

Abstract
Objective:
Facioscapulohumeral muscular dystrophy (FSHD) is a neuromuscular disease with an unclear genetic mechanism. Most patients have a contraction of the D4Z4 macrosatellite repeat array at 4qter, which is thought to cause partial demethylation (FSHD1) of the contracted allele. Demethylation has been surveyed at 3 restriction enzyme sites in the first repeat and only a single site across the entire array, and current models postulate that a generalized D4Z4 chromatin alteration causes FSHD. The background of normal alleles has confounded the study of epigenetic alterations; however, rare patients (FSHD2) have a form of the disease in which demethylation is global, i.e., on all D4Z4 elements throughout the genome. Our objective was to take advantage of the global nature of FSHD2 to identify where disease-relevant methylation changes occur within D4Z4.
Methods:
Using bisulfite sequencing of DNA from blood and myoblast cells, methylation levels at 74 CpG sites across 3 disparate regions within D4Z4 were measured in FSHD2 patients and controls.
Results:
We found that rates of demethylation caused by FSHD2 are not consistent across D4Z4. We identified a focal region of extreme demethylation within a 5′ domain, which we named DR1. Other D4Z4 regions, including the DUX4 ORF, were hypomethylated but to a much lesser extent.
Conclusions:
These data challenge the simple view that FSHD is caused by a broad “opening” of D4Z4 and lead us to postulate that the region of focal demethylation is the site of action of the key D4Z4 chromatin regulatory factors that go awry in FSHD.
Facioscapulohumeral muscular dystropy (FSHD [OMIM 158900]) is a dominant genetic disease with variable onset that affects certain muscle groups of the face and upper body as well as abdominal and foot-extensor muscles with atrophy, increasing with age.1 The incidence is at least 1 in 25,000 individuals.2,3 Ninety-five percent of cases (FSHD1) are caused by a contraction of the D4Z4 repeat array located at 4q35.2 from the usual number of approximately 100, down to ≤10,4 which is thought to alter the chromatin of the array. In the other 5% of cases, an unknown event alters D4Z4 chromatin, including both alleles of 4q35.2 arrays, which may contain >10 repeats (FSHD2 or phenotypic FSHD).5 The 2 forms of the disease have similar clinical severity scores.6–8
Each D4Z4 repeat encodes a gene, DUX4, which is toxic at high levels of expression.9–14 DUX4 also sensitizes to oxidative stress, and perturbs myoblast differentiation at low levels of expression.9 Recent genetic analysis has revealed that all patients with either form of FSHD inherit a pathogenic allele with a unique DUX4 "ATTAAA" polyadenylation signal downstream of the last D4Z4 repeat of chromosome 4, whereas nonpathogenic alleles lack this signal.15 10qter has a similar subtelomeric D4Z4 repeat array displaying 99% nucleotide identity to that of 4qter, but reductions in chromosome 10 D4Z4 repeats do not result in disease5,6 and chromosome 10 alleles do not contain the DUX4 polyadenylation signal.15 The correlation of the disease with the polyadenylation signal suggests a model where D4Z4 chromatin is altered, allowing transcription of polyadenylated DUX4 messenger RNA from the last repeat of the pathogenic allele, which is then translated into a toxic protein.15
The phenotypic similarity of FSHD1 and FSHD2, together with the observed FSHD-associated chromatin changes frequently associated with reduced methylation,16 suggest that understanding D4Z4 epigenetic changes is critical to understanding FSHD. Previous reports surveyed D4Z4 methylation in the blood of FSHD patients using methylation-sensitive DNA restriction enzymes and Southern blotting.5,6,8,17 Only 3 CpG loci within D4Z4 or 4 chromatin protein-bound ChIP-PCR sites5–8,16–22 have been examined. When a probe was used to survey the first (proximal) repeat in FSHD1, it was found that only the contracted chromosome 4 allele showed reduced methylation, whereas in FSHD2, both chromosomes 4 and 10 showed demethylation.5,6 Methylation of internal repeats has been examined at only a single site, CpoI, with FSHD2 patients showing demethylation on both chromosomes.5
We have taken advantage of the unique feature of global D4Z4 hypomethylation in FSHD2 to investigate epigenetic changes in D4Z4 using bisulfite sequencing. This allowed us to survey how methylation patterns are altered at 74 CpG sites across 3 distinct domains of D4Z4, a significant increase in resolution over prior studies in which only a single methylation-sensitive restriction enzyme was used to examine internal repeats in FSHD. This improved resolution brings to light unexpected features of chromatin changes in D4Z4, including a surprisingly moderate rate of demethylation of DUX4, and an intensely focused region of demethylation upstream of the DUX4 gene, which likely represents an important regulatory site.
METHODS
Samples were provided by Dr. John Day or Dr. Rabi Tawil and were taken with patient consent under IRB-approved protocols. All but 2 patients were classified as FSHD2 by detailed genetic analysis of chromosomes 4q and 10q for length and allele type and had below-threshold levels of methylation using Southern blotting methods.6 Furthermore, all had a permissive chromosome 4 allele.15 Two patients were related to tested probands and were not tested themselves. Biopsy samples were taken from quadriceps muscles. Myoblast cells were cultured in human myoblast medium (DMEM/F10; HyClone, Thermo Scientific, Logan, UT) supplemented with 20% HyClone fetal bovine serum, penicillin 5 U/mL, and streptomycin 5 μg/mL (Gibco, Invitrogen, Carlsbad, CA), human basic fibroblast growth factor 10 pg/mL, dexamethasone 40 pg/mL (Stem Cell Technology, Vancouver, BC), and β-mercaptoethanol 0.114 mM (Sigma, St. Louis, MO). DNA was isolated with a PureLink Genomic DNA Mini Kit (Invitrogen), or as previously described5 and 500 ng of DNA was converted with the EZ DNA Methylation Direct Kit (Zymo Research, Irvine, CA). DNA was amplified by PCR with primers and conditions as described, then cloned, sequenced, and analyzed (see e-Methods and table e-1 on the Neurology® Web site at www.neurology.org).
RESULTS
Using the observation that all D4Z4 elements on chromosomes 4 and 10 are altered in FSHD2 as a foundation, we designed a strategy to examine global average methylation levels at CpG sites within 3 different regions of D4Z4, taking advantage of the distinct etiology of FSHD2 to evaluate what specific D4Z4 chromatin changes might be most relevant to the deregulation of D4Z4 associated with the FSHD phenotype. Based on reported methylation/demethylation at the CpoI site assayed over internal repeats of chromosomes 4 and 10,5 we expected that the 3 regions would be approximately 70% to 90% methylated in controls and would undergo at most 30% to 40% demethylation in FSHD2. We prepared DNA from peripheral blood lymphocytes (PBLs) or cultured myoblast cells and treated it with bisulfite, which converts unmethylated cytosine to uracil but does not convert methylated cytosine. The treated DNA was amplified by PCR with 3 sets of primers to give fragments in 3 D4Z4 regions: 2 upstream of the DUX4 ORF and 1 within the ORF (DR1-3; figure 1). The first site, DR1, is 1 kb upstream of the DUX4 ORF, 71 base pairs (bp) 3′ of a reported CTCF binding site19 (figure 1). CTCF can function as an insulator to limit domains of DNA methylation, and DNA binding by CTCF can be methylation sensitive.23 The low complexity of D4Z4 sequence (73% G or C) makes it challenging to design PCR assays that are specific. Choosing primers for bisulfite sequencing of D4Z4 is even more difficult because CpGs are undesirable within primers. For this reason, we were unable to survey the CTCF binding site itself. The second site (DR2) is located downstream of the DR1 site and includes a previously surveyed BsaAI methylation-sensitive restriction enzyme site. Because the DUX4 ORF has been implicated as a causal agent in FSHD,9,15,21,24,25 we also evaluated methylation within the ORF. The DR3 site targets approximately the middle 223 bp of the 1,275 bp DUX4 ORF.
Figure 1. The structure of the D4Z4 repeat unit showing regions analyzed by bisulfate sequencing.
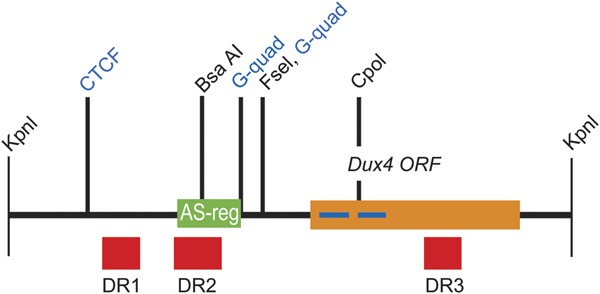
The DUX4 ORF is indicated in orange with homeodomains shown as blue bars. The regions surveyed in this study, DR1 to DR3, are shown in red. The 3 regions are at positions 563–814 (DR1), 997–1272 (DR2), and 2471–2692 (DR3) with respect to the start of the KpnI site. The locations of the previously surveyed methylation-sensitive restriction enzyme sites,5,6,8,17,18 the location of 2 previously identified adjacent G-quadruplex sites,26 and a previously identified CTCF binding site19 are indicated. The green region marked AS-reg has been shown by Block et al.27 to be important for bidirectional transcription from D4Z4.
Most CpGs are methylated in control PBLs
We first examined methylation rates in control PBL samples. All 3 surveyed sites averaged >50% in controls (DR1-3, figure 2). At DR2 and DR3, the average level of methylation was similar (69% and 63%, respectively) and not significantly different from DR1.
Figure 2. D4Z4 methylation in control and facioscapulohumeral muscular dystrophy (FSHD)2 peripheral blood lymphocyte samples.
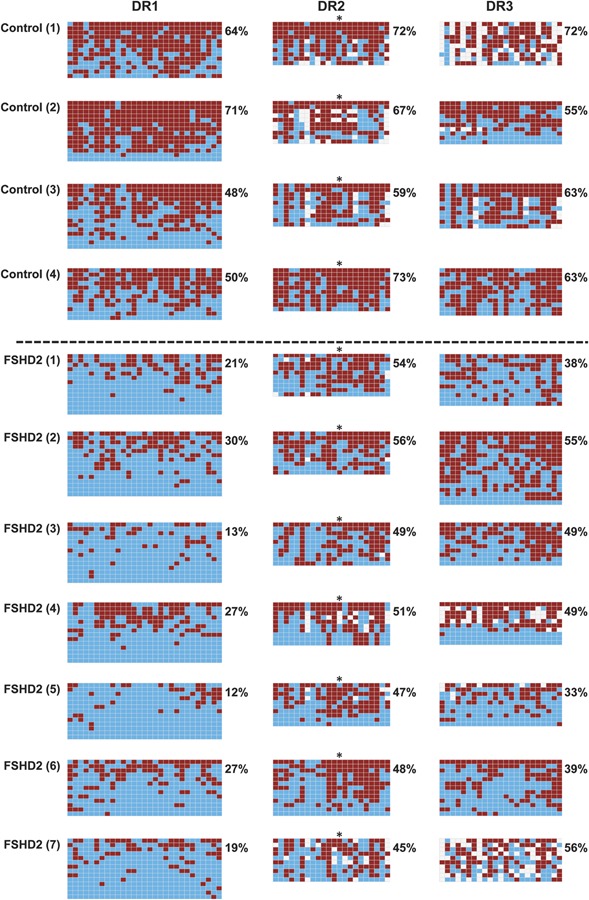
DNA was converted with sodium bisulfite, amplified by PCR and at least 10 individual clones were sequenced. Three D4Z4 regions (DR1–DR3) were surveyed. For each region surveyed, a panel is shown where red squares represent a methylated CpG within the sequenced DNA fragment and blue squares represent unmethylated CpGs. White squares are where the sequence of the clone did not correspond to a methylated or unmethylated CpG and are most likely due to polymorphisms with respect to the reference sequence (see figure e-1). The average methylation for all CpGs (excluding white squares) within the region for each sample is indicated to the right of each graph. A dotted line separates the controls from FSHD2 samples (below). An asterisk indicates the position of the CpG within the previously studied BsaAI restriction enzyme site.
In FSHD2 PBLs, demethylation is greatest at DR1
All 3 regions showed relative demethylation in PBL DNA of FSHD2 patients, however the demethylation at DR1 was remarkable (figure 2). The FSHD2 samples averaged only 21% methylation at DR1 whereas the controls averaged 55% (for averages, see figure 4), a reduction of >62%. These differences were found despite the fact that the results are based on bisulfite sequencing from the total PCR products, i.e., not exclusively products specific to chromosome 4. In some patients, every sequenced fragment was essentially unmethylated, which, because the vast majority of repeats are internal, suggests that DR1 is demethylated on the internal repeats of both chromosomes 4 and 10.
Figure 4. Average methylation at each region examined in blood and myoblast cells of facioscapulohumeral muscular dystrophy (FSHD)2 and control individuals.
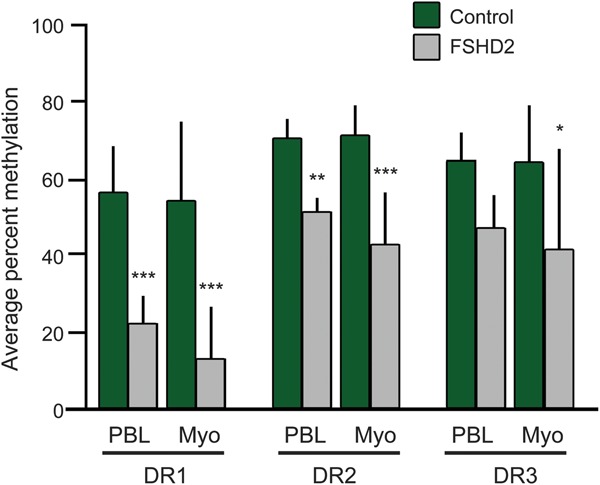
The percent methylation (y axis) of the averaged data for control compared with FSHD2 samples for blood DNA at each of the 3 tested sites (DR1–DR3) is shown. Differences between the means of control vs FSHD2 were tested using analysis of variance. The p values are as follows: DR1 peripheral blood lymphocyte (PBL) = 0.0002, Myo = 0.0002; DR2 PBL = 0.0019, Myo ≤0.001; DR3 PBL = 0.0749, Myo = 0.0300.
DR2 and DR3 are demethylated to a lesser extent
The average methylation at DR2 FSHD2 PBLs was also reduced compared with the control (figures 2 and 4). However, at DR3, this demethylation was not statistically significant (p = 0.0749, figure 4). This demonstrates a nonuniformity of FSHD-mediated demethylation along the repeat that has not been demonstrated previously because of the small number of CpGs surveyed.5
Methylation at DR1 is dramatically reduced in FSHD2 myoblasts
To expand our sample size, and to evaluate a muscle cell type, we sought to evaluate methylation levels in FSHD2 myoblasts. Some previous reports suggested that methylation levels were not different in PBL, biopsy, or myoblast samples5,17; however, D4Z4 methylation levels can vary among different cancer cell lines.26 We found that in myoblast samples, DR1 was even more extremely demethylated (figures 3 and 4). Controls ranged from 41% to 83% methylated, whereas FSHD2 samples ranged from 2% to 31% methylated. On average, DR1 had one-fourth as much methylation in FSHD2 as in unaffected controls.
Figure 3. D4Z4 methylation in control and facioscapulohumeral muscular dystrophy (FSHD)2 myoblasts.
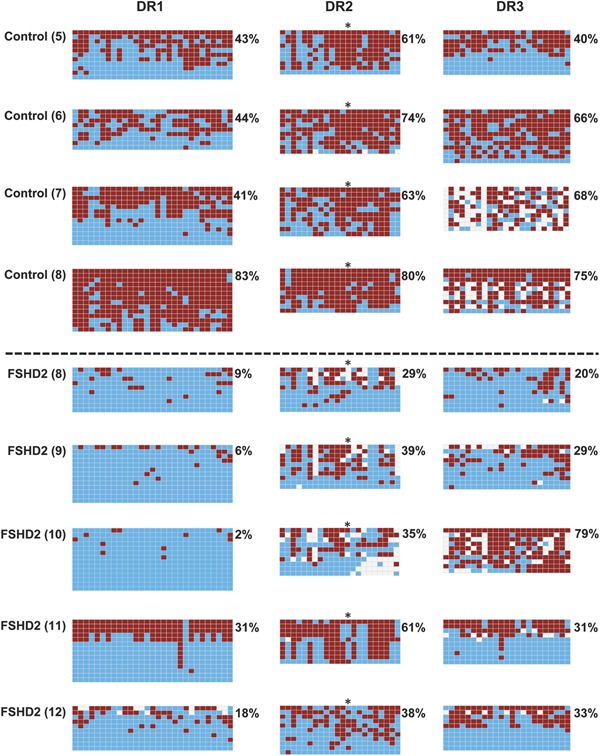
Format and annotation are as shown in figure 2.
At the DR2 and DR3 sites, FSHD2 myoblasts showed moderately lower methylation than controls; although this was highly significant at DR2 (p < 0.001), the DR3 site showed greater variability with a (still significant) p value of 0.03. Thus, the data from myoblast samples are broadly similar to the data from PBLs, with strong evidence for a focal demethylation at DR1.
Methylation at BsaAI vs DR2
The previously surveyed BsaAI site is within DR2. Looking at the methylation pattern across DR2, there was considerable variability at each individual CpG. At the specific CpG previously surveyed by BsaAI (number 13, marked by an asterisk), we found a modest reduction (from 62% methylation in controls to 45% in FSHD2 blood), similar to the average reduction for the whole of DR2. This compares with the 28% vs 50% average methylation found at the BsaAI site in the proximal repeat in FSHD2 and control individuals from PBL DNA reported previously using the Southern blot technique.6,15,17 In the myoblast samples, there was a greater difference between FSHD2 samples (31% methylation) and controls (83% methylation) at this site. These values show that the trends in methylation differences observed at this particular CpG in random D4Z4 repeats (our approach) were similar to those observed with BsaAI Southern blotting evaluating the first repeat only.
DISCUSSION
FSHD is a disease with a complicated molecular mechanism that is still not well understood. In the current molecular model, pathology results from ectopic expression of the DUX4 protein encoded by the last D4Z4 repeat unit.15 This is attributable to some event that causes a chromatin change allowing transcription within the normally silent somatic D4Z4.11,15,27,28 There are many unresolved questions, chief among them: What molecular mechanism accounts for this chromatin change? Studying epigenetic regulation in FSHD1 would require purification of the contracted permissive allele, which is technically challenging. Because in FSHD2, D4Z4 changes are not restricted to the permissive allele, cells from these rare individuals can be used to probe FSHD-related D4Z4 chromatin changes. They therefore represent an important resource to understand epigenetic mechanisms in both forms of the disease. Taking this latter approach, we have moved significantly beyond past studies of methylation in FSHD. Our data demonstrate that D4Z4 cannot be thought of as existing in 2 configurations, methylated or hypomethylated; rather, demethylation is focused on particular regions of D4Z4, e.g., DR1. This is the first observation of regional demethylation differences within D4Z4 that are FSHD specific. Such focally demethylated domains are likely to have a critical role in the overall accessibility of the factors that result in expression of the DUX4 gene.
Previous studies observed differences between D4Z4 and nearby external proximal sequences with one finding a region of decreased methylation5 and the other finding increased methylation.26 de Greef et al.5 found that high levels of methylation did not extend to a SmaI site outside of the D4Z4 array (30%–35%) compared to within the array (40%–90%) in PBLs and no FSHD-specific demethylation at this site. Tsumagari et al.26 found that in cancer cells, a 2.2-kb region proximal to the array was hypermethylated and a 1.4-kb region within the D4Z4 array was hypomethylated. These differing results suggest that even small differences in distance can result in very different methylation patterns and that different tissues can have different methylation patterns. Indeed, DR1 and DR2 are only 131 bp apart. A further consideration is that the repeats within the arrays might not be uniformly methylated. The proximal-most D4Z4 repeat was reported to be less methylated at the CpoI site than the internal repeats in both healthy and FSHD samples.5 When internal D4Z4 units were examined at the CpoI site, only FSHD2 individuals showed hypomethylation relative to controls. Because our study surveyed random clones from the genomic set of D4Z4 repeats, it is likely that internal repeats are considerably overrepresented relative to terminal repeats. Therefore, we conclude that the DR1 region is probably demethylated within the majority of internal repeats. Given that we discovered the greatest changes in methylation at DR1, we postulate that chromatin regulatory factors that go awry in FSHD2 may bind within or near this region.
Surprisingly, the DUX4 ORF site (DR3) showed only moderate demethylation and high variability. This result was unexpected, considering previous results showing demethylation of the CpoI site, which is also in the ORF but 410 bp upstream of DR3. Although this may be attributable to regional differences within the ORF, our data show that the status of the CpoI site cannot be assumed to equate to that of the DUX4 ORF overall. An intriguing possibility is that methylation instability at DR3 may influence DUX4 gene expression, which may tend toward either no expression or high expression in a small number of cells.21
Although hypomethylation at 3 methylation sensitive sites within D4Z4, BsaAI, FseI, and CpoI, was observed in a large cohort of FSHD2 patients and controls, there were a few control individuals who had reduced levels of methylation at 1 or more of these sites.6 An important question raised by that result is whether variation in methylation levels results from incomplete penetrance of the disease or from sporadic variation in methylation levels at a single site in an otherwise hypomethylated sequence. Our work suggests the latter and highlights the value of surveying a larger number of CpGs to assay local methylation changes.
The D4Z4 repeats from chromosomes 4 and 10 cannot be distinguished in the sequences we have amplified; therefore, our data represent global average changes at D4Z4, and it is possible that these are more extreme on one or the other chromosome. However, regarding DR1, where the greatest level of demethylation was seen, it is likely that chromosomes 4 and 10 are equally demethylated because the average can approach zero only if all D4Z4 repeats in the genome are subject to demethylation. Furthermore, the current model suggests that changes in the status of the last repeat are most critical to the phenotype, and to date even the methylation-sensitive restriction enzyme method cannot evaluate methylation of the terminal repeat. Therefore, both approaches, looking at global averages for D4Z4 (here) vs looking at the first or internal D4Z4 repeats of chromosome 4 (previous work), give approximations that need to be interpreted with their own caveats.
The extreme demethylation at DR1 leads to the question of how demethylation at this site could directly affect the D4Z4 repeat or DUX4 transcription. Just proximal to the DR1 site, within the D4Z4 repeat, is a CTCF binding site (figure 1). CTCF typically binds to DNA that is not methylated23 and its binding at D4Z4 can be blocked by methylation.19,29 The CTCF binding site was identified when Ottaviani et al.19 used transfected constructs to show that a single D4Z4 element was able to bind CTCF. However, CTCF binding was reduced with arrays of 4 repeats and was nearly absent with 12 repeats. Although we did not evaluate the CTCF binding site itself, we saw the most significant demethylation very near this posited binding site. Assuming CTCF does indeed bind this site when demethylated in FSHD, our data suggests that CTCF could actually be insulating from a silencer rather than from an enhancer.
Block et al.27 identified a region that controls bidirectional transcription of a reporter that replaced the DUX4 ORF within a D4Z4 repeat (NspI to AccIII, figure 1). Therefore, the FSHD-specific methylation patterns found within DR2 may have a direct role in transcriptional regulation and may be influenced by DR1 as well. In addition, a new noncoding RNA (DBE-T) has been described for D4Z4 starting in the proximal region and continuing until approximately the FseI site of D4Z4.28 Whether methylation at DR1 or DR2 would influence this noncoding RNA remains to be determined, but because DBE-T recruits polycomb repressors to D4Z4 and influences chromatin remodeling, it is possible that methylation patterns at DR1 and DR2 could be influenced by DBE-T.
Given the recent observation that the DUX4 polyadenylation signal explains the permissive alleles,15 and thus that DUX4 must have a central role in FSHD, it will be important to determine how methylation changes lead to the expression, splicing, or stability of DUX4 RNA. Examining the methylation status of the DR1 region of D4Z4 by bisulfite sequencing in cells throughout development and differentiation may help to clarify the underlying epigenetic mechanisms of FSHD.
Supplementary Material
ACKNOWLEDGMENT
The authors gratefully acknowledge the participation of patients and controls.
GLOSSARY
- bp
base pairs
- FSHD
facioscapulohumeral muscular dystrophy
- PBL
peripheral blood lymphocyte
Footnotes
Supplemental data at www.neurology.org
AUTHOR CONTRIBUTIONS
L.M. Hartweck: experimental design; collection, analysis, and interpretation of data; and drafting of the manuscript. L.J. Anderson: collection, analysis, and interpretation of data. R.J. Lemmers: sample collection and interpretation of data. E.A. Toso, A. Dandapat, and J.C. Dalton: collection and analysis of data. R. Tawil, J.W. Day, and S.M. van der Maarel: sample collection and interpretation of data. M. Kyba: study design, interpretation of data, and writing the manuscript.
STUDY FUNDING
This study was funded by NIH grants R01 AR055685 and RC2 AR058919 and the Dr. Bob and Jean Smith Foundation.
DISCLOSURE
L.M. Hartweck and L.J. Anderson report no disclosures. R.J. Lemmers receives support from The Netherlands Organization for Scientific Research (NOW) and the FSH Society, and is co-inventor on a patent application for FSHD. E.A. Toso, A. Dandapat, and J.C. Dalton report no disclosures. R. Tawil receives support from NIH (NINDS P01NS069539 [co-PI]). J.W. Day serves on the medical advisory committee for Muscular Dystrophy Association; receives research support from Genzyme Corporation, PTC Therapeutics, Inc., the NIH (NIAMS AR05722 [PI], NINDS NS056592 [PI], BUBDS BS040389 [co-PI], and NINDS NS056159 [co-PI]), and the Muscular Dystrophy Association; and receives royalties from patents on genetic testing for myotonic dystrophy type 2 and spinocerebellar ataxia type 5 that are licensed to Athena Diagnostics and receives institutional support for research from the University of Minnesota Medical School. S.M. van der Maarel receives research support from the NIH, MDA, AFM, IBL, FSH Society, Prinses Beatrix Fonds, NWO, Fields Center, Geraldi Norton Foundation, and the Eklund Family; and is co-inventor on a patent application for FSHD and receives royalties from testing for Myasthenia Gravis licensed to IBL. M. Kyba receives support from the NIH (NIAMS AR055685 [PI], NHLBI HL099478 Kyba [PI], NIAMS AR058919 [PI], NIA AG034370 [PI], NHLBI HL081186 [PI], NIGMS GM081627 [co-investigator], NHLBI HL100407 [co-PI]). Go to Neurology.org for full disclosures.
REFERENCES
- 1.van der Maarel SM, Frants RR, Padberg GW. Facioscapulohumeral muscular dystrophy. Biochim Biophys Acta 2007;1772:186–194 [DOI] [PubMed] [Google Scholar]
- 2.Padberg G. Facioscapulohumeral Disease. Leiden: University of Leiden; 1982 [Google Scholar]
- 3.Orphanet Prevalence of rare diseases: bibliographic data. Orphanet Report Series Rare Diseases Collection. 2010. Available at: http://www.orpha.net/orphacom/cahiers/docs/GB/Prevalence_of_rare_diseases_by_alphabetical_list.pdf Accessed December 7, 2012.
- 4.Wijmenga C, Hewitt JE, Sandkuijl LA, et al. Chromosome 4q DNA rearrangements associated with facioscapulohumeral muscular dystrophy. Nat Genet 1992;2:26–30 [DOI] [PubMed] [Google Scholar]
- 5.de Greef JC, Lemmers RJ, van Engelen BG, et al. Common epigenetic changes of D4Z4 in contraction-dependent and contraction-independent FSHD. Hum Mutat 2009;30:1449–1459 [DOI] [PubMed] [Google Scholar]
- 6.de Greef JC, Lemmers RJ, Camano P, et al. Clinical features of facioscapulohumeral muscular dystrophy 2. Neurology 2010;75:1548–1554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Padua L, Aprile I, Frusciante R, et al. Quality of life and pain in patients with facioscapulohumeral muscular dystrophy. Muscle Nerve 2009;40:200–205 [DOI] [PubMed] [Google Scholar]
- 8.van Overveld PG, Enthoven L, Ricci E, et al. Variable hypomethylation of D4Z4 in facioscapulohumeral muscular dystrophy. Ann Neurol 2005;58:569–576 [DOI] [PubMed] [Google Scholar]
- 9.Bosnakovski D, Xu Z, Gang EJ, et al. An isogenetic myoblast expression screen identifies DUX4-mediated FSHD-associated molecular pathologies. EMBO J 2008;27:2766–2779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gabriels J, Beckers MC, Ding H, et al. Nucleotide sequence of the partially deleted D4Z4 locus in a patient with FSHD identifies a putative gene within each 3.3 kb element. Gene 1999;236:25–32 [DOI] [PubMed] [Google Scholar]
- 11.Geng LN, Yao Z, Snider L, et al. DUX4 activates germline genes, retroelements, and immune mediators: implications for facioscapulohumeral dystrophy. Dev Cell 2012;22:38–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kowaljow V, Marcowycz A, Ansseau E, et al. The DUX4 gene at the FSHD1A locus encodes a pro-apoptotic protein. Neuromuscul Disord 2007;17:611–623 [DOI] [PubMed] [Google Scholar]
- 13.Vanderplanck C, Ansseau E, Charron S, et al. The FSHD atrophic myotube phenotype is caused by DUX4 expression. PLoS One 2011;6:e26820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wallace LM, Garwick SE, Mei W, et al. DUX4, a candidate gene for facioscapulohumeral muscular dystrophy, causes p53-dependent myopathy in vivo. Ann Neurol 2011;69:540–552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lemmers RJ, van der Vliet PJ, Klooster R, et al. A unifying genetic model for facioscapulohumeral muscular dystrophy. Science 2010;329:1650–1653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zeng W, de Greef JC, Chen YY, et al. Specific loss of histone H3 lysine 9 trimethylation and HP1gamma/cohesin binding at D4Z4 repeats is associated with facioscapulohumeral dystrophy (FSHD). PLoS Genet 2009;5:e1000559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.van Overveld PG, Lemmers RJ, Sandkuijl LA, et al. Hypomethylation of D4Z4 in 4q-linked and non-4q-linked facioscapulohumeral muscular dystrophy. Nat Genet 2003;35:315–317 [DOI] [PubMed] [Google Scholar]
- 18.de Greef JC, Wohlgemuth M, Chan OA, et al. Hypomethylation is restricted to the D4Z4 repeat array in phenotypic FSHD. Neurology 2007;69:1018–1026 [DOI] [PubMed] [Google Scholar]
- 19.Ottaviani A, Rival-Gervier S, Boussouar A, et al. The D4Z4 macrosatellite repeat acts as a CTCF and A-type lamins-dependent insulator in facio-scapulo-humeral dystrophy. PLoS Genet 2009;5:e1000394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ottaviani A, Schluth-Bolard C, Rival-Gervier S, et al. Identification of a perinuclear positioning element in human subtelomeres that requires A-type lamins and CTCF. EMBO J 2009;28:2428–2436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Snider L, Geng LN, Lemmers RJ, et al. Facioscapulohumeral dystrophy: incomplete suppression of a retrotransposed gene. PLoS Genet 2010;6:e1001181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bodega B, Ramirez GD, Grasser F, et al. Remodeling of the chromatin structure of the facioscapulohumeral muscular dystrophy (FSHD) locus and upregulation of FSHD-related gene 1 (FRG1) expression during human myogenic differentiation. BMC Biol 2009;7:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ohlsson R, Lobanenkov V, Klenova E. Does CTCF mediate between nuclear organization and gene expression? Bioessays 2010;32:37–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dixit M, Ansseau E, Tassin A, et al. DUX4, a candidate gene of facioscapulohumeral muscular dystrophy, encodes a transcriptional activator of PITX1. Proc Natl Acad Sci USA 2007;104:18157–18162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Snider L, Asawachaicharn A, Tyler AE, et al. RNA transcripts, miRNA-sized fragments and proteins produced from D4Z4 units: new candidates for the pathophysiology of facioscapulohumeral dystrophy. Hum Mol Genet 2009;18:2414–2430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tsumagari K, Qi L, Jackson K, et al. Epigenetics of a tandem DNA repeat: chromatin DNaseI sensitivity and opposite methylation changes in cancers. Nucleic Acids Res 2008;36:2196–2207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Block GJ, Petek LM, Narayanan D, et al. Asymmetric bidirectional transcription from the FSHD-causing D4Z4 array modulates DUX4 production. PLoS One 2012;7:e35532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cabianca DS, Casa V, Bodega B, et al. A long ncRNA links copy number variation to a polycomb/trithorax epigenetic switch in FSHD muscular dystrophy. Cell 2012;149:819–831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Filippova GN. Genetics and epigenetics of the multifunctional protein CTCF. Curr Top Dev Biol 2008;80:337–360 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.