

ABSTRACT
Objective:
To determine whether amyloid burden, as indexed by Pittsburgh compound B (PiB) retention, identifies patients with Parkinson disease with mild cognitive impairment (PD-MCI) compared to those with normal cognition (PD-nl). A related aim is to determine whether amyloid burden predicts cognitive decline in a cohort of subjects with PD without dementia.
Methods:
In this prospective cohort study, we examined 46 subjects with PD without dementia, of whom 35 had normal cognition and 11 met criteria for PD-MCI at study baseline. All subjects underwent standardized neurologic and neuropsychological examinations and PiB PET at baseline, and clinical examinations were conducted annually for up to 5 years.
Results:
At baseline, precuneus PiB retention did not distinguish PD-MCI from PD-nl. Subjects with PD-MCI declined more rapidly than PD-nl subjects on cognitive tests of memory, executive function, and activation retrieval. Of the 35 PD-nl subjects, 8 progressed to PD-MCI and 1 to dementia; of the 11 PD-MCI subjects, 5 converted to dementia. Both higher PiB retention and a diagnosis of PD-MCI predicted a greater hazard of conversion to a more severe diagnosis. Baseline PiB retention predicted worsening in executive function over time. The APOE ε4 allele also related to worsening in executive function, as well as visuospatial function, activation retrieval, and performance on the Mini-Mental State Examination. In contrast to its relation to cognitive decline, PiB retention did not affect progression of motor impairment.
Conclusions:
At baseline measurements, amyloid burden does not distinguish between cognitively impaired and unimpaired subjects with PD without dementia, but our data suggest that amyloid contributes to cognitive, but not motor, decline over time.
Cognitive impairments are common in Parkinson disease (PD) and increase in severity and frequency over time.1–3 Whether cognitive impairments that fall short of a dementia diagnosis herald impending dementia is an unresolved question, but longitudinal studies in PD support this possibility.4,5 Dementia is common in PD, with a relative risk two- to sixfold higher than the general population,6 corresponding to lifetime risk estimates of 30%–80%.7 Multiple pathologic processes have been linked to dementia in PD: degeneration of basal forebrain cholinergic nuclei,8 frontal-subcortical circuit deafferentation due to degeneration of brainstem dopaminergic neurons,9 diffuse cortical Lewy bodies associated with α-synuclein,10 and Alzheimer-like lesions with Aβ-laden senile plaques.11 Techniques to determine which of these is dominant have been limited until now. The advent of molecular neuroimaging with amyloid radioligand Pittsburgh compound B (PiB) affords an opportunity to examine in life the potential contribution of Aβ burden to the progression of cognitive impairments in patients with PD without dementia. We and others have previously shown that PiB uptake is higher in parkinsonian patients with dementia than in patients with PD without dementia.12–14 We report a longitudinal study of patients with PD without dementia and show that PiB uptake is associated with multiple indices of cognitive decline, including conversion to dementia.
METHODS
Standard protocol approval and patient consents
The Partners HealthCare System Institutional Review Board approved this study. Informed consent was obtained from all patients participating in the study.
Participants
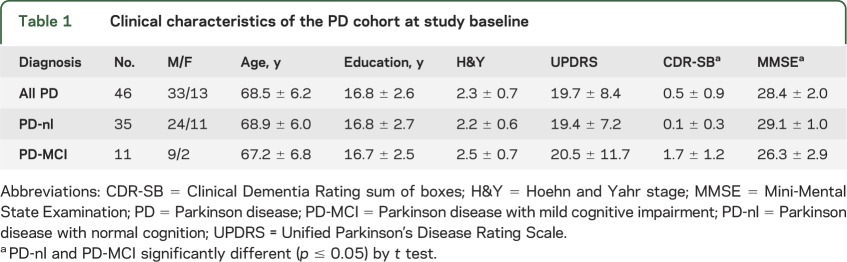
We enrolled 47 patients with PD without dementia between 2006 and 2011 into a prospective cohort study. A total of 46 met research diagnostic criteria for idiopathic PD15; 1 was excluded because of drug-induced parkinsonism. There were 33 men and 13 women, with mean ± SD age 68.5 ± 6.2 years at study baseline, education 16.8 ± 2.6 years, Hoehn & Yahr (H&Y) scale 2.3 ± 0.7, and Unified Parkinson’s Disease Rating Scale (UPDRS) motor subscore 19.7 ± 8.4 (table 1). All were taking optimal doses of dopaminergic medications prescribed by their neurologist and were tested in the “on” condition. None were taking drugs with prominent anticholinergic effects, such as trihexyphenidyl, that might be expected to impair cognition. Of the 46, 35 scored in normal ranges on a set of cognitive tests (see below) at study baseline, and 32 of these had no cognitive complaint. Eleven met criteria for mild cognitive impairment,16 with a cognitive complaint, such as memory loss, that was corroborated by an informant, plus at least 1 cognitive test score that fell in the impaired range. We used published normal cognitive test values with 1 SD thresholds.e1–e4 Patients with Parkinson disease with mild cognitive impairment (PD-MCI) had a global Clinical Dementia Rating (CDR) score of at least 0.5; 8/11 were amnestic, multidomain; 3/11 were nonamnestic, multidomain. The memory box score was at least 0.5 in 10 of these subjects. The mean Mini-Mental State Examination (MMSE) score was 29.1 ± 1.0 for the Parkinson disease with normal cognition (PD-nl) group, and 26.3 ± 2.9 for the PD-MCI group. Some subjects developed PD dementia (PDD) over the course of the study, as defined by the 2007 Movement Disorders criteria.6
Table 1.
Clinical characteristics of the PD cohort at study baseline
Clinical and neuropsychological examinations
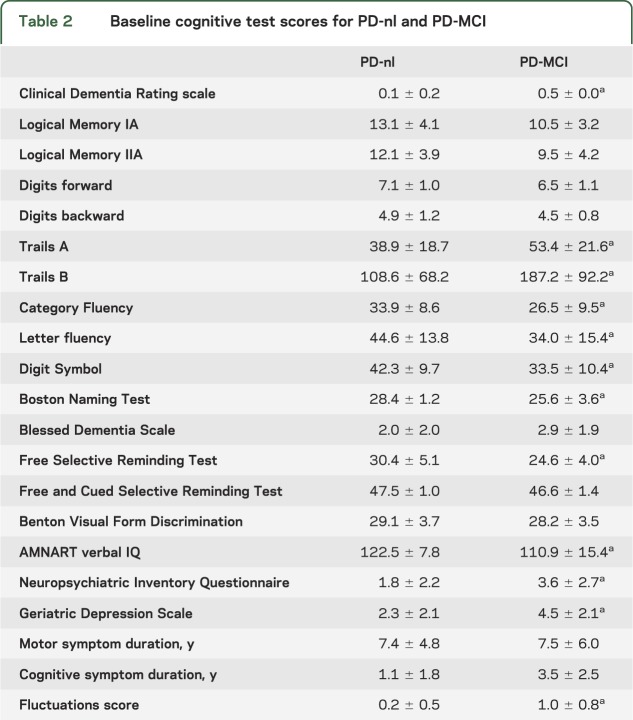
Neurologic examination included the H&Y scalee5 and 4-part UPDRS.e6 APOE genotypee7 was measured in 43/46 participants. Psychiatric assessments included the Neuropsychiatric Inventory Questionnairee8 and Geriatric Depression Scale.e9 Neuropsychological (NP) assessment included the MMSE,e10 American National Adult Reading Test,e11 Wechsler Memory Scale–Revised (WMS-R), Logical Memory I and IIA,e8 WMS-R Digit Span Forward and Backward,e12 Category Fluency,e13 Trailmaking A and B,e14 Wechsler Adult Intelligence Scale–Revised (WAIS-R) Digit Symbol,e15 Boston Naming,e16 Controlled Oral Word Fluency,e17 Benton Visual Form Discrimination,e17 and Free and Cued Selective Remindinge18 (table 2). Timed tests were considered in the context of bradykinesia. The CDR sum of boxes (CDR-SB) scoree19 was calculated from information provided by the subject's knowledgeable informant who rated the subject's abilities in the spheres of memory, orientation, judgment, community activities, home and hobbies, and personal care.
Table 2.
Baseline cognitive test scores for PD-nl and PD-MCI
Across all NP tests and visits, in 14 instances an NP score was missing due to severity of cognitive impairment. In these instances, we estimated the missing value with a multiple regression equation with the predictors of MMSE and CDR-SB, derived from the nonmissing NP data. Estimated values were constrained to the acceptable range for the NP. A perturbation of random normal error with variance estimated from the regression mean square error was added so that the new score variances were not artificially attenuated.
To reduce the number of variables, factor analyses based on performance grouped the NP tests into 4 distinct domains: episodic memory (Logical Memory IA and IIA); activation retrieval (verbal fluency, Category Fluency, Boston Naming, and Free and Cued Selective Reminding); executive function (Trails B and WAIS-R Digit Symbol); and visuospatial abilities (Benton Visual Form Discrimination). The executive function factor captures elements of attention and working memory. Cognitive composite scores were produced for each domain, where each composite was a mean of nonmissing z scores (relative to data from cognitively normal subjects with PD) for the set of NP tests. For each computed composite, no more than half of NP component variables were allowed to be missing for the factor score to be nonmissing.
PiB PET and image analyses
N-methyl-[11C]2-(4′-methylaminophenyl)-6-hydroxybenzothiazole (PiB) was prepared at Massachusetts General Hospital as previously described.17 Subjects were positioned in a Siemens HR+ scanner (3D mode; 63 image planes; 15.2-cm axial field of view; 5.6-mm transaxial resolution; 2.4-mm slice interval; 69 frames: 12 × 15 seconds, 57 × 60 seconds; Knoxville, TN). After a transmission scan, 8–15 mCi of 11C PiB was injected as a bolus, followed immediately by a 60-minute dynamic acquisition. PET data were reconstructed and corrected for attenuation. Each frame was evaluated to verify adequate count statistics and absence of head movement.
Each subject's precuneus, frontal cortex aggregate region of interest, and striatum were identified with the Automated Anatomic Labeling template following SPM spatial transformation, as described previously.12 PiB retention was calculated using the Logan graphical analysis method,18,19 with cerebellar cortex as reference tissue input function; specific PiB retention was expressed as the distribution volume ratio (DVR), as in previous studies.20,21 The precuneus is an early, common site of amyloid deposition and frequently carries the highest cortical amyloid burden measured with PiB PET. PiB retention in the precuneus correlated highly with mean cortical PiB retention (r = 0.94). The frontal aggregate's component regions are listed in appendix e-1 on the Neurology® Web site at www.neurology.org.
Protocol design
At study entry, subjects underwent comprehensive clinical and NP examinations and completed PiB PET imaging. Subjects returned for annual clinical and NP examinations for a mean of 2.5 ± 1.4 years (range 1–5 years). There were 156 total visits over this time: 41 subjects completed visit 2, 32 completed visit 3, 25 completed visit 4, and 12 completed visit 5.
Data analysis
We focused on 2 longitudinal measures: change in continuous numeric indicators of cognition (MMSE and the cognitive composites) and transition to more severe diagnoses. Change in continuous indicators was assessed with mixed random/fixed coefficient longitudinal regression models. Intercept and linear slope of change relative to years in the study were random subject terms, initially allowed to be correlated, occasionally forcing independence of random terms or the random slope to be fixed, in order to attain algorithm convergence. We conducted backward elimination of fixed terms, which included baseline diagnosis and its interaction with time, PiB DVR and its interaction with time, gender, years of education, baseline age, and baseline duration of motor symptoms. Residual distributions were checked to assess model fit and conformance with normality assumptions. Because of missing values, APOE ε4 status was assessed in a separate analysis. The relation of PiB DVR to time to transition to a more severe diagnosis was assessed with Cox proportional hazards regression models containing predictors of PiB DVR, baseline diagnosis and its interaction with PiB, baseline age/duration, years of education, and gender. Earliest transition from PD-nl to PD-MCI (or PDD) and from PD-MCI to PDD were both included in the same analysis to increase sample size. All subjects either remained stable in their diagnosis across time (considered “censored” observations in the model) or transitioned to a more severe diagnosis. In 1 case, a subject with baseline PD-MCI reverted to PD-nl for the subsequent 3 years. That subject was considered stable PD-nl for the analysis, which is why the distribution of baseline diagnoses reported in the Cox regression below differs slightly from what is reported elsewhere. The assumption of proportional hazards was checked and verified. SAS (version 9.3) and JMP Pro (version 9.0.2) software were used for analyses and graphs.
RESULTS
Baseline differences in clinical and amyloid biomarkers between PD-nl and PD-MCI
At baseline, the PD-MCI group performed more poorly than the PD-nl group on most cognitive tests (table 2), as well as on the cognitive factors of episodic memory (p = 0.0569), executive function (p = 0.0015), and activation retrieval (p < 0.0001), but not visuospatial function (p = 0.5066). In contrast, the 2 groups' mean H&Y scores (PD-nl 2.2 ± 0.6, PD-MCI 2.5 ± 0.7) and UPDRS scores (PD-nl 19.4 ± 7.2, PD-MCI 20.5 ± 11.7) did not differ. The mean PiB DVR in the precuneus region for the entire cohort was 1.14 ± 0.19. Mean cortical PiB retention was 1.15 ± 0.14. Although PD-MCI subjects were cognitively impaired, baseline PiB retention did not distinguish PD-nl (1.14 ± 0.22) from PD-MCI (1.13 ± 0.06) (figure 1). Neither precuneus nor frontal PiB retention pooled across the groups related to baseline performance on any of the cognitive factors (adjusting for age). The APOE ε4 allele was not strongly linked to PiB uptake. Its occurrence did not differ between PD-nl and PD-MCI. Amyloid burden in the striatum was similar in PD-nl, PD-MCI, and PDD subjects.
Figure 1. PiB retention did not differentiate PD-nl and PD-MCI subjects.
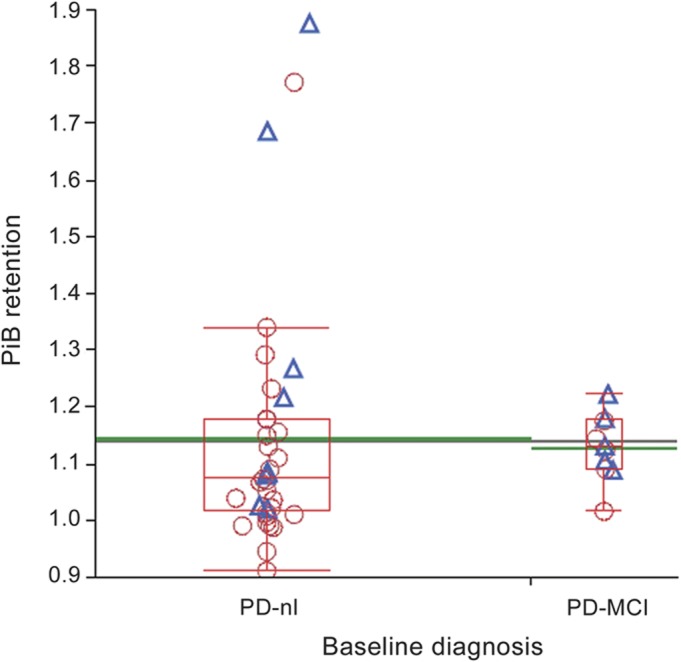
The black line extending across the whole figure represents the grand mean. The green line extending beyond the box for each group represents its mean. The red line for each group within the box represents the group median, box edges show the interquartile range, and box whiskers extend to as much as 1.5 interquartiles above and below the edges. Box width is proportional to group sample size. Subjects who subsequently progressed to a worse diagnosis are depicted as blue triangles, while those who remained stable are shown as red circles. PD-MCI = Parkinson disease with mild cognitive impairment; PD-nl = Parkinson disease with normal cognition; PiB = Pittsburgh compound B.
Longitudinal cognitive course of PD-nl and PD-MCI
Over time, cognitive symptoms progressed for the entire cohort. Of the 35 subjects with PD-nl, 26 remained stable, 8 progressed to PD-MCI, and 1 progressed to dementia. Of the 11 subjects with PD-MCI, 1 reverted to PD-nl, 5 remained stable, and 5 progressed to dementia. Thus, 14 of 46 patients with PD without dementia showed progression in cognitive impairments. The Cox regression analyses showed that subjects with PD-MCI at baseline converted to a more severe diagnosis sooner than baseline PD-nl subjects (hazard ratio = 3.45, 95% confidence interval [CI] = 1.016, 10.843; p = 0.035). Subjects with PD-MCI also worsened more quickly than subjects with PD-nl on the CDR-SB (random effects longitudinal model, interaction of baseline diagnosis and years in the study; p < 0.0001) and on the cognitive factors of executive function (p = 0.032), episodic memory (p < 0.012), and activation retrieval (p = 0.020).
Impact of amyloid biomarkers on longitudinal course in PD
The pooled group of subjects who transitioned to a more severe cognitive diagnosis (n = 14) had higher baseline PiB retention than the group who remained diagnostically stable (n = 32; p = 0.048, Mann-Whitney test). We performed a nonparametric survival analysis of 2 groups formed with a median split on PiB retention. The group with higher PiB retention transitioned to a more severe diagnosis significantly sooner than the group with lower PiB retention (p = 0.035; figure 2). This was not due to a systematic difference in baseline cognitive function, as the high PiB DVR group performed better at baseline than the low PiB DVR group on the visuospatial abilities factor (p = 0.023), worse on the activation retrieval factor (p = 0.005), and similarly (p > 0.05) on the remaining 2 factors. As a continuous numeric predictor, higher amyloid burden also increased the likelihood of transition to a more severe diagnosis in the Cox regression (p = 0.068; hazard ratio = 8.78 per unit PiB, 95% CI = 0.634, 80.792), additive to and independent of the baseline PD-nl vs PD-MCI diagnosis effect. In addition, higher PiB retention significantly (p = 0.035) related to worsening in executive function for the sample as a whole: across subjects, a 1 unit higher score on PiB was associated with an estimated decline in the linear slope of trajectory of about 0.4 SDs of the executive function factor per year. Higher PiB retention was also marginally related to faster decline in visuospatial function (p = 0.060). The presence of an APOE ε4 allele was independently associated with decline in MMSE (p = 0.005), executive function (p = 0.003), activation retrieval (p = 0.004), and visuospatial abilities (p = 0.013). Progression of motor impairment, as assessed with H&Y and UPDRS motor subscale scores, did not relate to precuneus or striatum PiB retention, or to the APOE ε4 allele.
Figure 2. Kaplan-Meier survival curves.

Subjects with Pittsburgh compound B (PiB) retention above the median for the sample converted to a more severe diagnosis sooner than those with values below the median. Parkinson disease with normal cognition and Parkinson disease with mild cognitive impairment groups were pooled together for this analysis. Shading around survival curves indicates 95% confidence bands. “Censored” indicates the last visit for subjects who had not transitioned to a more severe diagnosis.
Of the 3 subjects with highest amyloid burden (figure 1), 2 transitioned from PD-nl to PD-MCI over the course of the study. Of these 2, 1 remains PD-MCI; the other discontinued the study due to a traumatic subdural hemorrhage. The third subject remains PD-nl.
DISCUSSION
Diffuse cortical Lewy bodies and α-synuclein,10 loss of basal forebrain cholinergic neurons8 and medial nigral dopaminergic neurons,9 and Alzheimer disease (AD) pathology11 have all been implicated in the development of cognitive impairments and dementia in PD. The results of this study support the contribution of cortical amyloid Aβ deposits. The hypothesis that amyloid contributes to dementia initially came from the Alzheimer field, where excessive Aβ is viewed as an early, perhaps inciting, event in a cascade of pathologic changes leading to synaptic loss, neuronal degeneration, and clinical dementia.22,23 The exact Aβ moiety involved remains controversial, with research focused on the relative importance of monomers, oligomers, or fibrils in senile plaques. The 42 amino acid peptide (Aβ1-42) aggregates readily, is the principal component in the senile plaque, and seems to be the more toxic species.24 Mechanisms whereby Aβ causes neurodegeneration remain under active investigation. Nonetheless, excessive amyloid accompanies dementia in AD and in other conditions, including dementia with Lewy bodies, where we confirmed in a human postmortem case that PiB binds to Aβ deposits in senile plaques.25 Numerous subsequent investigators have confirmed this finding,26 leaving no doubt that PiB selectively and specifically binds to Aβ-pleated sheets and gives an accurate representation of cerebral Aβ burden.
In this study, PD-nl and PD-MCI had similar, generally modest PiB retention, with most subjects well below the high values characteristic of AD. Because sample size was limited, additional studies will be required to confirm these findings. There is substantial PiB uptake in ∼30% of healthy control subjects,27 which squares with our current experience with patients with PD without dementia.12 The significance of elevated PiB retention in cognitively normal individuals is uncertain, and it is an open research question whether their amyloidosis represents a disease process that will progress to dementia on some timescale or whether it represents a risk factor. Alternatively, these PiB-positive individuals may have yet-to-be-discovered mechanisms that blunt Aβ toxicity and spare neurons from degeneration. Data on this point are incomplete, although high PiB retention is common in mild cognitive impairment (MCI)28 and is a powerful predictor of subsequent conversion to AD.29 Our findings suggest that Aβ may play a similar role in PD: at baseline imaging Aβ may seem bland but deleterious effects emerge as patients decline cognitively over time to MCI and then dementia. Together, α-synuclein and Aβ-42 increase each other's toxicity.30 Amyloid may therefore contribute to cognitive impairment in PD both directly and indirectly, by enhancing α-synuclein toxicity. Thus, lower levels of amyloid may be necessary to cause dementia in the synucleinopathies than in AD.
Additional support for amyloid's association with cognitive decline in PD comes from CSF measurements of Aβ. Mean CSF Aβ1-42 in PD has been reported to be lower than in healthy controls and higher than in AD,31 with Aβ1-42 levels decreasing systematically from cognitively normal PD to PD-MCI to PDD, down to AD levels.32 In addition, a recent longitudinal study found that low CSF Aβ1-42 predicted progression to dementia in PD.33 The CSF formula of low Aβ1-42 and high tau is characteristic of AD and denotes subjects with MCI who are likely to progress to AD.34 Although the basis for decreased CSF Aβ is unclear, high PiB uptake is strongly associated with low CSF Aβ1-42.35 We did not measure CSF Aβ in our study, but anticipate that future studies will confirm this relationship in PD as well as in AD.
Aβ-related declines occurred in some cognitive domains and predicted conversion of diagnosis. Amyloid burden specifically related to progressive impairment of executive function and weakly to visuospatial function. These results are tantalizing, as frontal lobe functions and visuospatial skills are prominently affected in PD.6 APOE genotype also related to declining performance in most cognitive domains. APOE ε4 has been implicated in cognitive impairment in PD (reference 36, but see reference 37) and may have influenced amyloid burden in our cohort.38 Although we did not find a significant correlation between amyloid burden and APOE genotype, this may have been due to sample size. Because advanced age is also known to influence Aβ accumulation in the brain,39 we matched our groups for age and adjusted for age in our analyses. Consistent with prior results identifying MCI in PD as a risk factor for dementia,4,5 we also found that subjects with PD-MCI declined more rapidly on cognitive testing than subjects with PD-nl and converted to dementia more rapidly than subjects with PD-nl converted to PD-MCI or PDD.
Strengths of this study include its prospective design enrolling a well-characterized cohort of patients with PD who underwent annual comprehensive neurologic and NP examinations over 2.5 years. Additional strengths are careful PiB scans and sophisticated image analyses conducted by experienced personnel. Although the relatively small number of enrolled subjects could be seen as a weakness, the number was selected based on a power analysis at study onset and proved sufficient to demonstrate the deleterious long-term effects of cortical Aβ deposits on cognition in PD. Finding the association between amyloid burden and cognitive impairment in PD has clear therapeutic implications. Various drug strategies are evolving to decrease Aβ production, to prevent its aggregation, or to accelerate its clearance from the brain; clinical trials in AD are already underway based on these approaches. As a proof of principle, antibodies directed against Aβ1-42 have been shown to decrease senile plaque number and tau phosphorylation in human autopsy cases.40 Should this immune treatment or any of the other antiamyloid approaches under consideration prove effective in AD, similar treatments will become immediately relevant for treating PD with the expectation of preventing dementia, or at least slowing progression of cognitive impairments.
Supplementary Material
GLOSSARY
- AD
Alzheimer disease
- CDR
Clinical Dementia Rating
- CDR-SB
Clinical Dementia Rating sum of boxes
- CI
confidence interval
- DVR
distribution volume ratio
- H&Y
Hoehn & Yahr
- MCI
mild cognitive impairment
- MMSE
Mini-Mental State Examination
- NP
neuropsychological
- PD
Parkinson disease
- PD-MCI
Parkinson disease with mild cognitive impairment
- PD-nl
Parkinson disease with normal cognition
- PDD
Parkinson disease dementia
- PiB
Pittsburgh compound B
- UPDRS
Unified Parkinson’s Disease Rating Scale
- WAIS-R
Wechsler Adult Intelligence Scale–Revised
- WMS-R
Wechsler Memory Scale–Revised
Footnotes
Supplemental data at www.neurology.org
AUTHOR CONTRIBUTIONS
Dr. Gomperts: study concept and design, acquisition of data, data analysis and interpretation, writing the manuscript. Dr. Locascio: data analysis and interpretation, critical revision of the manuscript for important intellectual content. Dr. Rentz: data analysis and interpretation, critical revision of the manuscript for important intellectual content. Ms. Santarlasci: acquisition of data and data analysis. Dr. Marquie: critical revision of the manuscript for important intellectual content. Dr. Johnson: interpretation of data analyses, critical revision of the manuscript for important intellectual content. Dr. Growdon: study concept and design, acquisition of data, interpretation of data analyses, writing the manuscript, study supervision.
STUDY FUNDING
Supported by the Michael J. Fox Foundation for Parkinson's Research (to S.N.G. and J.H.G.), the National Institute of Neurological Disorders and Stroke (to K.A.J.), the National Institute on Aging (to K.A.J.), the Alzheimer's Disease Association (to K.A.J.), and the Caja Madrid Foundation scholarship for postgraduate studies (to M.M.). This study is not industry-sponsored.
DISCLOSURE
S. Gomperts is funded by NIH grants from NIMH (1K08MH81207), the NIA (NACC, expired), and National Institute of Neurological Disorders and Stroke (R21 NS060310, expired), and receives research support from the Michael J. Fox Foundation. J. Locascio reports no disclosures. D. Rentz receives research support from the Alzheimer Association IIRG-08-90934 and the Michael J. Fox Foundation. A. Santarlasci reports no disclosures. M. Marquie receives research support from the Caja Madrid Foundation scholarship for postgraduate studies. K. Johnson has received funding for travel and speaker honoraria from Pfizer Inc; serves as a consultant for GEHC Ltd, Avid Radiopharmaceuticals, Inc./Eli Lilly and Company, Bayer Schering Pharma, Pfizer Inc, Elan Corporation/Janssen, and Bristol-Myers Squibb; and receives research support from the Michael J. Fox Foundation, Avid Radiopharmaceuticals, Inc./Eli Lilly and Company, Bristol-Myers Squibb, Janssen (Janssen AI), Pfizer Inc, the NIH (NIA, National Institute of Neurological Disorders and Stroke), the Alzheimer Association, and the American Health Assistance Foundation. J. Growdon receives research support from the Michael J. Fox Foundation and the NIA (NACC, expired; ADRC P50 AG005134, current) and National Institute of Neurological Disorders and Stroke (R21 NS060310, expired). He serves on the Scientific Advisory Board of Neurimmune Therapeutics. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Locascio JJ, Corkin S, Growdon JH. Relation between clinical characteristics of Parkinson's disease and cognitive decline. J Clin Exp Neuropsychol 2003;25:94–109 [DOI] [PubMed] [Google Scholar]
- 2.Mortimer JA, Pirozzolo FJ, Hansch EC, Webster DD. Relationship of motor symptoms to intellectual deficits in Parkinson disease. Neurology 1982;32:133–137 [DOI] [PubMed] [Google Scholar]
- 3.Litvan I, Aarsland D, Adler CH, et al. MDS task force on mild cognitive impairment in Parkinson’s disease: critical review of PD-MCI. Mov Disord 2011;26:1814–1824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Janvin CC, Larsen JP, Aarsland D, Hugdahl K. Subtypes of mild cognitive impairment in Parkinson disease: progression to dementia. Mov Disord 2006;21:1343–1349 [DOI] [PubMed] [Google Scholar]
- 5.Williams-Gray CH, Evans JR, Goris A, et al. The distinct cognitive syndromes of Parkinson’s disease: 5 year follow-up of the CamPaIGN cohort. Brain 2009;132:2958–2969 [DOI] [PubMed] [Google Scholar]
- 6.Emre M, Aarsland D, Brown R, et al. Clinical diagnostic criteria for dementia associated with Parkinson’s disease. Mov Disord 2007;22:1689–1707 [DOI] [PubMed] [Google Scholar]
- 7.Aarsland D, Andersen K, Larsen JP, Lolk A, Kragh-Sorensen P. Prevalence and characteristics of dementia in Parkinson disease: an 8-year prospective study. Arch Neurol 2003;60:387–392 [DOI] [PubMed] [Google Scholar]
- 8.Tiraboschi P, Hansen LA, Alford M, et al. Cholinergic dysfunction in diseases with Lewy bodies. Neurology 2000;54:407–411 [DOI] [PubMed] [Google Scholar]
- 9.Rinne JO, Rummukainen J, Paljarvi L, Rinne UK. Dementia in Parkinson's disease is related to neuronal loss in the medial substantia nigra. Ann Neurol 1989;26:47–50 [DOI] [PubMed] [Google Scholar]
- 10.Harding AJ, Halliday GM. Cortical Lewy body pathology in the diagnosis of dementia. Acta Neuropathol 2001;102:355–363 [DOI] [PubMed] [Google Scholar]
- 11.Mattila PM, Röyttä M, Torikka H, Dickson DW, Rinne JO. Cortical Lewy bodies and Alzheimer-type changes in patients with Parkinson’s disease. Acta Neuropathol 1998;95:576–582 [DOI] [PubMed] [Google Scholar]
- 12.Gomperts SN, Rentz DM, Moran E, et al. Imaging amyloid deposition in Lewy body diseases. Neurology 2008;71:903–910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Edison P, Rowe CC, Rinne JO, et al. Amyloid load in Parkinson's disease dementia and Lewy body dementia measured with [11C]PIB positron emission tomography. J Neurol Neurosurg Psychiatry 2008;79:1331–1338 [DOI] [PubMed] [Google Scholar]
- 14.Rowe CC, Ng S, Ackermann U, et al. Imaging beta-amyloid burden in aging and dementia. Neurology 2007;68:1718–1725 [DOI] [PubMed] [Google Scholar]
- 15.Hughes AJ, Daniel SE, Blankson S, Lees AJ. A clinicopathologic study of 100 cases of Parkinson’s disease. Arch Neurol 1993;50:140–148 [DOI] [PubMed] [Google Scholar]
- 16.Winblad B, Palmer K, Kivipelto M, et al. Mild cognitive impairment–beyond controversies, towards a consensus: report of the International Working Group on Mild Cognitive Impairment. J Intern Med 2004;256:240–246 [DOI] [PubMed] [Google Scholar]
- 17.Mathis CA, Wang Y, Holt DP, et al. Synthesis and evaluation of 11C-labeled 6-substituted 2-arylbenzothiazoles as amyloid imaging agents. J Med Chem 2003;46:2740–2754 [DOI] [PubMed] [Google Scholar]
- 18.Logan J, Fowler JS, Volkow ND, et al. Distribution volume ratios without blood sampling from graphical analysis of PET data. J Cereb Blood Flow Metab 1996;16:834–840 [DOI] [PubMed] [Google Scholar]
- 19.Lopresti BJ, Klunk WE, Mathis CA, et al. Simplified quantification of Pittsburgh Compound B amyloid imaging PET studies: a comparative analysis. J Nucl Med 2005;46:1959–1972 [PubMed] [Google Scholar]
- 20.Fagan AM, Mintun MA, Mach RH, et al. Inverse relation between in vivo amyloid imaging load and cerebrospinal fluid Abeta-42 in humans. Ann Neurol 2006;59:512–519 [DOI] [PubMed] [Google Scholar]
- 21.Johnson KA, Gregas M, Becker JA, et al. Imaging of amyloid burden and distribution in cerebral amyloid angiopathy. Ann Neurol 2007;62:229–234 [DOI] [PubMed] [Google Scholar]
- 22.Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 2002;297:353–356 [DOI] [PubMed] [Google Scholar]
- 23.Hyman BT. Amyloid-dependent and amyloid independent stages of Alzheimer disease. Arch Neurol 2011;68:1062–1064 [DOI] [PubMed] [Google Scholar]
- 24.O’Brien RJ, Wong PC. Amyloid precursor protein processing and Alzheimer’s disease. Annu Rev Neurosci 2011;34:185–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Backsai BJ, Frosch MP, Freeman SH, et al. Molecular imaging with Pittsburgh Compound B confirmed at autopsy: a case report. Arch Neurol 2007;64:431–434 [DOI] [PubMed] [Google Scholar]
- 26.Ikonomovic MD, Klunk WE, Abrahamson EE, et al. Post-mortem correlates of in vivo PiB-PET amyloid imaging in a typical case of Alzheimer’s disease. Brain 2008;131:1630–1645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Morris JC, Roe CM, Xiong C, et al. APOE predicts amyloid-beta but not tau Alzheimer pathology in cognitively normal aging. Ann Neurol 2010;67:122–131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rowe CC, Ellis KA, Rimajova M, et al. Amyloid imaging results from the Australian Imaging, Biomarkers, and Lifestyle (AIBL) study of aging. Neurobiol Aging 2010;31:1275–1283 [DOI] [PubMed] [Google Scholar]
- 29.Zhang S, Han D, Tan X, Feng J, Guo Y, Ding Y. Diagnostic accuracy of (18)F-FDG and (11) C-PIB-PET for prediction of short-term conversion to Alzheimer’s disease in subjects with mild cognitive impairment. Int J Clin Pract 2012;66:185–198 [DOI] [PubMed] [Google Scholar]
- 30.Masliah E, Rockenstein E, Veinbergs I, et al. beta-amyloid peptides enhance alpha-synuclein accumulation and neuronal deficits in a transgenic mouse model linking Alzheimer's disease and Parkinson's disease. Proc Natl Acad Sci USA 2001;98:12245–12250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shi M, Bradner J, Hancock AM, et al. Cerebrospinal fluid biomarkers for Parkinson disease diagnosis and progression. Ann Neurol 2011;69:570–580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Montine TJ, Shi M, Quinn JF, et al. CSF Aβ(42) and tau in Parkinson’s disease with cognitive impairment. Mov Disord 2010;25:2682–2685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Siderowf A, Xie SX, Hurtig H, et al. CSF amyloid β 1-42 predicts cognitive decline in Parkinson’s disease. Neurology 2010;21:1055–1061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Herskowits AZ, Growdon JH. Sharpen that needle. Arch Neurol 2010;67:918–920 [DOI] [PubMed] [Google Scholar]
- 35.Jagust WJ, Landau SM, Shaw LM, et al. Relationships between biomarkers in aging and dementia. Neurology 2009;73:1193–1199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pankratz, Byder L, Halter C, et al. Presence of an APOE4 allele results in significantly earlier onset of Parkinson's disease and a higher risk with dementia. Mov Disord 2006;21:45–49 [DOI] [PubMed] [Google Scholar]
- 37.Jasinska-Myga B, Opala G, Goetz CG, et al. Apolipoprotein E gene polymorphism, total plasma cholesterol level, and Parkinson disease dementia. Arch Neurol 2007;64:261–265 [DOI] [PubMed] [Google Scholar]
- 38.Verghese PB, Castellano JM, Holtzman DM. Apolipoprotein E in Alzheimer's disease and other neurological disorders. Lancet Neurol 2011;10:241–252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Becker JA, Hedden T, Carmasin, et al. Amyloid-beta associated cortical thinning in clinically normal elderly. Ann Neurol 2011;69:1032–1042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Serrano-Pozo A, William CM, Ferrer I, et al. Beneficial effect of human anti-amyloid-beta active immunization on neurite morphology and tau pathology. Brain 2010;133:1312–1327 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.