

Abstract
Objective:
To identify brain regions with metabolic changes in DYT11 myoclonus-dystonia (DYT11-MD) relative to control subjects and to compare metabolic abnormalities in DYT11-MD with those found in other forms of hereditary dystonia and in posthypoxic myoclonus.
Methods:
[18F]-fluorodeoxyglucose PET was performed in 6 subjects with DYT11-MD (age 30.5 ± 10.1 years) and in 6 nonmanifesting DYT11 mutation carriers (NM-DYT11; age 59.1 ± 8.9 years) representing the parental generation of the affected individuals. These data were compared to scan data from age-matched healthy control subjects using voxel-based whole brain searches and group differences were considered significant at p < 0.05 (corrected, statistical parametric mapping). As a secondary analysis, overlapping abnormalities were identified by comparisons to hereditary dystonias (DYT1, DYT6, dopa-responsive dystonia) and to posthypoxic myoclonus.
Results:
We found significant DYT11 genotype-specific metabolic increases in the inferior pons and in the posterior thalamus as well as reductions in the ventromedial prefrontal cortex. Significant phenotype-related increases were present in the parasagittal cerebellum. This latter abnormality was shared with posthypoxic myoclonus, but not with other forms of dystonia. By contrast, all dystonia cohorts exhibited significant metabolic increases in the superior parietal lobule.
Conclusions:
The findings are consistent with a subcortical myoclonus generator in DYT11-MD, likely involving the cerebellum. By contrast, subtle increases in the superior parietal cortex relate to the additional presence of dystonic symptoms. Although reduced penetrance in DYT11-MD has been attributed to the maternal imprinting epsilon-sarcoglycan mutations, NM-DYT11 carriers showed significant metabolic abnormalities that are not explained by this genetic model.
Myoclonus-dystonia (MD) is a hyperkinetic movement disorder characterized by variable combinations of mild to moderate dystonia and predominant myoclonus, i.e., brief “lightning-like” jerks without other neurologic dysfunction.1 The most frequent genetic variant of MD, DYT11, has been related to various loss of function mutations in the epsilon-sarcoglycan gene (SGCE) located on chromosome 7q21.2,3
Inheritance of DYT11 is autosomal dominant with incomplete penetrance. Nearly all penetrant cases have been found to be paternally transmitted, consistent with maternal imprinting.2 In this vein, molecular studies have shown maternal imprint of the SGCE gene in human blood cells,4 murine embryogenic fibroblasts,5 and neonatal as well as embryonic brain tissue.6 However, in adult rodent brain tissue, the maternal imprint was found to be incomplete (figure 3 in reference 5). Despite the established link between SGCE mutations and DYT11-MD, the mechanisms by which the mutated protein produces the clinical manifestations of the disorder remain largely unknown.
To date, electrophysiologic and imaging studies in MD1,e1–e7 have suggested a subcortical origin for myoclonus. In this study, we used [18F]-fluorodeoxyglucose (FDG)-PET to assess genotypic and phenotypic metabolic changes in patients with DYT11-MD and in nonmanifesting carriers of this mutation (NM-DYT11). We hypothesized that significant subcortical abnormalities are present in DYT11-MD affecteds. However, because of maternal imprinting of the SGCE mutation, we hypothesized that corresponding changes are not evident in nonmanifesting carriers of this gene. We also performed several secondary analyses to determine which of the metabolic features of DYT11-MD are shared with the primary hereditary dystonias (DYT1, DYT6),e8 dopa-responsive dystonia (DRD),e9 and posthypoxic myoclonus.e10
METHODS
Subjects
We studied 6 subjects with DYT11-MD (age 30.5 ± 10.1 years; 3 male/3 female) and 6 NM-DYT11 carriers (age 59.1 ± 8.9 years; 6 male/0 female) from 5 families. The DYT11 families were recruited through the Mirken Department of Neurology at Beth Israel Medical Center in New York. Three of the DYT11-MD patients and 2 of the NM-DYT11 carriers were on chronic antidepressant treatment with selective serotonin reuptake inhibitors (SSRIs). Informed consent was obtained from all participants under protocols approved by the institutional review boards of the participating institutions.
These families carried different mutations of the SGCE gene (table 1). Two of these families have been reported previously with a c.1151_1152delT mutation (family 2)7 and a deletion of exons 2–5 (patient 1).8 The third family had a previously reported c.304C>T mutation.9 The remaining 2 families had novel mutations: c.1037+5G>C and c.198_199insGAGAATA. Review of the pedigrees confirmed that all affected subjects had inherited the mutation from their fathers. Of note, the NM-DYT11 carriers constituted the paternal generation of the DYT11-MD affecteds. Thus, an age difference was inherent across the 2 gene-positive groups (p < 0.01). Maternal inheritance of the SGCE mutation in the NM-DYT11 subjects was confirmed in 4 of the 6 gene carriers (2 had clinically affected mothers and 2 had nonmanifesting mothers in whom the mutation was molecularly confirmed). Scans from 24 healthy volunteer subjects were used as controls for group comparison. Scans from the 12 older members of this group (Cold: age 56.5 ± 12.5 years; 5 female/7 male) were compared to those from the NM-DYT11 carriers (age p = 0.6; gender p = 0.12). Likewise, scans from the 12 younger healthy volunteers (Cyoung: age 28.2 ± 4.8 years; 3 female/9 male) were compared to those from the DYT11-MD affecteds (age p = 0.6; gender p = 0.3). Because of the low frequency of SGCE mutations,e11 inadvertent inclusion of SGCE mutation carriers among the control subjects was considered unlikely.
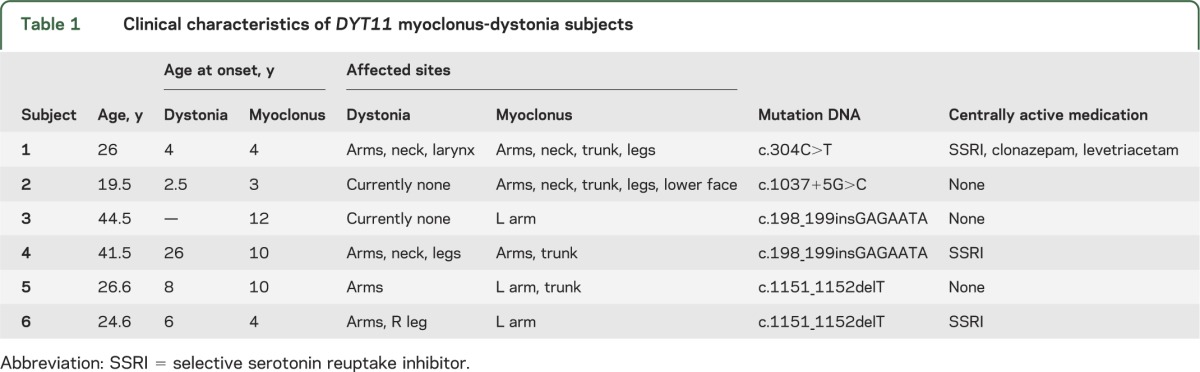
Table 1.
Clinical characteristics of DYT11 myoclonus-dystonia subjects
In secondary analyses, we determined the topographic overlap between the regional metabolic abnormalities observed in the DYT11-MD subjects and those previously described in patients with primary hereditary dystonia (DYT1: n = 18; age 40.3 ± 14.2 years; 7 female, 11 male; DYT6: n = 13; age 33.6 ± 15.0 years; 8 female, 5 male; see references 10 and e8), with dopa-responsive dystonia (DRD; n = 9; age 49.8 ± 15.0 years; 8 female, 1 male; see reference e9), and posthypoxic myoclonus (n = 7; age 47.7 ± 10.1 years; 3 female, 4 male; see table 1 in reference e10). FDG-PET data from these comparison groups have been published previously.10,e8–e10
Clinical characteristics of the manifesting subjects are presented in table 1. Exclusion criteria for all subjects were 1) past history of neurologic illnesses other than the movement disorder leading to study inclusion (i.e., MD, dystonia, or posthypoxic dystonia); 2) prior or current exposure to neuroleptic agents or drug use; 3) past medical history of severe hypertension, cardiovascular disease, or diabetes mellitus; and 4) abnormal MRI. For controls and nonmanifesting subjects, the following additional exclusion criteria were applied: current use of psychotropic medication, abnormal neurologic examination, and past history of dystonic symptoms.
PET
All subjects fasted overnight before PET imaging. Medications (see table 1) were withheld in the affected subjects for at least 12 hours before scanning. FDG-PET studies were performed in 3-dimensional mode using the GE Advance tomograph (General Electric; Milwaukee, WI) at North Shore University Hospital, Manhasset, New York. Subjects were scanned during the resting state in a relaxed and comfortable position. The details of the scanning procedure have been presented elsewhere.11
Image analysis
Image data processing and preprocessing were applied using Statistical Parametric Mapping 5 software (SPM5; Institute of Neurology, London, UK; ucl.ac.uk/spm/software/spm5/) following established standards (realignment, spatial normalizing, smoothing with 10 _ 10 _ 10 mm).
Voxel-wise comparisons were performed using the full factorial model implemented in SPM5. To control for potential confounds resulting from the variability in age and gender, these factors were entered as covariates of no interest. To reduce intersubject variability, regional metabolic measurements were normalized by global hemispheric values. SPM{t} maps were generated to assess the effects of DYT11 mutation status (conjunction analysis of DYT11-MD vs Cyoung and NM-DYT11 vs Cold), as well as the effects of penetrance in carriers of this genotype (DYT11-MD vs NM-DYT11). These comparisons were conducted using the full factorial model with the DYT11 carriers and the control scans. In the secondary analyses, the model was expanded to include data from groups of patients with DYT1 or DYT6 dystonia, DRD, or posthypoxic myoclonus. In this model, shared metabolic abnormalities were identified using conjunction analysis of the respective group contrasts with control images.
For all voxel-based analyses, results were considered significant at p < 0.05 corrected for multiple comparisons at the cluster level, for contiguous clusters of [ke] > 50 voxels. The results of the conjunction analyses were considered significant at p < 0.05, family-wise error rate (FWE)–corrected without spatial constraint.12 Given the rarity of the DYT11 mutation and the small number of mutation carriers available for study, we also reported the results of group contrasts thresholded at p < 0.001, voxel-level uncorrected. While considered exploratory, the resulting clusters were constrained by a spatial extent threshold of [ke] > 80 voxels to reduce Type I error. Post hoc analyses on adjusted rCMRGlu values from significant prefrontal and cerebellar clusters were used to test for the effect of SSRIs on group differences. Coordinates were reported in the standard anatomical space developed at the Montreal Neurological Institute (MNI).
Standard protocol approvals, registrations, and patient consents
This study was approved by the institutional review boards of the participating institutions. Written consent was obtained from each subject following detailed explanation of the procedures.
RESULTS
Metabolic abnormalities related to the DYT11 genotype
We found genotype-related metabolic increases in the ventral inferior pons and in the right posterior thalamus (table 2Aiii, figure 1A). These changes were present in both DYT11-MD patients (table 2Ai) and NM-DYT11 carriers (table 2Aii) when separately compared to age-matched control subjects. Genotype-related metabolic reductions were present bilaterally in the ventromedial prefrontal cortex (table 2B, figure 1B). No effect of SSRIs was evident in this area: the metabolic reduction remained significant even after excluding the 5 DYT11 subjects on SSRIs (DYT11: rCMRGlu (adjusted): 15.8 ± 0.28 mg/dL; Controls: rCMRGlu (adjusted): 17.1 ± 0.17; p > 0.001).
Table 2.
Genotype-related metabolic changes in DYT11 mutation carriersa
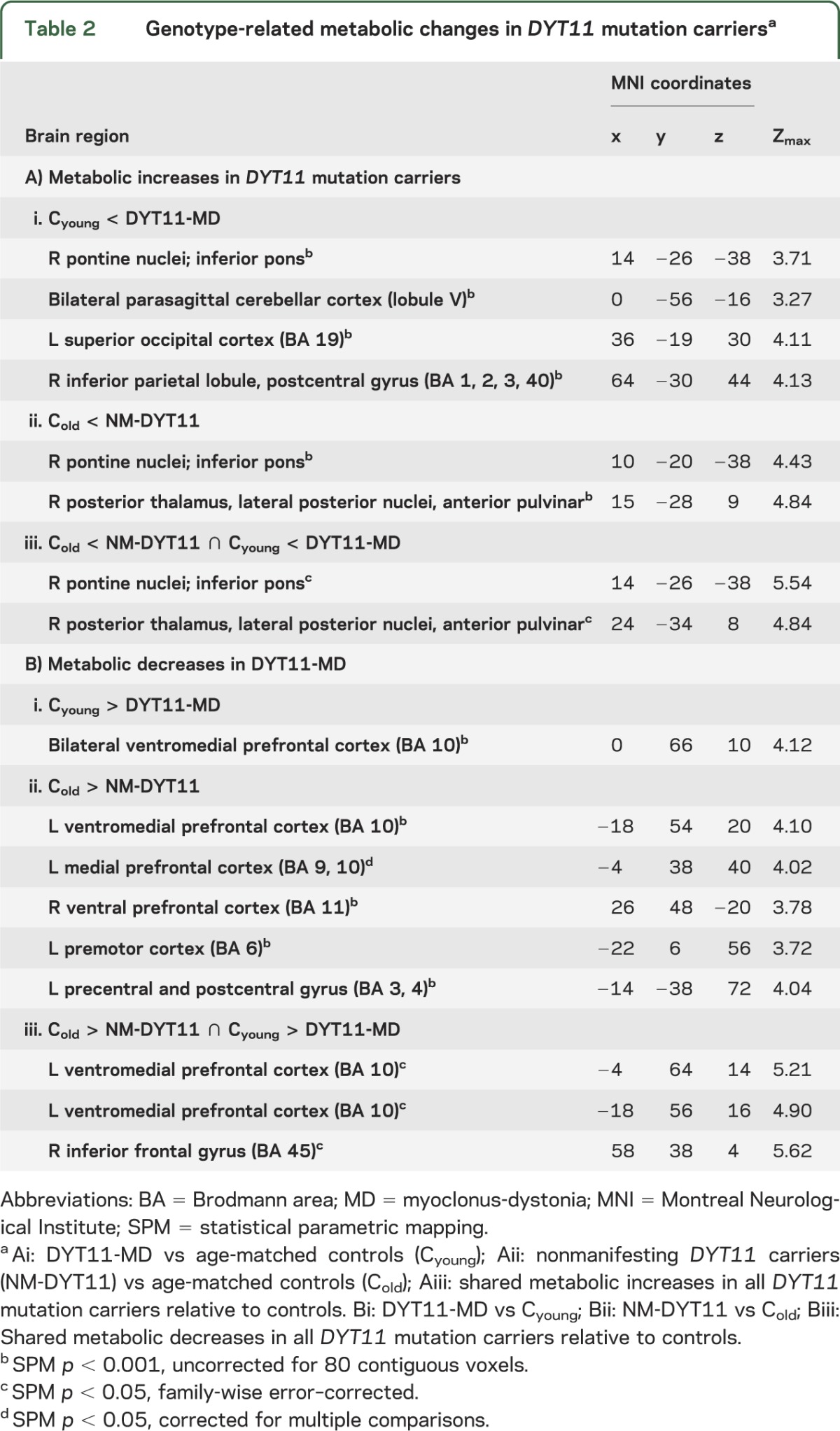
Figure 1. Metabolic abnormalities in DYT11 mutation carriers regardless of symptoms.
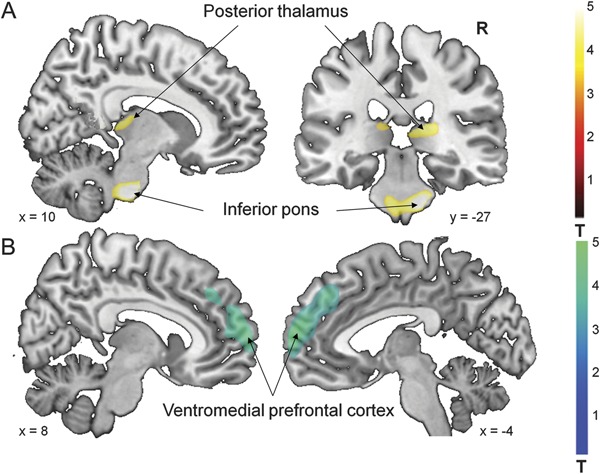
Statistical parametric maps (SPM) comparing [18F]-fluorodeoxyglucose PET scans from DYT11 mutation carriers to control subjects (table 2). (A) Increased regional metabolism was found in the inferior pons and posterior thalamus (table 2Aiii). (B) Decreased regional metabolism was found in the ventromedial prefrontal cortex (table 2Biii). (SPM{t} maps were superimposed on a single-subject MRI T1 template. Coordinates indicate the position of the slice in Montreal Neurological Institute standard space. The color scales represent t scores thresholded at t = 3.5, SPM corrected p < 0.05.)
Metabolic abnormalities related to phenotype
Group comparison of DYT11-MD compared to NM-DYT11 and healthy control subjects revealed metabolic increases in the left parasagittal cerebellum (lobule V) (table 3, figure 2A). No significant effect of SSRIs was present in this region. In a model that included age and SSRI treatment as covariates, we found an effect of group (DYT11-MD vs NM-DYT11 p = 0.0004), but no contribution from SSRI (p = 0.12).
Table 3.
Phenotype-related metabolic increases in DYT11 myoclonus-dystoniaa
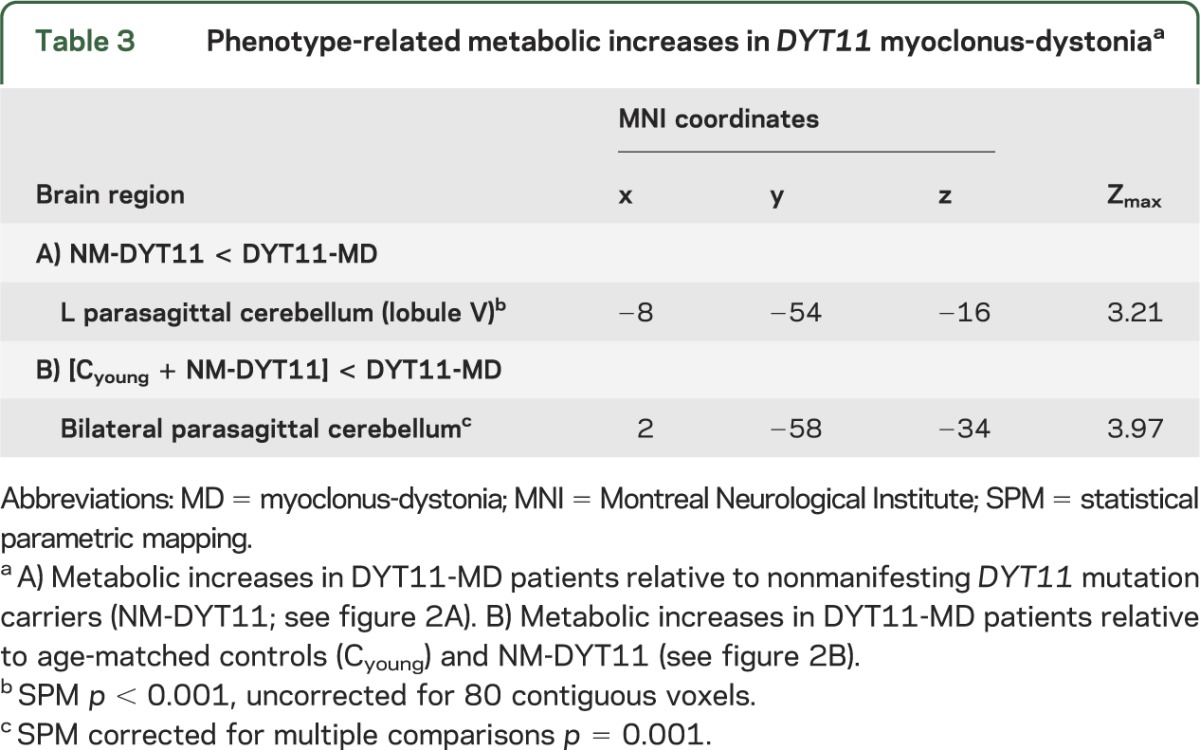
Figure 2. Metabolic abnormalities in symptomatic DYT11 mutation carriers.
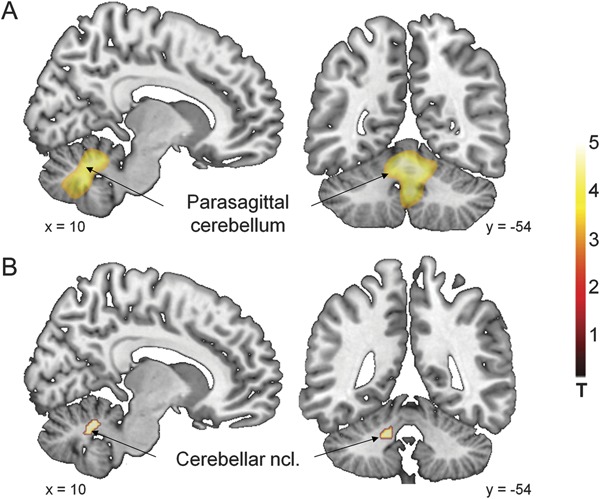
(A) Metabolic increases in DYT11 myoclonus-dystonia (MD) relative to nonmanifesting DYT11 mutation carriers and to age-matched controls were present in the parasagittal cerebellum bilaterally (see table 3). (B) Metabolic increases of parasagittal cerebellar nuclei were a shared characteristic of DYT11-MD and posthypoxic myoclonus. (Statistical parametric maps [SPM{t}] were superimposed as in figure 1 [with identical slice positions of figure 2, A and B]. The color scales represent t scores thresholded at t = 3.0, SPM corrected p < 0.001.)
No metabolic reductions were present in this comparison.
Shared abnormalities: Myoclonus-dystonia and other forms of dystonia
Dystonia patients of all subtypes exhibited abnormal metabolic increases in the superior parietal lobule (Brodmann area [BA] 7/40; MNI coordinates −30, −70, 44; Zmax 3.05; SPM p = 0.001 uncorrected for 80 contiguous voxels). Abnormally reduced metabolic activity was evident in the ventromedial prefrontal cortex of both DYT11-MD and DYT1 dystonia patients (BA 10; MNI coordinates 6, 62, 2; Zmax 4.76; SPM p = 0.05, FWE-corrected). Conjunction analysis of metabolic abnormalities in DYT11-MD and posthypoxic myoclonus disclosed the presence of shared increases in the parasagittal cerebellar nuclei bilaterally (figure 2B; right: MNI coordinates 6, −54, −24; Zmax 5.18; left: MNI coordinates −14, −44, −32; Zmax 4.98; SPM p = 0.05, FWE-corrected). Both groups, similar to the overlap with DYT1 dystonia (see above), also exhibited shared metabolic reductions in the ventromedial prefrontal cortex bilaterally (BA 10; right: MNI coordinates 6, 62, 2; Zmax 4.76; left: MNI coordinates −2, −58, 12; Zmax 4.70; SPM p = 0.05, FWE-corrected).
DISCUSSION
This study showed that SGCE mutations are associated with abnormal metabolic increases in the pontine nuclei and in the posterior thalamus, in conjunction with metabolic decreases in the ventromedial prefrontal cortex. Importantly, these changes were present irrespective of clinical penetrance. That said, additional changes were present in the parasagittal cerebellum, where DYT11-MD affecteds exhibited higher metabolic activity than their nonmanifesting counterparts as well as healthy control subjects.
These findings are difficult to reconcile with the notion of maternal imprinting of the SGCE gene in that the pontocerebellar and ventromedial prefrontal metabolic changes seen in the affecteds were also present in nonmanifesting parental gene carriers. Indeed, abnormal motor activation responses have recently been reported in NM-DYT11 carriers, consistent with the presence of an underlying functional change in these subjects. It is likely, however, that maternal imprinting is not a binary, all-or-nothing regulator of gene penetrance. The molecular evidence for maternal imprinting stems from data from human blood cells and rodent neurons4–6; evidence demonstrating methylation of the promotor region of SGCE in human neurons is currently lacking. In fact, imprinting varies by species, tissue, and developmental stage.13–16 Of note, imprinted genes have been shown to play an important role during embryogenesis. This notion has been expanded to include their important functional role in neurodevelopment.15 While imprinted genes are labeled in all tissues and during all stages of development, their expression is not invariably maintained, and can change considerably during development, differentiation, and disease.13,17 In particular, it has been noted that imprinted expression can be lost during cellular differentiation in a tissue-specific manner. This applies particularly to SGCE, for which weak maternal expression, in addition to the paternal expression, has been shown in adult rodent neuronal cells,5 even though the maternal imprint was maintained in neonatal6 and embryonic brain tissue.5 Thus, it is possible that a biallelic expression of SGCE is needed during certain periods of neurodevelopment, during which a loss-of-function mutation in the maternal allele (as is posited for DYT11-MD) gives rise to the consistent regional metabolic changes that were observed.
The metabolic increases found in our cohort of DYT11-MD are highly concordant with earlier imaging studies in single cases of DYT11-MD.e5,e6 These reports described abnormalities in parasagittal cerebellar and posterior thalamic activation responses18,e5 and in resting-state glucose metabolism.e6 While SGCE expression has been demonstrated in both of these brain regions, particularly high expression has been described in cerebellar Purkinje cells.19 SGCE was identified as a homolog of α-sarcoglycan, but unlike this muscle-specific protein, SGCE is highly expressed in the brain, as well as in other tissues.20 In general, sarcoglycans form one element of a large multiprotein complex, the transmembrane dystrophin-glycoprotein (DG) complex.21 The participation of SGCE in the formation of DG-like complexes in central nervous tissue, however, remains hypothetical. There is evidence for a function of DG-like complexes at central neuronal synapses, in particular at GABAergic synapses of the cerebellum. Moreover, the DG-like complex has been associated with neurodevelopment.21 SGCE has been found in neuronal cells of rodents in the cerebral cortex, striatum, thalamus, pons, midbrain, and cerebellum,2,19,22,23 with particularly high expression in monoaminergic midbrain neurons and in cerebellar Purkinje cells.19 A recent study of the human brain showed highest expression of a brain-specific isoform of SGCE in Purkinje cells and neurons in the dentate nucleus with moderate to low expression in the caudate, putamen, and substantia nigra.24
These neurochemical findings accord with our observation relating the myoclonic component of MD to increased metabolic activity in the parasagittal cerebellum, which was evident in comparisons of DYT11-MD patients with either unaffected NM-DYT1 carriers or healthy control subjects.
We note that hypermetabolism of the fastigial nuclei was a shared feature of DYT11-MD and posthypoxic myoclonus. While the cerebellum is implicated in primary dystonia,25,e8 this structure has also been associated with various myoclonus syndromes.26,e12 Hypermetabolism of the parasagittal cerebellum distinguished manifesting from nonmanifesting DYT11 carriers, and was also found to link the metabolic landscape of posthypoxic myoclonus with that of MD. It is therefore likely that the parasagittal cerebellar changes point to a role of the cerebellum in DYT11-MD, as suggested in a recent neurophysiologic study.27 Several case reports have also linked pathology in the parasagittal cerebellum,e12,e13 the dentate nucleus,e14 or Purkinje cellse15,e16 to myoclonus and phenomenologic similarities have been noted between MD and spinocerebellar ataxia type 14.e17 Indeed, experimental animal models of posthypoxic myoclonus have been shown to exhibit Purkinje cell loss.e18
The localization of the genotype-specific changes in pons and thalamus is consistent with the expression of SGCE in these areas.22 While the DYT11 carriers included in this study did not share a common genotype, each of the mutations interferes with SGCE synthesis by the insertion of stop codons that result in a truncated protein coding sequence. We note that metabolic changes in the inferior pons have been found in essential blepharospasm28 and may point to a shared mechanism for the alteration in brainstem reflexes that has been found in both MD and this disorder.29,30 Indeed, a study of DYT11-MD found increased responsivity of the blink reflex.e3 Thalamic lesions have been associated with secondary MD,31 and thalamic deep brain stimulation is used to treat MD.32,33 However, lesions and interventions target mainly the ventral tier thalamic nuclei, whereas the current study localized the metabolic changes in this region to the posterior thalamus, particularly the lateralis posterior and anterior pulvinar nuclei. Although these regions are not considered part of the motor thalamus, a recent retrograde tracer study in the monkey suggested a role for the posterior thalamus in mediating complex motor behavior.34 Complex motor acts, in particular species-typical motor behaviors (such as reaching, grasping, or defensive acts), can be elicited from distinct functional zones of the posterior parietal cortex in primates.35,36 Interestingly, Gharbawie et al.34 showed functionally organized projections from the posterior thalamus to these functional zones. Alternatively, it is also possible that the posterior thalamic changes in our study reflect altered metabolism of the reticular thalamic nucleus. Combined lesions of brainstem and reticular thalamic nucleus were present in an animal model of posthypoxic myoclonus, suggesting these regions as possible myoclonus generators.37 Due to its shell-like form, a clear separation of the reticular nucleus from adjacent posterior thalamic nuclei is not possible.
The current study confirms our earlier finding of parietal hypermetabolism in manifesting dystonia patients, irrespective of etiology.10 By contrast, medial prefrontal hypometabolism in DYT11-MD had more restricted overlap with the other disorders in that this feature was present only in manifesting DYT1 and in posthypoxic myoclonus. Functional changes in this region have been implicated consistently in mood disorders,38 to which DYT1 and DYT11 mutation carriers may be particularly susceptible.39 The psychiatric comorbidity of posthypoxic myoclonus has not been formally reported. Nonetheless, increased rates of anxiety and depression have been reported in survivors of resuscitation, which include individuals with posthypoxic myoclonus.40
Our study has several limitations. Due to the rarity of DYT11-MD and the invasive nature and high costs of the PET procedures, we were able to only include a small number of subjects. Indeed these subjects were derived from only 5 families and the results therefore may not be generalizable to other DYT11 mutations. Moreover, the control subjects should ideally have been recruited as noncarrier relatives of the manifesting DYT11 mutation carriers. It is also noteworthy that the MD phenotype was not identical in all subjects. Even though all 6 manifesting carriers had myoclonus at the time of PET, only 4 additionally exhibited dystonia. This may have contributed to the lack of significant overlap of MD with other dystonias. Furthermore, for ethical reasons we refrained from full medication washout from medications with a longer half-life than 12 hours in all cohorts used in this and earlier studies (which were used for comparative reasons here). Systematic effects of medications have been excluded through post hoc analyses in each of the studies.
Our data support the idea of a role of the cerebellum in conjunction with thalamus and pons in DYT11-MD, particularly for myoclonic symptoms. Given the presence of significant metabolic changes in NM-DYT11 carriers localized to brain regions with high SGCE expression during adulthood, it is likely that maternal imprinting during adulthood is not the sole regulator of penetrance in this disorder.
Supplementary Material
ACKNOWLEDGMENT
The authors thank all patients and family members who participated in this study; Dr. Thomas Chaly for radiochemistry support, Claude Margouleff for technical support, Toni Fitzpatrick for study coordination and editorial support, as well as Noam Gerber for help with manuscript preparation; and the Neurogenetics DNA Diagnostic Laboratory at Massachusetts General Hospital, in particular Drs. Katherine Sims and Winnie Xin, for clinical genetic testing, Dr. Zoltan Mari of Johns Hopkins University for genotyping of individual 2 from our series, and Drs. Cordelia Schwarz and Marta San Luciano for examination of study subjects.
Glossary
- BA
Brodmann area
- DG
dystrophin-glycoprotein
- DRD
dopa-responsive dystonia
- DYT11-MD
DYT11 myoclonus-dystonia
- FDG
18F-fluorodeoxyglucose
- FWE
family-wise error
- MD
myoclonus-dystonia
- MNI
Montreal Neurological Institute
- NM-DYT11
nonmanifesting DYT11 mutation carriers
- SSRI
selective serotonin reuptake inhibitor
Footnotes
Supplemental data at www.neurology.org
AUTHOR CONTRIBUTIONS
S.B. and D.E. directed the study. S.B. received funding for the study. M.C., D.R., L.O., S.B., and D.E. participated in the study design. D.R., R.S.-P., and S.F. participated in subject recruitment. L.O. contributed to sample collection, preparation, and genotyping. V.D. collected imaging data. R.S.-P., S.F., and S.B. performed subject assessments. M.C. performed analysis of imaging data. M.C. wrote the manuscript, which was reviewed and approved by all authors.
STUDY FUNDING
The Constance W. and James W. Brown Family Foundation provided the financial support for all scans in DYT11 carriers.
DISCLOSURE
M. Carbon received research support from the NIH (Dystonia Coalition U54 NS 065701). D. Raymond receives research support from the Michael J. Fox Foundation for Parkinson's Research and the Marcled Foundation. L. Ozelius serves on the scientific advisory boards of the Benign Essential Blepharospasm Research Foundation, the National Spasmodic Dysphonia Association, and the Tourette Syndrome Association. She receives research support from the NIH and the Michael J. Fox Foundation for Parkinson's Research and patent royalties from Athena Diagnostics. R. Saunders-Pullman serves on the Scientific Advisory Board of the Dystonia Medical Research Foundation. She has received research support from the NIH/NINDS (K23NS047256 and K02NS073836), the Michael J. Fox Foundation for Parkinson's Research, the Thomas Hartman Foundation for Parkinson's Research, the Bachmann-Strauss Dystonia Parkinson's Foundation, and the Marcled Foundation. S. Frucht has received travel funding/speaker honoraria from Merz Pharmaceuticals and Impax Pharmaceuticals, receives publishing royalties from Humana Press, Inc., has served as consultant for UCB Pharmaceuticals, Jazz Pharmaceuticals, Merz Pharmaceuticals, GE Healthcare, and Allergan, Inc., and has received lecture sponsorship from the American Academy of Neurology Movement Disorders Society. V. Dhawan has received research support from Neurologix, Inc., the NIH (NIDCD, NIAID, National Institute of Neurological Disorders and Stroke), and the Dana Foundation. S. Bressman serves on scientific advisory boards for the Bachmann Strauss Dystonia and Parkinson's Foundation, the Michael J. Fox Foundation for Parkinson's Research, and the Dystonia Medical Research Foundation. She receives research support from the NIH and the Michael J. Fox Foundation for Parkinson's Research and patent royalties from Athena Diagnostics. D. Eidelberg currently serves as a scientific advisory board member and has received honoraria from the Michael J. Fox Foundation and The Bachmann-Strauss Dystonia and Parkinson Foundation; is listed as coinventor of United States Patent No. 5,632,276 (filed January 27, 1995, granted May 27, 1997) and No. 5,873,823 (filed September 4, 1996, granted February 23, 1999), without financial gain. He served as scientific director of the Thomas Hartman Foundation for Parkinson's Research, Inc.; was a paid consultant for Neurologix, Inc. and Merck & Co., Inc; received research support from the NIH (National Institute of Neurological Disorders and Stroke, NCRR, NIDCD, NIAID), the Dana Foundation, and the Bachmann-Strauss Dystonia and Parkinson Foundation; and has served as an expert in medico-legal cases. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Roze E, Apartis E, Clot F, et al. Myoclonus-dystonia: clinical and electrophysiologic pattern related to SGCE mutations. Neurology 2008;70:1010–1016 [DOI] [PubMed] [Google Scholar]
- 2.Zimprich A, Grabowski M, Asmus F, et al. Mutations in the gene encoding epsilon-sarcoglycan cause myoclonus-dystonia syndrome. Nat Genet 2001;29:66–69 [DOI] [PubMed] [Google Scholar]
- 3.Valente EM, Edwards MJ, Mir P, et al. The epsilon-sarcoglycan gene in myoclonic syndromes. Neurology 2005;64:737–739 [DOI] [PubMed] [Google Scholar]
- 4.Muller B, Hedrich K, Kock N, et al. Evidence that paternal expression of the epsilon-sarcoglycan gene accounts for reduced penetrance in myoclonus-dystonia. Am J Hum Genet 2002;71:1303–1311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Piras G, El Kharroubi A, Kozlov S, et al. Zac1 (Lot1), a potential tumor suppressor gene, and the gene for epsilon-sarcoglycan are maternally imprinted genes: identification by a subtractive screen of novel uniparental fibroblast lines. Mol Cell Biol 2000;20:3308–3315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang X, Sun Q, McGrath SD, Mardis ER, Soloway PD, Clark AG. Transcriptome-wide identification of novel imprinted genes in neonatal mouse brain. PLoS One 2008;3:e3839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Raymond D, Saunders-Pullman R, de Carvalho Aguiar P, et al. Phenotypic spectrum and sex effects in eleven myoclonus-dystonia families with epsilon-sarcoglycan mutations. Mov Disord 2008;23:588–592 [DOI] [PubMed] [Google Scholar]
- 8.Luciano MS, Ozelius L, Sims K, Raymond D, Liu L, Saunders-Pullman R. Responsiveness to levodopa in epsilon-sarcoglycan deletions. Mov Disord 2009;24:425–428 [DOI] [PubMed] [Google Scholar]
- 9.Kinugawa K, Vidailhet M, Clot F, Apartis E, Grabli D, Roze E. Myoclonus-dystonia: an update. Mov Disord 2009;24:479–489 [DOI] [PubMed] [Google Scholar]
- 10.Carbon M, Su S, Dhawan V, Raymond D, Bressman S, Eidelberg D. Regional metabolism in primary torsion dystonia: effects of penetrance and genotype. Neurology 2004;62:1384–1390 [DOI] [PubMed] [Google Scholar]
- 11.Feigin A, Fukuda M, Dhawan V, et al. Metabolic correlates of levodopa response in Parkinson's disease. Neurology 2001;57:2083–2088 [DOI] [PubMed] [Google Scholar]
- 12.Friston KJ, Stephan KE, Lund TE, Morcom A, Kiebel S. Mixed-effects and fMRI studies. Neuroimage 2005;24:244–252 [DOI] [PubMed] [Google Scholar]
- 13.Hudson QJ, Kulinski TM, Huetter SP, Barlow DP. Genomic imprinting mechanisms in embryonic and extraembryonic mouse tissues. Heredity 2010;105:45–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Taniguchi T, Sullivan MJ, Ogawa O, Reeve AE. Epigenetic changes encompassing the IGF2/H19 locus associated with relaxation of IGF2 imprinting and silencing of H19 in Wilms tumor. Proc Natl Acad Sci USA 1995;92:2159–2163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Davies W, Isles AR, Wilkinson LS. Imprinted gene expression in the brain. Neurosci Biobehav Rev 2005;29:421–430 [DOI] [PubMed] [Google Scholar]
- 16.Schulz R, Menheniott TR, Woodfine K, Wood AJ, Choi JD, Oakey RJ. Chromosome-wide identification of novel imprinted genes using microarrays and uniparental disomies. Nucleic Acids Res 2006;34:e88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Monk M, Salpekar A. Expression of imprinted genes in human preimplantation development. Mol Cell Endocrinol 2001;183(suppl 1):S35–S40 [DOI] [PubMed] [Google Scholar]
- 18.Beukers RJ, Foncke EM, van der Meer JN, et al. Disorganized sensorimotor integration in mutation-positive myoclonus-dystonia: a functional magnetic resonance imaging study. Arch Neurol 2010;67:469–474 [DOI] [PubMed] [Google Scholar]
- 19.Chan P, Gonzalez-Maeso J, Ruf F, Bishop DF, Hof PR, Sealfon SC. Epsilon-sarcoglycan immunoreactivity and mRNA expression in mouse brain. J Comp Neurol 2005;482:50–73 [DOI] [PubMed] [Google Scholar]
- 20.Ettinger AJ, Feng G, Sanes JR. epsilon-Sarcoglycan, a broadly expressed homologue of the gene mutated in limb-girdle muscular dystrophy 2D. J Biol Chem 1997;272:32534–32538 [DOI] [PubMed] [Google Scholar]
- 21.Waite A, Tinsley CL, Locke M, Blake DJ. The neurobiology of the dystrophin-associated glycoprotein complex. Ann Med 2009;41:344–359 [DOI] [PubMed] [Google Scholar]
- 22.Nishiyama A, Endo T, Takeda S, Imamura M. Identification and characterization of epsilon-sarcoglycans in the central nervous system. Brain Res Mol Brain Res 2004;125:1–12 [DOI] [PubMed] [Google Scholar]
- 23.Xiao J, LeDoux MS. Cloning, developmental regulation and neural localization of rat epsilon-sarcoglycan. Brain Res Mol Brain Res 2003;119:132–143 [DOI] [PubMed] [Google Scholar]
- 24.Ritz K, van Schaik BD, Jakobs ME, et al. SGCE isoform characterization and expression in human brain: implications for myoclonus-dystonia pathogenesis? Eur J Hum Genet 2011;19:438–444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jinnah HA, Hess EJ. A new twist on the anatomy of dystonia: the basal ganglia and the cerebellum? Neurology 2006;67:1740–1741 [DOI] [PubMed] [Google Scholar]
- 26.Shibasaki H, Thompson PD. Milestones in myoclonus. Mov Disord 2011;26:1142–1148 [DOI] [PubMed] [Google Scholar]
- 27.Hubsch C, Vidailhet M, Rivaud-Pechoux S, et al. Impaired saccadic adaptation in DYT11 dystonia. J Neurol Neurosurg Psychiatry 2011;82:1103–1106 [DOI] [PubMed] [Google Scholar]
- 28.Hutchinson M, Nakamura T, Moeller JR, et al. The metabolic topography of essential blepharospasm: a focal dystonia with general implications. Neurology 2000;55:673–677 [DOI] [PubMed] [Google Scholar]
- 29.Muller J, Rinnerthaler M, Poewe W, Kofler M. Auditory startle reaction in primary blepharospasm. Mov Disord 2007;22:268–272 [DOI] [PubMed] [Google Scholar]
- 30.Tolosa E, Montserrat L, Bayes A. Blink reflex studies in focal dystonias: enhanced excitability of brainstem interneurons in cranial dystonia and spasmodic torticollis. Mov Disord 1988;3:61–69 [DOI] [PubMed] [Google Scholar]
- 31.Lehericy S, Grand S, Pollak P, et al. Clinical characteristics and topography of lesions in movement disorders due to thalamic lesions. Neurology 2001;57:1055–1066 [DOI] [PubMed] [Google Scholar]
- 32.Gruber D, Kuhn AA, Schoenecker T, et al. Pallidal and thalamic deep brain stimulation in myoclonus-dystonia. Mov Disord 2010;25:1733–1743 [DOI] [PubMed] [Google Scholar]
- 33.Kuncel AM, Turner DA, Ozelius LJ, Greene PE, Grill WM, Stacy MA. Myoclonus and tremor response to thalamic deep brain stimulation parameters in a patient with inherited myoclonus-dystonia syndrome. Clin Neurol Neurosurg 2009;111:303–306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gharbawie OA, Stepniewska I, Burish MJ, Kaas JH. Thalamocortical connections of functional zones in posterior parietal cortex and frontal cortex motor regions in New World monkeys. Cereb Cortex 2010;20:2391–2410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cooke DF, Taylor CS, Moore T, Graziano MS. Complex movements evoked by microstimulation of the ventral intraparietal area. Proc Natl Acad Sci USA 2003;100:6163–6168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stepniewska I, Fang PC, Kaas JH. Microstimulation reveals specialized subregions for different complex movements in posterior parietal cortex of prosimian galagos. Proc Natl Acad Sci USA 2005;102:4878–4883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tai KK, Truong DD. Post-hypoxic myoclonus induces Fos expression in the reticular thalamic nucleus and neurons in the brainstem. Brain Res 2005;1059:122–128 [DOI] [PubMed] [Google Scholar]
- 38.Price JL, Drevets WC. Neurocircuitry of mood disorders. Neuropsychopharmacology 2010;35:192–216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Heiman GA, Ottman R, Saunders-Pullman RJ, Ozelius LJ, Risch NJ, Bressman SB. Increased risk for recurrent major depression in DYT1 dystonia mutation carriers. Neurology 2004;63:631–637 [DOI] [PubMed] [Google Scholar]
- 40.Wachelder EM, Moulaert VR, van Heugten C, Verbunt JA, Bekkers SC, Wade DT. Life after survival: long-term daily functioning and quality of life after an out-of-hospital cardiac arrest. Resuscitation 2009;80:517–522 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.