

Abstract
Ryanodine receptors (RyRs), located in the sarcoplasmic/endoplasmic reticulum (SR/ER) membrane, are required for intracellular Ca2+ release that is involved in a wide range of cellular functions. In addition to Ca2+-induced Ca2+ release in cardiac cells and voltage-induced Ca2+ release in skeletal muscle cells, we recently identified another mode of intracellular Ca2+ mobilization mediated by RyR, i.e., nitric oxide-induced Ca2+ release (NICR), in cerebellar Purkinje cells. NICR is evoked by neuronal activity, is dependent on S-nitrosylation of type 1 RyR (RyR1) and is involved in the induction of long-term potentiation (LTP) of cerebellar synapses. In this addendum, we examined whether peroxynitrite, which is produced by the reaction of nitric oxide with superoxide, may also have an effect on the Ca2+ release via RyR1 and the cerebellar LTP. We found that scavengers of peroxynitrite have no significant effect either on the Ca2+ release via RyR1 or on the cerebellar LTP. We also found that an application of a high concentration of peroxynitrite does not reproduce neuronal activity-dependent Ca2+ release in Purkinje cells. These results support that NICR is induced by endogenous nitric oxide produced by neuronal activity through S-nitrosylation of RyR1.
Keywords: nitric oxide, S-nitrosylation, ryanodine receptor, calcium, Purkinje cell, synaptic plasticity
There are an increasing number of studies showing that protein S-nitrosylation, the addition of a nitric oxide (NO) group to cysteine thiols, provides important roles in a wide range of signaling pathways.1,2 Because the ryanodine receptor (RyR), a Ca2+ release channel expressed in the SR/ER, has about 100 cysteine residues per subunit and almost half of these thiols are kept in a reduced state under the resting condition,3 RyR is thought to be one of the substrate proteins for S-nitrosylation. In accordance with this notion, the open probability of RyR1 measured in lipid bilayers is increased by NO donors,4-7 and this increase is accompanied by an increase in S-nitrosylation of the Ca2+ release channel at cysteine 3635.4,7-9 In addition, NO-induced activation of RyR1 in lipid bilayers and in SR vesicles is influenced by pO2 levels: RyR1 is activated by submicromolar levels of NO in the tissue pO2 level (~10 mmHg) but not in the ambient O2 level (~150 mmHg).4 In these previous studies, the redox regulations of RyRs were extensively studied using in vitro experimental systems, especially in lipid bilayers and SR samples isolated from skeletal muscles.1,10,11 Although S-nitrosylation of RyR1 is implicated in the enhancement of Ca2+ leak from the skeletal muscle SR in pathological conditions,12,13 physiological significance of S-nitrosylation-mediated modulation of RyR1 activity by endogenous NO remained elusive.
We previously found that burst stimulation (BS) of parallel fibers (PFs) generates a localized increase in NO concentration in Purkinje cell (PC) dendrites, as well as NO-dependent long-term potentiation (LTP) at the PF-PC synapse in the cerebellum.14 Because this LTP is also sensitive to the intracellular Ca2+ concentration, we examined a crosstalk between NO and Ca2+ signaling in PCs. Bath application of 1-Hydroxy-2-oxo-3-(N-methyl-3-aminopropyl)-3-methyl-1-triazene (NOC7), a NO donor, induced an increase in the intracellular Ca2+ concentration in PCs. The NO-induced Ca2+ increase was independent of Ca2+ influx from the extracellular medium, but was dependent on the activity of RyR1 and the sarco/endoplasmic reticulum Ca2+ ATPase (SERCA), suggesting that the Ca2+ increase is mediated by Ca2+ release from the intracellular store through RyR1. In addition, our study using RyR1-expressing HEK293 cells indicated that S-nitrosylation of RyR1 at cysteine 3635 is both necessary and sufficient for the NO-induced Ca2+ increase, which is hereafter referred to as “NO-induced Ca2+ release (NICR).”15 We then showed that BS to PFs induces intracellular Ca2+ release in PC dendrites. When BS — which induces NO-dependent-LTP — was applied to PF, Ca2+ levels were transiently but clearly elevated in PC dendrites. This BS-induced Ca2+ increase was indicated to be due to Ca2+ release mediated by RyR1, because the Ca2+ increase was abolished by the application of thapsigargin and cyclopiazonic acid, SERCA inhibitors and dantrolene, a RyR antagonist.16,17 In addition, the Ca2+ release was inhibited by the application of NG-Nitro-l-arginine methyl ester (L-NAME), a broad inhibitor of NO synthases (NOS), and was not observed in the cerebellum of mice deficient in neuronal type NOS (nNOS).18 These results strongly suggest that BS to PFs induces NICR in the PC dendrites.
NO signaling has two types of downstream pathways: activation of soluble guanylyl cyclase (sGC) and S-nitrosylation of target proteins. Because BS-induced NICR was inhibited by intracellular application of ascorbic acid, a reducing reagent, to Purkinje cells, but not by 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one (ODQ), an inhibitor of SGC, NICR was suggested to be dependent on S-nitrosylation of proteins including RyR1. Correspondingly, biochemical analysis demonstrated that NICR induced by the application of NOC7 is accompanied with S-nitrosylation of RyR1 in cerebellar slice preparations. Furthermore, all manipulations that we used to inhibit NICR also abolished PF-LTP. Taken together, our study indicated that NICR is a novel Ca2+ mobilizing mechanism in neuronal cells and is essential for the induction of PF-LTP.
However, neuronal activity in conjunction with certain forms of synaptic plasticity may be associated with superoxide generation,19 and NO generation leads to peroxynitrite formation when superoxide is simultaneously generated.20 Because peroxynitrite is known to regulate cell signaling via molecular modifications, including protein nitration,20 it seems possible that NICR is affected by peroxynitrite produced from endogenous NO in the presence of superoxide. We, therefore, examined the potential role of peroxynitrite in the BS-induced Ca2+ release and LTP. Bath application of the peroxynitrite scavenger, uric acid,21 and the peroxynitrite decomposition catalyst, 5,10,15,20-Tetrakis(4-sulfonatophenyl) porphyrinato iron (III) (FeTPPS),22 had no effect on BS-induced Ca2+ increase (Fig. 1), although the concentration of uric acid (100 μM) and FeTPPS (10 μM) were thought to be high enough.23,24 We subsequently examined effects of uric acid and FeTPPS on the induction of PF-LTP. In accordance with the insensitivity of NICR to these reagents, neither uric acid nor FeTPPS impaired the induction of PF-LTP induced by BS (Fig. 2). These results do not support the involvement of peroxynitrite in NICR and PF-LTP induction, although there remains a possibility that the scavengers did not completely antagonizing the effect of peroxynitrite. We therefore examined the effect of an application of peroxynitrite on the intracellular Ca2+ concentration of Purkinje cells in cerebellar slice preparations. Because our previous NO imaging experiments during BS-induced NO generation from parallel fibers indicates that the peak NO concentration in the center of response is about 5 µM,14 we applied 10 µM peroxynitrite to the cerebellar slice preparations. However, even at this very high concentration, peroxynitrite induced only very little changes in the Ca2+ concentration (Fig. 1D). The slow increase in the Ca2+ concentration became absent in the presence of uric acid or FeTPPS. However, it was resistant to an intracellular application of 10 mM ascorbic acid, which completely abolishes BS-induced NICR.15 Therefore, the peroxynitrite-dependent slow response seems to have a mechanism different from that of NICR and might be accounted for by an irreversible effect of peroxynitrite on the endoplasmic reticulum that has been reported previously.5 These results suggest that the role of peroxynitrite, if any, in BS-induced Ca2+ release is very little.

Figure 1. NO-induced Ca2+ release (NICR) is not affected by peroxynitrite. (A-C) Ca2+ response, induced by a series of 60 BS at 1 Hz (BS; Gray bar) in the standard ACSF (A; data were obtained from 10 cells in 10 slices), was not inhibited by peroxynitrite scavenger, uric acid (B; 100 µM, n = 7 cells in 7 slices), or peroxynitrite decomposition catalyst, FeTPPS (C; 10 µM, n = 6 cells in 6 slices). Uric acid or FeTPPS was present in bathing solution from 30 min before the recording. (D) Ca2+ response to bath application of 10 μM peroxynitrite (ONOO-) (open circle; n = 6 cells, 6 slices). It was abolished by 100 µM uric acid (open triangle; n = 7 cells, 7 slices) or 10 µM FeTPPS (closed triangle; n = 7 cells, 7 slices), but was insensitive to 10 mM ascorbic acid applied intracellularly through the patch pipette (closed circle; n = 8 cells; 8 slices). (E) Summary of data in A–D. The peak Ca2+ responses are shown. UA: uric acid; Fe: FeTPPS; Asc: ascorbic acid. Data are presented as Mean ± SEM *p < 0.05, ANOVA / Tukey-Kramer test.

Figure 2. Cerebellar LTP is not affected by peroxynitrite. (A-C) Cerebellar LTP, induced by BS in the standard ACSF (A; n = 8 cells in 8 slices), was not inhibited by 100 µM uric acid (B; n = 5 cells in 5 slices) or 10 µM FeTPPS (C; n = 5 cells in 5 slices). (D) Effects of uric acid (UA) and FeTPPS on the magnitude of LTP (normalized EPSC averaged between 21–30 min after BS). Data are presented as Mean ± SEM.
In conclusion, in combination with the observations in our previous study,15 it is suggested that NICR is dependent on the NO-dependent S-nitrosylation of RyR1 rather than on indirect actions of peroxynitrite or cyclic GMP (Fig. 3). Biochemical analysis of RyR1 purified from the cerebellar cortex after the LTP induction may further confirm specific involvement of endogenous NO in NICR and PF-LTP.
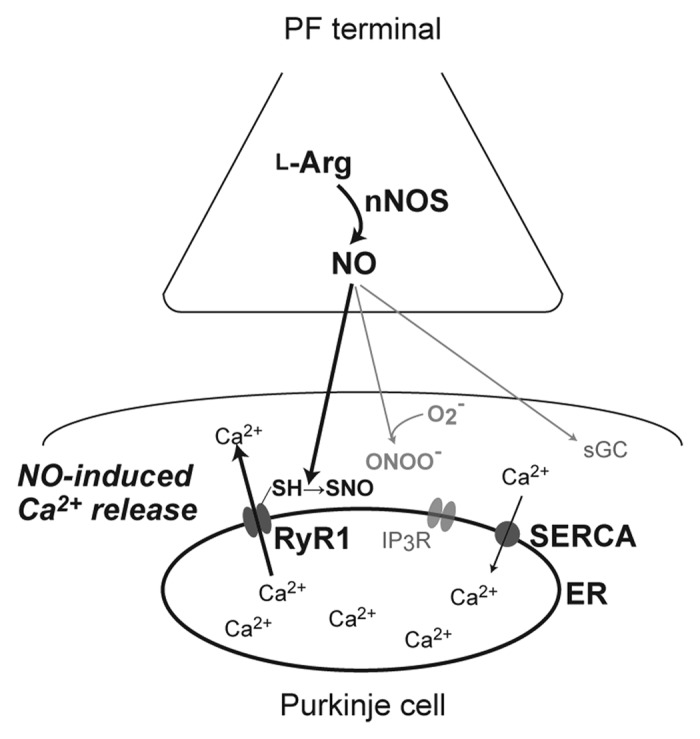
Figure 3. A schematic diagram of signaling pathways for nitric oxide-induced Ca2+ release (NICR) in cerebellar Purkinje cells. Nitric oxide (NO) is produced at the parallel-fiber terminal in response to neuronal activity, and diffuses into the cerebellar Purkinje cell. Then, type 1 ryanodine receptors (RyR1), located in the endoplasmic reticulum (ER) membrane, are S-nitrosylated and activated, resulting in Ca2+ release from the ER. Cyclic GMP and peroxynitrite, which is produced by the reaction of NO and superoxide, are unlikely to be involved in NICR. nNOS: neuronal NO synthase; IP3R1: inositol 1,4,5-trisphosphate receptor; SERCA: sarco/endoplasmic reticulum Ca2+ ATPase.
Materials and Methods
Preparation of cerebellar slices, Ca2+ imaging and electrophysiological analysis were performed essentially as described previously.15,25 C57BL/6 mice (postnatal days 24–32) were sacrificed by cervical dislocation under anesthesia with diethyl ether. The cerebellum was excised, and parasagittal cerebellar slices (250 µm thick) were prepared from the vermis. Whole-cell recordings were obtained from visually identified PCs at room temperature (23–25°C).
For intracellular Ca2+ imaging in PCs, a Ca2+-sensitive dye, Oregon Green 488 BAPTA-1 (100 µM), was introduced into Purkinje cells through the patch pipette. Five to nine sequential confocal images (excitation at 488 nm), obtained at 3–4 µm z-axis intervals, were acquired every 0.8 sec using an upright microscope (BX51WI; Olympus) equipped with a confocal scanning unit and an argon laser (FV300, Olympus), and projected onto a plane to obtain dendrite images at 10 sec intervals.
For focal stimulation of PFs, a stimulation pipette was filled with standard bathing solution and used to apply square pulses (0.1 ms in duration, 0–20 V in amplitude) to the molecular layer, specifically in the middle portion of the molecular layer approximately one-third of the distance below the pial surface. The intensity of each stimulus was adjusted to evoke PF-EPSCs with an amplitude of 60–120 pA. The ionic current was recorded from PCs using a patch-clamp amplifier (EPC-9, HEKA, Lambrecht/Pfalz, Germany) at a holding potential of –90 or –80 mV, after compensating for liquid junction potential. The signals were filtered at 2 kHz and digitized at 20 kHz. After obtaining a stable initial recording for at least 10 min, 60 burst stimulations (1 burst stimulation: 5 pulses at 50 Hz) were repeatedly applied at 1 Hz to induce LTP. Series resistance and membrane resistance were monitored throughout the experiments, and the data were discarded when either of these resistances varied by more than 10%. The data were also discarded when the slope of PF-EPSC amplitude averaged every minute during the initial recording for 10 min was larger than 2% or when the amplitude did not become stable within 20 min after the onset of whole-cell configuration.14
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/channels/article/22555
References
- 1.Hess DT, Matsumoto A, Kim SO, Marshall HE, Stamler JS. Protein S-nitrosylation: purview and parameters. Nat Rev Mol Cell Biol. 2005;6:150–66. doi: 10.1038/nrm1569. [DOI] [PubMed] [Google Scholar]
- 2.Shahani N, Sawa A. Nitric oxide signaling and nitrosative stress in neurons: role for S-nitrosylation. Antioxid Redox Signal. 2011;14:1493–504. doi: 10.1089/ars.2010.3580. [DOI] [PubMed] [Google Scholar]
- 3.Zalk R, Lehnart SE, Marks AR. Modulation of the ryanodine receptor and intracellular calcium. Annu Rev Biochem. 2007;76:367–85. doi: 10.1146/annurev.biochem.76.053105.094237. [DOI] [PubMed] [Google Scholar]
- 4.Eu JP, Sun JH, Xu L, Stamler JS, Meissner G. The skeletal muscle calcium release channel: coupled O2 sensor and NO signaling functions. Cell. 2000;102:499–509. doi: 10.1016/S0092-8674(00)00054-4. [DOI] [PubMed] [Google Scholar]
- 5.Stoyanovsky D, Murphy T, Anno PR, Kim YM, Salama G. Nitric oxide activates skeletal and cardiac ryanodine receptors. Cell Calcium. 1997;21:19–29. doi: 10.1016/S0143-4160(97)90093-2. [DOI] [PubMed] [Google Scholar]
- 6.Sun JH, Xu L, Eu JP, Stamler JS, Meissner G. Nitric oxide, NOC-12, and S-nitrosoglutathione modulate the skeletal muscle calcium release channel/ryanodine receptor by different mechanisms. An allosteric function for O2 in S-nitrosylation of the channel. J Biol Chem. 2003;278:8184–9. doi: 10.1074/jbc.M211940200. [DOI] [PubMed] [Google Scholar]
- 7.Xu L, Eu JP, Meissner G, Stamler JS. Activation of the cardiac calcium release channel (ryanodine receptor) by poly-S-nitrosylation. Science. 1998;279:234–7. doi: 10.1126/science.279.5348.234. [DOI] [PubMed] [Google Scholar]
- 8.Porter Moore C, Zhang JZ, Hamilton SL. A role for cysteine 3635 of RYR1 in redox modulation and calmodulin binding. J Biol Chem. 1999;274:36831–4. doi: 10.1074/jbc.274.52.36831. [DOI] [PubMed] [Google Scholar]
- 9.Sun JH, Xin CL, Eu JP, Stamler JS, Meissner G. Cysteine-3635 is responsible for skeletal muscle ryanodine receptor modulation by NO. Proc Natl Acad Sci U S A. 2001;98:11158–62. doi: 10.1073/pnas.201289098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hidalgo C, Donoso P, Carrasco MA. The ryanodine receptors Ca2+ release channels: cellular redox sensors? IUBMB Life. 2005;57:315–22. doi: 10.1080/15216540500092328. [DOI] [PubMed] [Google Scholar]
- 11.Lanner JT, Georgiou DK, Joshi AD, Hamilton SL. Ryanodine receptors: structure, expression, molecular details, and function in calcium release. Cold Spring Harb Perspect Biol. 2010;2:a003996. doi: 10.1101/cshperspect.a003996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bellinger AM, Reiken S, Carlson C, Mongillo M, Liu X, Rothman L, et al. Hypernitrosylated ryanodine receptor calcium release channels are leaky in dystrophic muscle. Nat Med. 2009;15:325–30. doi: 10.1038/nm.1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Durham WJ, Aracena-Parks P, Long C, Rossi AE, Goonasekera SA, Boncompagni S, et al. RyR1 S-nitrosylation underlies environmental heat stroke and sudden death in Y522S RyR1 knockin mice. Cell. 2008;133:53–65. doi: 10.1016/j.cell.2008.02.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Namiki S, Kakizawa S, Hirose K, Iino M. NO signalling decodes frequency of neuronal activity and generates synapse-specific plasticity in mouse cerebellum. J Physiol. 2005;566:849–63. doi: 10.1113/jphysiol.2005.088799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kakizawa S, Yamazawa T, Chen Y, Ito A, Murayama T, Oyamada H, et al. Nitric oxide-induced calcium release via ryanodine receptors regulates neuronal function. EMBO J. 2011;31:417–28. doi: 10.1038/emboj.2011.386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kobayashi S, Yano M, Suetomi T, Ono M, Tateishi H, Mochizuki M, et al. Dantrolene, a therapeutic agent for malignant hyperthermia, markedly improves the function of failing cardiomyocytes by stabilizing interdomain interactions within the ryanodine receptor. J Am Coll Cardiol. 2009;53:1993–2005. doi: 10.1016/j.jacc.2009.01.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhao F, Li P, Chen SR, Louis CF, Fruen BR. Dantrolene inhibition of ryanodine receptor Ca2+ release channels. Molecular mechanism and isoform selectivity. J Biol Chem. 2001;276:13810–6. doi: 10.1074/jbc.M006104200. [DOI] [PubMed] [Google Scholar]
- 18.Huang PL, Dawson TM, Bredt DS, Snyder SH, Fishman MC. Targeted disruption of the neuronal nitric oxide synthase gene. Cell. 1993;75:1273–86. doi: 10.1016/0092-8674(93)90615-W. [DOI] [PubMed] [Google Scholar]
- 19.Kishida KT, Klann E. Sources and targets of reactive oxygen species in synaptic plasticity and memory. Antioxid Redox Signal. 2007;9:233–44. doi: 10.1089/ars.2007.9.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pacher P, Beckman JS, Liaudet L. Nitric oxide and peroxynitrite in health and disease. Physiol Rev. 2007;87:315–424. doi: 10.1152/physrev.00029.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Becker BF. Towards the physiological function of uric acid. Free Radic Biol Med. 1993;14:615–31. doi: 10.1016/0891-5849(93)90143-I. [DOI] [PubMed] [Google Scholar]
- 22.Stern MK, Jensen MP, Kramer K. Peroxynitrite decomposition catalysts. J Am Chem Soc. 1996;118:8735–6. doi: 10.1021/ja961279f. [DOI] [Google Scholar]
- 23.Lancel S, Tissier S, Mordon S, Marechal X, Depontieu F, Scherpereel A, et al. Peroxynitrite decomposition catalysts prevent myocardial dysfunction and inflammation in endotoxemic rats. J Am Coll Cardiol. 2004;43:2348–58. doi: 10.1016/j.jacc.2004.01.047. [DOI] [PubMed] [Google Scholar]
- 24.Tsukada K, Hasegawa T, Tsutsumi S, Katoh H, Kuwano H, Miyazaki T, et al. Effect of uric acid on liver injury during hemorrhagic shock. Surgery. 2000;127:439–46. doi: 10.1067/msy.2000.104486. [DOI] [PubMed] [Google Scholar]
- 25.Kakizawa S, Yamasaki M, Watanabe M, Kano M. Critical period for activity-dependent synapse elimination in developing cerebellum. J Neurosci. 2000;20:4954–61. doi: 10.1523/JNEUROSCI.20-13-04954.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]