desguerre: analysis of data, revising of manuscript. D. Rodriguez: analysis of data, revising of manuscript. C. Wilson: analysis of data, revising of manuscript. A. Diedrich: analysis of data, revising of manuscript. S. Green: analysis of data, revising of manuscript. H. Tran: analysis of data, revising of manuscript. L. Reese: analysis of data, revising of manuscript. R.L. Woltjer: design of study, analysis and interpretation of data, drafting and revising of manuscript. S.J. Hayflick: design of study, analysis and interpretation of data, drafting and revising of manuscript.
STUDY FUNDING
Supported by P30AG008017, NINDS UWXY3099.
DISCLOSURE
P. Hogarth has received research support from Schering-Plough Research Institute (a division of Merck), The European Commission, the NBIA Disorders Association, CHDI Foundation, and the Michael J. Fox Foundation. A. Gregory reports no disclosures. M. Kruer receives research support from EMD-Serono and has received consulting fees from Clinical Genetics, EMedicine, and the Mayo Clinic. He is funded by NIH NINDS grant UWXY3099 and receives support from the American Academy of Neurology Foundation and the American Philosophical Society. L. Sanford reports no disclosures. W. Wagoner owns stock in Exelixis, Inc. and Orasure Technologies, Inc. M. Natowicz and R.T. Egel report no disclosures. S.H. Subramony serves in the speakers' bureau for Athena Diagnostics. He receives research support from NIH/NINDS grant 1RC1NS068897, the National Ataxia Foundation, and the University of Florida Opportunity Award. J. Goldman has received research support from NIH/NINDS and the Parkinson's Disease Foundation; honoraria from the Movement Disorders Society, American Academy of Neurology, American Geriatrics Society, and Johns Hopkins Dystonia and Spasticity Practicum; and has been the site Principal Investigator for a trial sponsored by EMD Serono. E. Berry-Kravis receives research support from Novartis, Roche, and Seaside Therapeutics and serves on the advisory boards for autism and fragile X syndrome therapeutics for Novartis and Roche. N. Foulds, S. Hammans, I. Desguerre, D. Rodriguez, C. Wilson, A. Diedrich, S. Green, H. Tran, and L. Reese report no disclosures. R. Woltjer is funded by NIH grant AG008017 and receives research support from the Oregon Alzheimer's Disease Center. S. Hayflick has received research support from the NBIA Disorders Association, Hoffnungsbaume e.V., and the Associazione Italiana Sindromi Neurodegenerative da Accumulo di Ferro. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Hayflick SJ, Westaway SK, Levinson B, et al. Genetic, clinical, and radiographic delineation of Hallervorden-Spatz syndrome. N Engl J Med 2003;348:33–40 [DOI] [PubMed] [Google Scholar]
- 2.Hartig MB, Iuso A, Haack T, et al. Absence of an orphan mitochondrial protein, c19orf12, causes a distinct clinical subtype of neurodegeneration with brain iron accumulation. Am J Hum Genet 2011;89:543–550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ng PC, Henikoff S. SIFT: predicting amino acid changes that affect protein function. Nucleic Acids Res 2003;31:3812–3814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods 2010;7:248–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ferrer-Costa C, Gelpi JL, Zamakola L, Parraga I, de la Cruz X, Orozco M. PMUT: a web-based tool for the annotation of pathological mutations on proteins. Bioinformatics 2005;21:3176–3178 [DOI] [PubMed] [Google Scholar]
- 6.Capriotti E, Calabrese R, Casadio R. Predicting the insurgence of human genetic diseases associated to single point protein mutations with support vector machines and evolutionary information. Bioinformatics 2006;22:2729–2734 [DOI] [PubMed] [Google Scholar]
- 7.Kruer MC, Hiken M, Gregory A, et al. Novel histopathologic findings in molecularly-confirmed pantothenate kinase-associated neurodegeneration. Brain 2011;134:947–958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kurian MA, Morgan NV, MacPherson L, et al. Phenotypic spectrum of neurodegeneration associated with mutations in the PLA2G6 gene (PLAN). Neurology 2008;70:1623–1629 [DOI] [PubMed] [Google Scholar]
- 9.Sethi NK, Sethi PK. Eye-of-the-tiger sign. J Assoc Physicians India 2003;51:486. [PubMed] [Google Scholar]
- 10.Ebrahimi-Fakhari D, Cantuti-Castelvetri I, Fan Z, et al. Distinct roles in vivo for the ubiquitin-proteasome system and the autophagy-lysosomal pathway in the degradation of alpha-synuclein. J Neurosci 2011;31:14508–14520 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.