

Abstract
Ischemic stroke is one of the leading causes of disability and death in the world. Elucidation of the underlying mechanisms associated with neuronal death during this detrimental process has been of significant interest in the field of research. One principle component vital to the maintenance of cellular integrity is the cytoskeleton. Studies suggest that abnormalities at the level of this fundamental structure are directly linked to adverse effects on cellular well-being, including cell death. In recent years, evidence has also emerged regarding an imperative role for the transient receptor potential (TRP) family member TRPM7 in the mediation of excitotoxic-independent neuronal demise. In this review, we will elaborate on the current knowledge and unique properties associated with the functioning of this structure. In addition, we will deliberate the involvement of distinct mechanistic pathways during TRPM7-dependent cell death, including modifications at the level of the cytoskeleton.
Keywords: TRPM7, cytoskeleton, calcium, ischemia, neuron, stroke, OGD, cofilin, kinase, anoxia
Introduction
For a fundamental process that is an inevitable part of the day-to-day function of living organisms, the precise classification of cell death has been the subject of much debate.1 Initial strategies relied principally on morphological characteristics (such as cell rounding, alterations in cellular and organelle volume, and nuclear fragmentation), largely due to limitations in the availability of assays employing suitable biochemical markers.2 Recently, the Nomenclature Committee on Cell Death (NCCD) released an updated set of guidelines that incorporates current advancements in the literature as well as modern developments in the use of biochemical tests such as the monitoring of caspase and reactive oxygen species activity.3
In recent years, evidence has emerged implicating dysfunction of the cellular cytoskeleton in a variety of neurodegenerative diseases including amyotrophic lateral sclerosis, Alzheimer disease and Parkinson disease.4 The cytoskeleton is composed of filamentous organizations of microfilaments (or actin filaments), intermediate filaments (which can comprise of one of several proteins including vimentin, keratin, lamin or neurofilaments) and cylindrical microtubules (consisting of α and β-tubulin subunits), as well as multiple regulatory binding partners. The cytoskeleton has been implicated in a variety of critical cellular processes, including maintenance of basic architecture and integrity, organelle and vesicular trafficking, motility and cell division.5-12 Hence, this protein-based framework is vital to the preservation of cellular well-being, playing a key role in growth, differentiation and proliferation. More recently, cytoskeletal proteins have been shown to be active participants in signal transduction and signal regulation downstream of a variety of receptors and channels.5-12 Therefore, it is no surprise that abnormalities at the level of the cytoskeleton have been implicated in certain forms of cell death.5,13-18 Indeed, evidence suggests that cytoskeletal integrity is intrinsically associated with key mediators of cell death including intracellular calcium (Ca2+) regulation.19-21 Ca2+ is a vital second messenger for the propagation of information and has been a mainstay in theories explaining cytotoxicity.22-28 The abnormal build-up of cellular Ca2+ has also been demonstrated as a major factor during the initiation of ischemic death.22-24
Research into the molecular mechanisms of brain neuronal death has been a source of promise and disappointment in stroke research over the last two decades.29-33 Several key players including NMDA (N-Methyl-D-aspartate) and AMPA (α-amino-3-hydroxy-5-methyl-4- isoxazole-propionic acid) glutamatergic receptors have been identified with the spotlight given to the role of Ca2+ influx in neuronal death.29,34-40 Unfortunately, many of these studies have concentrated on the individual effects of neuronal channels and proteins, rather than examining the cell death process as whole. To date, neuronal cell demise remains poorly defined and viable therapeutic strategies for treating neurodegeneration following ischemic brain injury are still lacking. This review takes a new look at neuronal cell death including roles for both excitotoxic and non-excitotoxic processes (see Table 1). We will highlight recent insights into the function of the TRPM7 cation channel which has been implicated in ischemia-related neuronal death,41-47 and elaborate on the multiple mechanistic pathways that TRPM7 may utilize in contributing to the cell death process, including regulation of the cellular cytoskeleton.
Table 1. Prominent structures implicated in the mediation of excitotoxic and non-excitotoxic neuronal death.
| Excitotoxic components involved in ischemic neuronal death | ||
|---|---|---|
|
Name |
Known regulators |
Key findings |
| NMDA receptors |
Glutamate |
NMDA receptor antagonists block glutamate neurotoxicity (Choi, 1987) |
| AMPA receptors |
Glutamate |
AMPA receptor antagonists diminished loss of ischemic neuronal demise (Buchan et al., 1991) |
| EAATs |
Sodium electrochemical gradient |
Antisense oligonucleotide-mediated disruption of glutamate transporter synthesis produced enhanced glutamate levels and neurodegeneration (Rothstein et al., 1996) |
|
Non-excitotoxic structures involved in ischemic neuronal death | ||
|
Name |
Known regulators |
Key findings |
| ASICs |
Acidosis |
Cerebroventricular injections of ASIC inhibitors or ASIC knockout provided a protective effect on neurons from ischemic insult (Xiong et al., 2004) |
| Gap junction channels |
Calcium and pH levels |
Gap junction hemichannels expressed in neurons were activated following OGD (Thompson et al., 2006) |
| TRP channels | TRPM2: Hydrogen peroxide TRPM7: See Table 2 | TRPM2: Hydrogen peroxide induced neuronal death in cultures was averted in the presence of TRPM2-siRNA (Kaneko et al., 2006) TRPM7: See Table 2 |
Abbreviations: NMDA, N-Methyl-D-aspartate; AMPA, α-amino-3-hydroxy-5-methyl-4- isoxazole-propionic acid; EAATs, excitatory amino acid transporters; ASICs, acid-sensing ion channels; OGD, oxygen glucose deprivation; TRPM2, transient receptor potential melastatin 2; TRPM7, transient receptor potential melastatin 7; siRNA, small interfering RNA
Understanding Excitotoxicity: The Role of Glutamatergic Receptors
To better appreciate TRPM7 activity and the ischemic process as a whole, it is necessary to first discuss the phenomenon of excitotoxicity. Glutamate is the major excitatory neurotransmitter in the CNS and plays a fundamental role in critical processes from neuronal development and survival to sensory perception and the ability to learn new tasks.48 Glutamate signaling via ionotropic receptors, in particular, the NMDA and AMPA subtypes, have received considerable attention for their roles in synaptic function.49-51 Ca2+ influx via these structures can instigate numerous kinase signaling pathways, including protein kinase C (PKC) and calcium/calmodulin-dependent kinase II (CaMKII).49 These cascades act to stimulate overall AMPA receptor activity by means of phosphorylation and fortifying their relative numbers at the postsynaptic membrane, bolstering synaptic function and plasticity.25,26 In pathological situations, inordinate levels of glutamate release can exceedingly stimulate both NMDA and AMPA receptors to allow an abnormally high level of Ca2+ ion influx. This elevated ion flow results in the activation of several enzyme cascades such as phospholipases, endonucleases and proteases that are detrimental to cellular well-being in a process that is referred to as “excitotoxicity,” where glutamate acts as an “excitotoxin.”25-28 The ensuing injury to critical structures such as the membrane and cytoskeleton can finally lead to cellular death. Another possible mechanism for excitotoxicity may result from abnormalities at the level of excitatory amino-acid transporters (EAATs). These structures play an essential role in maintaining extracellular concentrations of glutamate within normal parameters via membrane transport to prevent detrimental damage as a consequence of receptor overstimulation.52 Disruption of EAAT production has been shown to induce excitotoxic degeneration under both in vivo and in vitro settings.53 Excitotoxicity has been demonstrated to occur in acute neuronal damage during stroke and brain or spinal cord trauma, as well as neurodegenerative ailments such as multiple sclerosis, amyotrophic lateral sclerosis, Alzheimer disease, Parkinson disease and Huntington's disease.26-30
Non-Excitotoxic Cell Death
The association of excitotoxicity with pathological situations led to the logical supposition of targeting glutamatergic receptors as a therapeutic countermeasure during ischemia. Unfortunately, anti-excitotoxic therapy (AET) has not lived up to expectations, with the presence of serious side-effects (including increased early mortality rates) and other detrimental consequences that result from blocking physiological NMDA receptor activity.54-57 Therefore, researchers sought to explore glutamate-independent mechanisms as alternative therapeutic targets (see Table 1). Ongoing research shows that Ca2+ signaling plays a central role in neuronal death in the absence of excitotoxic sources. Examination of the underlying mechanisms associated with ischemic-related acidosis revealed the activation of Ca2+ permeable acid-sensing ion channels (ASICs).58-60 Inhibiting the function of these ASICs via pharmacological or genetic manipulations resulted in robust protection of neurons from ischemic damage. Additionally, the exploration of intercellular gap junction hemichannels during anoxic processes has also yielded intriguing results.61-64 In interneurons, these channels are aggregates of proteins named connexins that permit communication between opposing cell membranes. Knockout mice specifically lacking connexin 32 demonstrated an increased susceptibility to induced ischemia in comparison to wild-type animals.61 Moreover, neuronally expressed gap junction channels comprising of pannexin proteins were shown to open in response to oxygen glucose deprivation (OGD), a well known model of experimental ischemia.63 Other primary candidates for the mediation of non-excitotoxic mechanisms of neuronal death have been members of the transient receptor potential (TRP) family of channels.41,65-67 Henceforth, this review will focus on the discussion of recent developments and potential mechanisms associated with the actions of one particular member of this family, i.e., TRPM7.
Transient Receptor Potential (TRP) Channels and TRPM7
In comparison to other families of ionic channels, TRP channels stand out in respect to their varied modes of activation and diverse functionalities.68-74 The need for activity-inducing regulatory mechanisms vary greatly among different TRP channels, ranging from not being required to the specific necessity of factors such as calcium concentration state, pH levels, oxygenation status, osmolarity and mechanosensitivity. To date, around 33 subunits belonging to this superfamily have been identified that have been further classified into two groups subdivided into seven smaller families relative to homology. These include TRPC (canonical), TRPM (melastatin), TRPV (vanilloid), TRPA (ankyrin), TRPML (mucolipin), TRPN (no mechanoreceptor potential C) and TRPP (polycystin). In contrast to some TRP members (TRPA, TRPC, TRPN and TRPV), TRPM channels do not possess numerous N-terminal ankyrin repeats, instead comprising of four analogous domains termed the TRPM homology regions. These regions have been speculated to be involved in channel formation and membrane transport mediation.75
TRPM7 is one of eight members of the TRPM subfamily that was first discovered over a decade ago. This structure has been uniquely coined a “chanzyme,” being distinctive among proteins of its class (including most belonging to the same superfamily) in that it possesses both cationic ion channel activity as well as an inherent protein kinase domain.76 Interestingly, TRPM7 was first identified and cloned in 2001 by multiple studies under diverse settings. One report investigating phospholipase C interacting partners employed yeast two-hybrid screening to initially determine and then clone TRPM7 via polymerase chain reaction (PCR) using mouse brain cDNA.77 In exploring novel putative members of the α kinase class of proteins through a combination of cloning and sequencing, another group utilized mouse melanoma and kidney cDNA libraries to ascertain large regions sharing homology with the TRP family that were expressed over a wide range of tissues.78 A further study aiming to demonstrate previously unidentified cation channels in hematopoietic cells provided functional evidence of characteristic TRPM7 activity following cloning.79 Therefore, the diverse nature of these initial studies only served to demonstrate a taste of the varied functionality that would eventually be linked with TRPM7. To date, these broad range of activities have also encompassed the proliferation of retinoblastoma,80 head and neck carcinoma cell lines,81 and gastric cancer cells.82,83 TRPM7 subunits comprise of 1863 amino acid residues with an expected molecular weight of around 212kDa, with the mouse gene being over 90% homologous to human TRPM7.
The Distinct Composition of TRPM7
Structurally, the TRPM7 channel is made up of six membrane-traversing domains (a characteristic shared by multiple voltage-dependent channels) along with a pore-forming loop (P-loop) located between the last two regions (see Fig. 1). Channel function was demonstrated to be intrinsically linked with Mg2+ and Mg-ATP levels,79 and was proposed to be necessary for the homeostasis of divalent cations (including Ca2+ and Mg2+) within the cell.84 In keeping with these key characteristics, TRPM7 currents detected in cell lines were referred to as Magnesium-Nucleotide-regulated Metal (MagNuM) currents.79,85 Alongside the channel region, the TRPM7 membrane domains are flanked by lengthy intracellular N- and C-terminals that are thought to represent areas of critical function and activities. Evidence from other TRP family members has suggested that the N-terminal may play a role in channel efficacy, where expression of either a curtailed version of this region or isolated N-terminals disrupted normal channel function.86,87 TRPM7 has also been previously localized to synaptic vesicle membranes of sympathetic neurons, where it can complex with proteins such as synapsin I and synaptotagmin I.88,89 Furthermore, immunoprecipitation experiments revealed a direct interaction between the TRPM7 N-terminal and snapin,88 a protein that is vital for neurotransmitter release through mediation of soluble N-ethylmaleimide-sensitive factor activating protein receptor (SNARE) activity.90
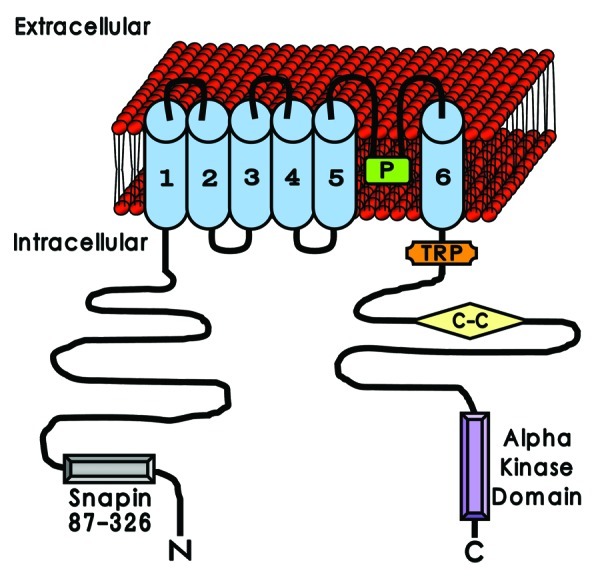
Figure 1. The unique structural composition of TRPM7. TRPM7 is composed of six membrane-spanning domains (blue), with a pore-forming loop (P, in green) located between domains 5 and 6. Intracellularly, TRPM7 comprises of substantial terminal areas that are suggested to be important for its function. The N-terminal (N) is proposed to contribute to neurotransmitter release via binding interactions with proteins such as snapin (Snapin 87–326, in gray). Identified structures with putative roles on the C-terminal (C) include the TRP binding domain (TRP, in orange), the coiled-coiled region (C-C, in yellow) and the α kinase domain (Alpha kinase domain, in purple). The probable interplay between these structural areas in relation to TRPM7 activity has been the subject of continued debate and analysis.
With regards to the TRPM7 C-terminal, multiple inherent components have been identified that have been predicted to contribute to cellular activity. Among these, a compact TRP binding domain (or TRP box, an area also observed in the TRPC and TRPV subfamilies) is purported to be located in close proximity or even within the sixth transmembrane domain. Functionally, the TRP box has been contended to act as a mediator that regulates between the open and closed states of the channel based on thermoreceptive studies of TRPM8 in the presence of phosphatidylinositol 4,5-bisphosphate [PtdIns(4,5)P2].91 Distal to the TRP box, a coiled-coil domain is also present on the C-terminal. These are ubiquitous structural motifs made up of several α helices that recur systematically in what is known as a heptad repeat. Examination of the TRPM7 crystal structure at high resolution has revealed a symmetrical four-stranded coiled-coil domain that is likely to be involved in the formation of antiparallel tetrameric assemblies.57 The furthest intracellular region recognized on the TRPM7 C-terminal with a highly anticipated level of importance is an atypical α kinase domain. Characteristically, kinases belonging to eukaryotic systems share similar homology in their catalytic domains (as in the case of serine/threonine and tyrosine kinases) and are collectively called conventional protein kinases (CPKs).92,93 However, a smaller number of distinct kinases have been identified94-96 that do not possess analogous sequencing with CPKs. Members of this unique grouping include phosphatidylinositol 3-kinase related kinases and α kinases, and hence have been termed atypical protein kinases (APKs). The α kinase subgroup was named after the fact that the targeted residues of these proteins are situated largely within α helices97 as opposed to the loopy and irregular structures associated with CPK phosphorylation sites.93,98,99 Interestingly, irrespective of the sequencing differences observed, a number of similarities between α kinases and CPKs were ascertained during structural alignment investigations. This included the presence of two-lobed folding, uniformity in the conservation of structural components involving subdomains 1–8 and shared positioning of fundamental catalytic residues.93
The TRPM7 α kinase region has been demonstrated by cloning studies in various laboratories.77-79 Interactions at the level of this domain can include the phospholipase C enzyme group,100 the motor protein myosin IIA,101,102 the calcium and phospholipid-associated protein annexin 1,103 and the translational determinant eukaryotic elongation factor 2.104 The most likely candidate to mediate phosphotransferase activity within the kinase domain is Mg2+.105-107 It has been postulated that a “linked uptake and sensor” mechanism may explain this modulation whereby Mg2+ flow through an open TRPM7 channel could activate the catalytic activity of its kinase region as required via a regulatory feedback loop.69 Initially, it was proposed that TRPM7 kinase activity may be a necessary prerequisite for channel function, where the use of kinase mutants exhibited a decrease in current level and requirement for intracellular adenosine triphosphate (ATP) during whole-cell studies.77 However, later investigations contradicted these findings. One report found that while TRPM7 could functionally regulate Mg2+ homeostasis to affect channel activity through contribution of the kinase domain, this action was independent of autophosphorylation.105 These results were echoed in another study where site-specific disruption of either TRPM7 self-phosphorylation or catalytic activity did not significantly modify measurements of Ca2+ influx or whole-cell current-voltage relationships.107 Therefore, the involvement of phosphotransferase activity in the regulation of the TRPM7 channel remains open to debate.
TRPM7 in Neuronal Cell Death
The use of AET was initially thought to be a promising approach to combat the detrimental effects of glutamate-mediated excitotoxicity.54-57 However, favorable outcomes in relation to the prevention of neuronal cell death through the utilization of glutamate-receptor inhibitors were limited to a 1 h time-frame following the initiation of ischemic insult during experimentation.41 A widely recognized model to mimic stroke and ischemic conditions has been through the use of OGD.31,108 This is usually accomplished by subjecting primary neuronal cultures to a temperature regulated and controlled environment via an anaerobic chamber.
We utilized this methodology in order to divulge the causative factor(s) behind the persistence of cell demise in spite of blocked excitotoxicity. Toward achieving this goal, several factors were kept in mind. Primarily, the perseverance of Ca2+ signaling during the process despite the presence of MK-801, CNQX and Nimodipine (inhibitors for NMDA, AMPA and L-type calcium channels, respectively) suggested an atypical source of ion flow. Second, patch-clamp recordings under these conditions detected the existence of a cation current whose current-voltage relationship was sensitive to relative Ca2+ levels and demonstrated a non-selective cation conductance in response to polyvalent cations such as gadolinium (Gd3+) and zinc (Zn2+).41 This current was accordingly termed IOGD, and demonstrated a dependence on reactive oxygen species (ROS) production and the nitric oxide signaling pathway during antioxidant and nitric oxide synthase (NOS) inhibitor studies respectively. Given the above evidence, the TRP family of proteins were considered as leading contenders for IOGD mediation, with TRPM7 being at the forefront. Expression of TRPM7 in HEK-293 cells displayed outwardly rectifying current-voltage curves during electrophysiology, a trait also shared by IOGD. Taking advantage of known TRPM7 modulators PtdIns(4,5)P2 (activator) and Gd3+ (blocker) in addition to small interfering RNA (siRNA) inhibition as experimental tools, the plausible role of this channel in anoxic neuronal death was further investigated in culture systems. Critically, blockage of TRPM7 function abolished key IOGD-associated attributes, including calcium uptake, generation of ROS and most importantly, cell death.41,109,110 These results were consistent with previous studies that suggested the involvement of TRPM7 in cell viability during overexpression experiments.79,84
Another study focused on TRPM7 functionality in the CA1 region of the hippocampus,42 an area known to be vital for learning and memory.49-51 Here, reduction in the levels of extracellular Ca2+ and Mg2+ presented a cation current in neurons with similar electrical characteristics previously assigned to TRPM7. Boosting channel function in this manner was seen to provoke death in HEK-293 cells, while lowering TRPM7 expression diminished current activity induced by attenuation of divalent cation concentrations.42 This data bolstered the notion of TRPM7 acting as a putative sensor of cation levels under certain settings. Subsequently, these findings were extended to examine the possible advantageous effects of in vivo neuronal TRPM7 suppression in the same area.43 The use of viral vector injections bearing TRPM7-specific shRNA into the hippocampus did not alter the normal well-being of adult rodents, with neuronal structure and electrical activity being preserved. Ischemia was subsequently induced in these animals via occlusion of the common carotid and vertebral arteries (also known as 4-vessel occlusion). Following this insult, TRPM7 shRNA possessing cells exhibited an increased resistance to cell death and demonstrated enhanced recovery from the detrimental effects of ischemia in relation to synaptic plasticity, contextual fear conditioning and Morris Water Maze-based spatial navigation.43 Another pathway implicated in neuronal demise is through lipopolysaccharides (LPS). A recent report revealed that either LPS or hydrogen peroxide treatment of differentiated PC12 neurons resulted in an augmentation of TRPM7 expression levels.44 Moreover, siRNA inhibition of TRPM7 in primary hippocampal neuronal cultures and PC12 neurons safeguarded these cells against deterioration. Similarly, TRPM7 involvement during oxidant insult was also observed in mouse cortical neurons, where channel inhibition via 2-aminoethoxydiphenyl borate or siRNA offered protection from hydrogen peroxide-mediated damage.45
While investigations into channel-mediated Ca2+ overload have been at the forefront of cell death research, studies have also suggested that TRPM7 is capable of inflicting injury via homeostasis of other cations. One report demonstrated that Zn2+-influenced damage caused by either direct application of the ion or OGD required the actions of TRPM7 in mouse neuronal cultures.46 Another group verified that neuronal levels of Mg2+ were indeed elevated by means of a TRPM7-dependent mechanism following OGD or chemically-induced ischemia in rat hippocampal cultures.47
Though the several studies above have demonstrated a clear role for TRPM7 during ischemic cell death, data concerning the precise signaling pathways engaged in this process has been sparse, including the possible involvement of cytoskeletal components.
TRPM7 and the Cytoskeleton
The sizeable composition of the cellular cytoskeleton along with its inherent capacity to sequester a diverse network of integral components has greatly contributed to its role as a region of cellular enrichment.5,6,8,10 In addition to the probable functional roles outlined previously, a significant amount of research has been devoted to the possible role of TRPM7 in regulation of the cytoskeleton. Based on earlier work that implicated myosin heavy chain (MHC) α kinases111-113 in mediating Dictyostelium amoebae cytoskeletal components, one group explored the possibility of the TRPM7 kinase domain performing a similar role in mammals.101 Through utilization of the N1E-115 neuroblastoma cell line, the authors found that expressed TRPM7 promoted cell spreading and cell-matrix adhesion through channel rather than kinase domain activity. Confocal imaging revealed a scattered distribution of the contractility-inducing motor protein myosin IIA and enhanced podosome formation in TRPM7 expressing vs. control cells.101 Podosomes are dynamic actin-enriched sites of adhesion that can interact with the extracellular matrix.114 In this case, podosome development through TRPM7 stimulation via the actomyosin relaxing agent bradykinin was dependent on the kinase region by means of myosin II suppression. The authors also discovered that TRPM7 could directly complex with both β-actin and myosin IIA, and cause phosphorylation of the latter at the COOH-terminus.101,102 Furthermore, the same group determined that such substrate identification and subsequent phosphorylation was greatly enhanced in the presence of TRPM7 autophosphorylation.115
Another group employed the use of inducible TRPM7 overexpression in HEK-293 cell lines to study its potential ability in regulating members of the calpain protease family during cell adhesion.116 In this experimental set-up, an increase in cellular rounding and loss of adhesive properties were observed within a day. In contrast, a generated cell line with reduced endogenous expression of TRPM7 demonstrated the opposite results, with an enhancement in cell adherence. Through the use of TRPM7 mutants, the above findings were attributed to channel rather than kinase function, with cell rounding decreasing appreciably in cell lines completely lacking or deficient in channel activity.116 Further investigations with short hairpin RNA (shRNA) and the calpain inhibitor ALLM confirmed m-calpain as a key mediator of these TRPM7-dependent morphological alterations.116 M-calpain is a non-lysosomal protease that is responsive to millimolar concentrations of cytosolic Ca2+ and has been previously implicated in the destabilization of cellular adhesion.117 Moreover, this TRPM7-associated activation of m-calpain was correlated with the actions of the p38 MAPK (mitogen-activated protein kinase) and JNK (c-Jun N-terminal kinase) signaling cascades in response to stress-related increases in nitric oxide and ROS.118
Recently, the ability of TRPM7 to regulate Mg2+ homeostasis has also been suggested to play a possible modulatory role during its cytoskeletal effects.119 Fibroblast cell lines lacking TRPM7 exhibited notable alterations in cell morphology, with modification of focal adhesions, diminished actin fibers and elevated cortical levels of actin and myosin IIA.119 These effects were reversible by adenoviral expression of TRPM7 constructs with intact channel activity (suggesting a lack of functional involvement of the kinase domain during this process). In addition, the presence of a selective Mg2+ transporter SLC41A2 (solute carrier family 41 member 2) was also able to rescue the observed deficits and restore cytoskeletal integrity.119 Another intriguing perspective on TRPM7-related cell death may involve its presence in vascular smooth muscle cells (VSMCs). Here, it has been suggested that TRPM7 is intrinsically involved in responding to blood vessel wall damage during shear stress.120 The authors of this study propose that shear stress results in a rapid upregulation of TRPM7 activity at the plasma membrane via the actions of cytoskeletal components and motor proteins. Therefore, TRPM7 may also have an effect on cellular viability by means of the vascular network in relation to acute changes in blood flow.
Recent data from our laboratory (Bent et al., publication under review) has also revealed an integral role for TRPM7-dependent cytoskeletal regulation in the mediation of ischemic neuronal cell death. Specifically, OGD-influenced instigation of TRPM7 resulted in the activation of the key actin-binding protein cofilin in rat cortical cultures. Cofilin is affiliated with the ADF/cofilin (actin depolymerization factor) family and has been associated with reorganization of the actin network, a process vital to filament recycling and turnover.121-124 The utilization of Gd3+ and siRNA also indicated that the stimulation of this protein was correlated with a TRPM7-dependent inactivation of LIMK1 (Lin-11, Isl-1 and Mec-3 kinase 1), a known suppressor of cofilin actuation.125-127 More interestingly, RNA silencing of cofilin significantly reduced neuronal demise following the detrimental effects of OGD, reflecting the importance of this protein in cellular viability under these circumstances. These results offer a rare glimpse into the largely unknown signaling pathways initiated exclusively by TRPM7 activation.
Taken together, the above studies provide compelling evidence for TRPM7-guided governance of cytoskeletal components under a diverse range of experimental settings.
TRPM7-Dependent Ischemic Cell Death: Lingering Questions and Future Directions
The discovery of the unique composition of TRPM7 has been somewhat of a double-edged sword in the analysis of this structure. On one front, it has offered the exciting possibility of exploring distinct and previously unknown mechanisms due to the rare bifunctional nature of this protein.77,78,104,106,107 Furthermore, the ubiquitous distribution of TRPM7 has alluded to its possible role in a myriad of processes in various regions of the body. On the other hand, the dual presence of both a channel pore and a kinase domain in close proximity has greatly complicated the delineation of specific functional roles for each region.
While data accumulated from our and other laboratories clearly reflect the significance of TRPM7 activity during pathological situations, it is also likely that this structure may display considerable influence over neuronal physiology. It has been observed that activation or overactivation of NMDA receptors can lead to physiological (induction of synaptic plasticity)49,50 or pathophysiological (excitotoxicity)25-28 consequences respectively. In a similar manner, the hospitable bifunctional composition of TRPM7 may also be associated with basic but essential neuronal activities in relation to relevant stimuli or cellular status. In particular, a detailed examination of the possible role of TRPM7 in common forms of synaptic plasticity (such as long-term potentiation and long-term depression)49,50 and associated behavioral tasks is lacking. One report recently implicated TRPM7 channel activity in sensory neurons to preferentially modulate touch-induced escape in zebrafish, potentially through central synapse neurotransmitter release.128 Furthermore, while global knockout mice for TRPM7 have shown to be embryonically lethal,129 specific interference of this factor in the neural crest impeded derivation of dorsal root ganglion sensory neurons,130 indicating a clear developmental role.
We and others have provided conclusive evidence of an essential mediatory role for TRPM7 during anoxic neuronal death (see Table 2). This has been demonstrated through the use of at least three different experimental models of ischemia (OGD, 4-vessel occlusion and chemically-induced) encompassing both in vitro and in vivo settings, indicating the ubiquitous nature of TRPM7 involvement.41-47 However, a number of important questions still linger in this regard. While TRPM7 channel activity has proven essential for neuronal demise, a defined role for the kinase region has yet to be determined. This includes the possibilities that the kinase domain may coordinate/facilitate channel function during cell death, or alternatively participate in the process in an independent manner. The resourceful use of various TRPM7 channel and kinase mutants during past studies in cell lines may provide a viable means to approach questions regarding region-specific roles in neurons.101,102,115,116,118,119 Another important factor to consider when assessing ischemic-dependent TRPM7 activation is the putative role of environmental pH. It is widely accepted that ischemia and its ensuing cellular damage is associated with a reduction in pH levels.131 Since a number of studies have linked TRPM7 functionality to pH status,132-134 it can be assumed that decreasing pH may contribute to the enhancement of channel activity under anoxic conditions. However, specific data examining this relationship in the context of neuronal cell death has been lacking.
Table 2. Notable TRPM7-related findings during neuronal studies examining ischemia.
| Neuronal preparations | Ischemic or stress model utilized | TRPM7 modulators employed | Major results |
|---|---|---|---|
| Rat cortical cultures |
OGD |
Gd3+ (inhibitor), TRPM7 siRNA |
Suppression of TRPM7 presented decreased Ca2+ uptake, reduction in ROS levels, and averted cell death (Aarts et al., 2003) |
| Mouse hippocampal cultures |
Attenuation of divalent cation concentration |
Reduction in Ca2+ levels (activator), TRPM7 shRNA |
Diminishing Ca2+ levels augmented TRPM7 current in neurons and increased cell death in HEK-293 cells. Current was abolished in the presence of TRPM7 shRNA (Wei et al., 2007) |
| Rat hippocampal sections, cultures and acute slices |
4-vessel occlusion |
In vivo intrahippocampal stereotaxic injections of recombinant serotype 1 adeno-associated virus containing TRPM7 shRNA |
TRPM7 shRNA attenuated neuronal cell death and improved recovery from ischemic damage in relation to synaptic plasticity, Morris Water Maze performance and contextual fear memory (Sun et al., 2009) |
| Rat hippocampal cultures and neurons differentiated from pheochromocytoma PC12 cells |
LPS or hydrogen peroxide |
TRPM7 siRNA |
Elevated TRPM7 expression was observed following either LPS or hydrogen peroxide administration. Both primary and PC12 derived neurons were protected from LPS-mediated cell death in the presence of TRPM7 siRNA (Nunez-Villena et al., 2011) |
| Mouse cortical cultures |
Hydrogen peroxide |
2-aminoethoxydiphenyl borate (inhibitor), TRPM7 siRNA |
Application of either 2-aminoethoxydiphenyl borate or TRPM7 siRNA safeguarded neurons from hydrogen peroxide-induced damage (Coombes et al., 2011) |
| Mouse cortical cultures |
Zn2+ treatment and OGD |
Gd3+ (inhibitor), 2-aminoethoxydiphenyl borate (inhibitor) or TRPM7 siRNA |
Utilization of TRPM7 inhibitors or siRNA silencing protected neurons from Zn2+-mediated injury under direct ion administration or OGD (Inoue et al., 2010) |
| Rat hippocampal cultures | Acute chemical ischemia (KCN administration) or OGD | Gd3+ (inhibitor), 2-aminoethoxydiphenyl borate (inhibitor) or TRPM7 shRNA | OGD or chemically-induced increases in neuronal Mg2+ levels were inhibited by TRPM7 blockers or shRNA (Zhang et al., 2011) |
Abbreviations: TRPM7, transient receptor potential melastatin 7; OGD, oxygen glucose deprivation; Gd3+, gadolinium; Ca2+, calcium; siRNA, small interfering RNA; shRNA, short hairpin RNA; LPS, lipopolysaccharide; Zn2+, zinc; Mg2+, magnesium; KCN, potassium thiocyanate
In addition to elucidating the factors behind TRPM7 activation during ischemia, another pending issue remains the clarification of the underlying mechanisms that this chanzyme may instigate. In this case, the observed elevation of Zn2+ or Mg2+ following OGD in primary cultures46,47 suggests the involvement of distinct downstream signaling pathways from those normally associated with NMDA-receptor mediated excitotoxicity and Ca2+ overload.28 The demonstrated ability of TRPM7 to interact with cytoskeletal structures during cell line studies may also provide another viable avenue for exploration. While results pertaining to the nature of TRPM7 involvement in cellular adhesion have been conflicting (likely due to variances in relative expression levels and type of cell lines utilized),101,116 a comprehensive assessment of this phenomenon during ischemic neuronal death would be of significant interest. In this regard, recent reports have revealed the breakdown of Neural Cell Adhesion Molecule (NCAM) as a functional consequence of injury caused by middle cerebral artery occlusion.135,136 A noteworthy finding in the N1E-115 neuroblastoma cell line was the involvement of TRPM7 in the development of podosomes. While these actin-based protrusions have been suggested to contribute toward tissue invasiveness during malignancy,137 they are also thought to be mediators of cell adhesion.114 The relatively recent discovery that synaptically-localized podosomes can also exist in neural structures may be indicative of their potential roles in neuronal systems.138 In this case, podosomes appeared as dynamic adhesive structures that possessed a multitude of prominent proteins (including actin, dynamin, myosin IIA, talin, vinculin and paxillin) and contributed functionally to postsynaptic maturation. With regards to the neuronal cytoskeleton, our findings that cofilin is a crucial effector of TRPM7-related signaling suggests a vital interplay between these two factors for the realization of anoxic cell death. Further studies in this area would be imperative to shed light on the TRPM7/cytoskeleton rapport. For these purposes, the utilization of preexisting knockout mice for the cofilin regulator LIMK and its upstream mediators may prove fruitful.126,139,140 Interestingly, modifications in the level of intracellular Ca2+ ions are observed during both TRPM7-mediated cell injury41-47 and disruption of the cytoskeleton19-21 respectively. Therefore, a more detailed examination of factors associated with this second messenger pathway may yield additional evidence in the intriguing relationship between TRPM7 and the cytoskeleton. In consideration of the numerous processes involved during cerebral ischemia, the ubiquitous nature of TRPM7 (for example its presence in VSMCs) may also suggest broader and as yet unspecified roles for its functionality beyond synaptic capabilities. Hence, investigating the contribution of non-neuronal sources of TRPM7 (such as the cerebral vasculature) to these conditions may be of considerable interest.
Recent studies have also yielded promising results in relation to the development of pharmacological modulators for this chanzyme. TRPM7 activity was shown to be enhanced in the presence of phospholipase C-coupled receptor agonists such as bradykinin, lysophosphatidic acid and thrombin under physiological settings.141 In addition to cation blockers, reports have identified 5-lipoxygenase inhibitors (such as AA861, NDGA and MK886) as robust attenuators of TRPM7 current.83,142 Administration of AA861 in HEK-293 cells also prevented the occurrence of death from the actions of apoptotic agents (such as staurosporine) to an extent comparable to TRPM7 knock-down.142 Another study demonstrated that the SK (small conductance calcium-activated potassium) channel regulator NS8593 was an effective suppressor of TRPM7 current in a variety of different cell lines and preparations.143 Furthermore, an intriguing mediation of TRPM7 activity was exhibited by the protease inhibitor nafamostat mesylate in accordance with variations in the extracellular concentrations of Ca2+ and Mg2+.144 Research dedicated to the discovery of such drug classes represents an exciting endeavor in the continuing attempts to discern more specific TRPM7 inhibitors.145 It is our hope that ongoing explorations into the underlying mechanisms and pharmacology associated with this unique chanzyme will result in the development of effective countermeasures that may be utilized during clinical trials of ischemic therapy.
Acknowledgments
This work was supported by funding from the Canadian Stroke Network (CSN) and the Natural Sciences and Engineering Research Council of Canada (NSERC). We thank Russell Bent and other members of the Aarts lab for their support. The authors declare that they have no conflicts of interest.
Glossary
Abbreviations:
- ADF
actin depolymerization factor
- AET
anti-excitotoxic therapy
- AMPA
alpha-amino-3-hydroxy-5-methyl-4- isoxazole-propionic acid
- APKs
atypical protein kinases
- ATP
adenosine triphosphate
- ASICs
acid sensing ion channels
- Ca2+
calcium
- CaMKII
calcium/calmodulin-dependent kinase II
- CPKs
conventional protein kinases
- EAATs
excitatory amino acid transporters
- Gd3+
gadolinium
- JNK
c-Jun N-terminal kinase
- KCN
potassium thiocyanate
- LIMK1
Lin-11, Isl-1 and Mec-3 kinase 1
- LPS
lipopolysaccharides
- MagNuM
magnesium-nucleotide-regulated metal
- MAPK
mitogen-activated protein kinase
- Mg2+
magnesium
- Mg-ATP
magnesium-adenosine triphosphate
- MHC
myosin heavy chain
- NCAM
neural cell adhesion molecule
- NCCD
Nomenclature Committee on Cell Death
- NMDA
N-Methyl-D-aspartate
- NOS
nitric oxide synthase
- OGD
oxygen-glucose deprivation
- PCR
polymerase chain reaction
- PtdIns(4,5)P2
phosphatidylinositol 4,5-bisphosphate
- PKC
protein kinase C
- ROS
reactive oxygen species
- shRNA
short hairpin RNA
- siRNA
small interfering RNA
- SK
small conductance calcium-activated potassium
- SLC41A2
solute carrier family 41 member 2
- SNARE
soluble N-ethylmaleimide-sensitive factor activating protein receptor
- TRP
transient receptor potential
- TRPA
transient receptor potential ankyrin
- TRPC
transient receptor potential canonical
- TRPM
transient receptor potential melastatin
- TRPM7
transient receptor potential melastatin 7
- TRPM8
transient receptor potential melastatin 8
- TRPML
transient receptor potential mucolipin
- TRPN
transient receptor potential no mechanoreceptor potential C
- TRPP
transient receptor potential polycystin
- TRPV
transient receptor potential vanilloid
- VSMCs
vascular smooth muscle cells
- Zn2+
zinc
Footnotes
Previously published online: www.landesbioscience.com/journals/channels/article/22824
References
- 1.Kroemer G, El-Deiry WS, Golstein P, Peter ME, Vaux D, Vandenabeele P, et al. Nomenclature Committee on Cell Death Classification of cell death: recommendations of the Nomenclature Committee on Cell Death. Cell Death Differ. 2005;12(Suppl 2):1463–7. doi: 10.1038/sj.cdd.4401724. [DOI] [PubMed] [Google Scholar]
- 2.Kroemer G, Galluzzi L, Vandenabeele P, Abrams J, Alnemri ES, Baehrecke EH, et al. Nomenclature Committee on Cell Death 2009 Classification of cell death: recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ. 2009;16:3–11. doi: 10.1038/cdd.2008.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Galluzzi L, Vitale I, Abrams JM, Alnemri ES, Baehrecke EH, Blagosklonny MV, et al. Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ. 2012;19:107–20. doi: 10.1038/cdd.2011.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cairns NJ, Lee VM-Y, Trojanowski JQ. The cytoskeleton in neurodegenerative diseases. J Pathol. 2004;204:438–49. doi: 10.1002/path.1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Janmey PA. The cytoskeleton and cell signaling: component localization and mechanical coupling. Physiol Rev. 1998;78:763–81. doi: 10.1152/physrev.1998.78.3.763. [DOI] [PubMed] [Google Scholar]
- 6.Carpenter CL. Actin cytoskeleton and cell signaling. Crit Care Med. 2000;28(Suppl):N94–9. doi: 10.1097/00003246-200004001-00011. [DOI] [PubMed] [Google Scholar]
- 7.Sheng M, Pak DT. Ligand-gated ion channel interactions with cytoskeletal and signaling proteins. Annu Rev Physiol. 2000;62:755–78. doi: 10.1146/annurev.physiol.62.1.755. [DOI] [PubMed] [Google Scholar]
- 8.Dillon C, Goda Y. The actin cytoskeleton: integrating form and function at the synapse. Annu Rev Neurosci. 2005;28:25–55. doi: 10.1146/annurev.neuro.28.061604.135757. [DOI] [PubMed] [Google Scholar]
- 9.Mazzochi C, Benos DJ, Smith PR. Interaction of epithelial ion channels with the actin-based cytoskeleton. Am J Physiol Renal Physiol. 2006;291:F1113–22. doi: 10.1152/ajprenal.00195.2006. [DOI] [PubMed] [Google Scholar]
- 10.Fletcher DA, Mullins RD. Cell mechanics and the cytoskeleton. Nature. 2010;463:485–92. doi: 10.1038/nature08908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dominguez R. Structural insights into de novo actin polymerization. Curr Opin Struct Biol. 2010;20:217–25. doi: 10.1016/j.sbi.2009.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Penzes P, Rafalovich I. Regulation of the actin cytoskeleton in dendritic spines. Adv Exp Med Biol. 2012;970:81–95. doi: 10.1007/978-3-7091-0932-8_4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Laster SM, Mackenzie JM., Jr. Bleb formation and F-actin distribution during mitosis and tumor necrosis factor-induced apoptosis. Microsc Res Tech. 1996;34:272–80. doi: 10.1002/(SICI)1097-0029(19960615)34:3<272::AID-JEMT10>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 14.Levee MG, Dabrowska MI, Lelli JL, Jr., Hinshaw DB. Actin polymerization and depolymerization during apoptosis in HL-60 cells. Am J Physiol. 1996;271:C1981–92. doi: 10.1152/ajpcell.1996.271.6.C1981. [DOI] [PubMed] [Google Scholar]
- 15.van Engeland M, Kuijpers HJ, Ramaekers FC, Reutelingsperger CP, Schutte B. Plasma membrane alterations and cytoskeletal changes in apoptosis. Exp Cell Res. 1997;235:421–30. doi: 10.1006/excr.1997.3738. [DOI] [PubMed] [Google Scholar]
- 16.Gourlay CW, Carpp LN, Timpson P, Winder SJ, Ayscough KR. A role for the actin cytoskeleton in cell death and aging in yeast. J Cell Biol. 2004;164:803–9. doi: 10.1083/jcb.200310148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kavanagh E, Vlachos P, Emourgeon V, Rodhe J, Joseph B. p57(KIP2) control of actin cytoskeleton dynamics is responsible for its mitochondrial pro-apoptotic effect. Cell Death Dis. 2012;3:e311. doi: 10.1038/cddis.2012.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pawlik A, Szczepanski MA, Klimaszewska A, Gackowska L, Zuryn A, Grzanka A. Phenethyl isothiocyanate-induced cytoskeletal changes and cell death in lung cancer cells. Food Chem Toxicol. 2012;50:3577–94. doi: 10.1016/j.fct.2012.07.043. [DOI] [PubMed] [Google Scholar]
- 19.van Haelst C, Rothstein TL. Cytochalasin stimulates phosphoinositide metabolism in murine B lymphocytes. J Immunol. 1988;140:1256–8. [PubMed] [Google Scholar]
- 20.Rosales C, Brown EJ. Signal transduction by neutrophil immunoglobulin G Fc receptors. Dissociation of intracytoplasmic calcium concentration rise from inositol 1,4,5-trisphosphate. J Biol Chem. 1992;267:5265–71. [PubMed] [Google Scholar]
- 21.Lange K, Brandt U. Calcium storage and release properties of F-actin: evidence for the involvement of F-actin in cellular calcium signaling. FEBS Lett. 1996;395:137–42. doi: 10.1016/0014-5793(96)01025-3. [DOI] [PubMed] [Google Scholar]
- 22.Schlaepfer WW, Bunge RP. Effects of calcium ion concentration on the degeneration of amputated axons in tissue culture. J Cell Biol. 1973;59:456–70. doi: 10.1083/jcb.59.2.456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schanne FA, Kane AB, Young EE, Farber JL. Calcium dependence of toxic cell death: a final common pathway. Science. 1979;206:700–2. doi: 10.1126/science.386513. [DOI] [PubMed] [Google Scholar]
- 24.Choi DW. Glutamate neurotoxicity in cortical cell culture is calcium dependent. Neurosci Lett. 1985;58:293–7. doi: 10.1016/0304-3940(85)90069-2. [DOI] [PubMed] [Google Scholar]
- 25.Beal MF. Mechanisms of excitotoxicity in neurologic diseases. FASEB J. 1992;6:3338–44. [PubMed] [Google Scholar]
- 26.Hazell AS. Excitotoxic mechanisms in stroke: an update of concepts and treatment strategies. Neurochem Int. 2007;50:941–53. doi: 10.1016/j.neuint.2007.04.026. [DOI] [PubMed] [Google Scholar]
- 27.Dong XX, Wang Y, Qin ZH. Molecular mechanisms of excitotoxicity and their relevance to pathogenesis of neurodegenerative diseases. Acta Pharmacol Sin. 2009;30:379–87. doi: 10.1038/aps.2009.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Szydlowska K, Tymianski M. Calcium, ischemia and excitotoxicity. Cell Calcium. 2010;47:122–9. doi: 10.1016/j.ceca.2010.01.003. [DOI] [PubMed] [Google Scholar]
- 29.Olney JW. Brain lesions, obesity, and other disturbances in mice treated with monosodium glutamate. Science. 1969;164:719–21. doi: 10.1126/science.164.3880.719. [DOI] [PubMed] [Google Scholar]
- 30.Simon RP, Griffiths T, Evans MC, Swan JH, Meldrum BS. Calcium overload in selectively vulnerable neurons of the hippocampus during and after ischemia: an electron microscopy study in the rat. J Cereb Blood Flow Metab. 1984;4:350–61. doi: 10.1038/jcbfm.1984.52. [DOI] [PubMed] [Google Scholar]
- 31.Goldberg MP, Weiss JH, Pham PC, Choi DW. N-methyl-D-aspartate receptors mediate hypoxic neuronal injury in cortical culture. J Pharmacol Exp Ther. 1987;243:784–91. [PubMed] [Google Scholar]
- 32.Choi DW, Maulucci-Gedde M, Kriegstein AR. Glutamate neurotoxicity in cortical cell culture. J Neurosci. 1987;7:357–68. doi: 10.1523/JNEUROSCI.07-02-00357.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sattler R, Charlton MP, Hafner M, Tymianski M. Distinct influx pathways, not calcium load, determine neuronal vulnerability to calcium neurotoxicity. J Neurochem. 1998;71:2349–64. doi: 10.1046/j.1471-4159.1998.71062349.x. [DOI] [PubMed] [Google Scholar]
- 34.Rothman S. Synaptic release of excitatory amino acid neurotransmitter mediates anoxic neuronal death. J Neurosci. 1984;4:1884–91. doi: 10.1523/JNEUROSCI.04-07-01884.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Choi DW. Ionic dependence of glutamate neurotoxicity. J Neurosci. 1987;7:369–79. doi: 10.1523/JNEUROSCI.07-02-00369.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Buchan AM, Li H, Cho S, Pulsinelli WA. Blockade of the AMPA receptor prevents CA1 hippocampal injury following severe but transient forebrain ischemia in adult rats. Neurosci Lett. 1991;132:255–8. doi: 10.1016/0304-3940(91)90314-J. [DOI] [PubMed] [Google Scholar]
- 37.Sheardown MJ, Suzdak PD, Nordholm L. AMPA, but not NMDA, receptor antagonism is neuroprotective in gerbil global ischaemia, even when delayed 24 h. Eur J Pharmacol. 1993;236:347–53. doi: 10.1016/0014-2999(93)90470-3. [DOI] [PubMed] [Google Scholar]
- 38.Liu S, Lau L, Wei J, Zhu D, Zou S, Sun H-S, et al. Expression of Ca(2+)-permeable AMPA receptor channels primes cell death in transient forebrain ischemia. Neuron. 2004;43:43–55. doi: 10.1016/j.neuron.2004.06.017. [DOI] [PubMed] [Google Scholar]
- 39.Besancon E, Guo S, Lok J, Tymianski M, Lo EH. Beyond NMDA and AMPA glutamate receptors: emerging mechanisms for ionic imbalance and cell death in stroke. Trends Pharmacol Sci. 2008;29:268–75. doi: 10.1016/j.tips.2008.02.003. [DOI] [PubMed] [Google Scholar]
- 40.Li MH, Inoue K, Si HF, Xiong ZG. Calcium-permeable ion channels involved in glutamate receptor-independent ischemic brain injury. Acta Pharmacol Sin. 2011;32:734–40. doi: 10.1038/aps.2011.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Aarts M, Iihara K, Wei W-L, Xiong Z-G, Arundine M, Cerwinski W, et al. A key role for TRPM7 channels in anoxic neuronal death. Cell. 2003;115:863–77. doi: 10.1016/S0092-8674(03)01017-1. [DOI] [PubMed] [Google Scholar]
- 42.Wei W-L, Sun H-S, Olah ME, Sun X, Czerwinska E, Czerwinski W, et al. TRPM7 channels in hippocampal neurons detect levels of extracellular divalent cations. Proc Natl Acad Sci USA. 2007;104:16323–8. doi: 10.1073/pnas.0701149104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sun H-S, Jackson MF, Martin LJ, Jansen K, Teves L, Cui H, et al. Suppression of hippocampal TRPM7 protein prevents delayed neuronal death in brain ischemia. Nat Neurosci. 2009;12:1300–7. doi: 10.1038/nn.2395. [DOI] [PubMed] [Google Scholar]
- 44.Nuñez-Villena F, Becerra A, Echeverría C, Briceño N, Porras O, Armisén R, et al. Increased expression of the transient receptor potential melastatin 7 channel is critically involved in lipopolysaccharide-induced reactive oxygen species-mediated neuronal death. Antioxid Redox Signal. 2011;15:2425–38. doi: 10.1089/ars.2010.3825. [DOI] [PubMed] [Google Scholar]
- 45.Coombes E, Jiang J, Chu X-P, Inoue K, Seeds J, Branigan D, et al. Pathophysiologically relevant levels of hydrogen peroxide induce glutamate-independent neurodegeneration that involves activation of transient receptor potential melastatin 7 channels. Antioxid Redox Signal. 2011;14:1815–27. doi: 10.1089/ars.2010.3549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Inoue K, Branigan D, Xiong Z-G. Zinc-induced neurotoxicity mediated by transient receptor potential melastatin 7 channels. J Biol Chem. 2010;285:7430–9. doi: 10.1074/jbc.M109.040485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang J, Zhao F, Zhao Y, Wang J, Pei L, Sun N, et al. Hypoxia induces an increase in intracellular magnesium via transient receptor potential melastatin 7 (TRPM7) channels in rat hippocampal neurons in vitro. J Biol Chem. 2011;286:20194–207. doi: 10.1074/jbc.M110.148494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rapport RL. Nerve Endings: The Discovery Of The Synapse. W.W. Norton; 2005. [Google Scholar]
- 49.Lynch MA. Long-term potentiation and memory. Physiol Rev. 2004;84:87–136. doi: 10.1152/physrev.00014.2003. [DOI] [PubMed] [Google Scholar]
- 50.Malenka RC, Bear MF. LTP and LTD: an embarrassment of riches. Neuron. 2004;44:5–21. doi: 10.1016/j.neuron.2004.09.012. [DOI] [PubMed] [Google Scholar]
- 51.Collingridge GL, Peineau S, Howland JG, Wang YT. Long-term depression in the CNS. Nat Rev Neurosci. 2010;11:459–73. doi: 10.1038/nrn2867. [DOI] [PubMed] [Google Scholar]
- 52.Amara SG, Fontana ACK. Excitatory amino acid transporters: keeping up with glutamate. Neurochem Int. 2002;41:313–8. doi: 10.1016/S0197-0186(02)00018-9. [DOI] [PubMed] [Google Scholar]
- 53.Rothstein JD, Dykes-Hoberg M, Pardo CA, Bristol LA, Jin L, Kuncl RW, et al. Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron. 1996;16:675–86. doi: 10.1016/S0896-6273(00)80086-0. [DOI] [PubMed] [Google Scholar]
- 54.Davis SM, Albers GW, Diener HC, Lees KR, Norris J. Termination of Acute Stroke Studies Involving Selfotel Treatment. ASSIST Steering Committed. Lancet. 1997;349:32. doi: 10.1016/S0140-6736(05)62166-6. [DOI] [PubMed] [Google Scholar]
- 55.Morris GF, Bullock R, Marshall SB, Marmarou A, Maas A, Marshall LF, The Selfotel Investigators Failure of the competitive N-methyl-D-aspartate antagonist Selfotel (CGS 19755) in the treatment of severe head injury: results of two phase III clinical trials. J Neurosurg. 1999;91:737–43. doi: 10.3171/jns.1999.91.5.0737. [DOI] [PubMed] [Google Scholar]
- 56.Lees KR, Asplund K, Carolei A, Davis SM, Diener HC, Kaste M, et al. GAIN International Investigators Glycine antagonist (gavestinel) in neuroprotection (GAIN International) in patients with acute stroke: a randomised controlled trial. Lancet. 2000;355:1949–54. doi: 10.1016/S0140-6736(00)02326-6. [DOI] [PubMed] [Google Scholar]
- 57.Sacco RL, DeRosa JT, Haley EC, Jr., Levin B, Ordronneau P, Phillips SJ, et al. Glycine Antagonist in Neuroprotection Americas Investigators Glycine antagonist in neuroprotection for patients with acute stroke: GAIN Americas: a randomized controlled trial. JAMA. 2001;285:1719–28. doi: 10.1001/jama.285.13.1719. [DOI] [PubMed] [Google Scholar]
- 58.Xiong Z-G, Zhu X-M, Chu X-P, Minami M, Hey J, Wei W-L, et al. Neuroprotection in ischemia: blocking calcium-permeable acid-sensing ion channels. Cell. 2004;118:687–98. doi: 10.1016/j.cell.2004.08.026. [DOI] [PubMed] [Google Scholar]
- 59.Xiong Z-G, Chu X-P, Simon RP. Acid sensing ion channels--novel therapeutic targets for ischemic brain injury. Front Biosci. 2007;12:1376–86. doi: 10.2741/2154. [DOI] [PubMed] [Google Scholar]
- 60.Li M, Kratzer E, Inoue K, Simon RP, Xiong Z-G. Developmental change in the electrophysiological and pharmacological properties of acid-sensing ion channels in CNS neurons. J Physiol. 2010;588:3883–900. doi: 10.1113/jphysiol.2010.192922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Oguro K, Jover T, Tanaka H, Lin Y, Kojima T, Oguro N, et al. Global ischemia-induced increases in the gap junctional proteins connexin 32 (Cx32) and Cx36 in hippocampus and enhanced vulnerability of Cx32 knock-out mice. J Neurosci. 2001;21:7534–42. doi: 10.1523/JNEUROSCI.21-19-07534.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Contreras JE, Sánchez HA, Véliz LP, Bukauskas FF, Bennett MVL, Sáez JC. Role of connexin-based gap junction channels and hemichannels in ischemia-induced cell death in nervous tissue. Brain Res Brain Res Rev. 2004;47:290–303. doi: 10.1016/j.brainresrev.2004.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Thompson RJ, Zhou N, MacVicar BA. Ischemia opens neuronal gap junction hemichannels. Science. 2006;312:924–7. doi: 10.1126/science.1126241. [DOI] [PubMed] [Google Scholar]
- 64.Bargiotas P, Krenz A, Hormuzdi SG, Ridder DA, Herb A, Barakat W, et al. Pannexins in ischemia-induced neurodegeneration. Proc Natl Acad Sci USA. 2011;108:20772–7. doi: 10.1073/pnas.1018262108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hara Y, Wakamori M, Ishii M, Maeno E, Nishida M, Yoshida T, et al. LTRPC2 Ca2+-permeable channel activated by changes in redox status confers susceptibility to cell death. Mol Cell. 2002;9:163–73. doi: 10.1016/S1097-2765(01)00438-5. [DOI] [PubMed] [Google Scholar]
- 66.Kaneko S, Kawakami S, Hara Y, Wakamori M, Itoh E, Minami T, et al. A critical role of TRPM2 in neuronal cell death by hydrogen peroxide. J Pharmacol Sci. 2006;101:66–76. doi: 10.1254/jphs.FP0060128. [DOI] [PubMed] [Google Scholar]
- 67.Verma S, Quillinan N, Yang Y-F, Nakayama S, Cheng J, Kelley MH, et al. TRPM2 channel activation following in vitro ischemia contributes to male hippocampal cell death. Neurosci Lett. 2012;530:41–6. doi: 10.1016/j.neulet.2012.09.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Montell C, Birnbaumer L, Flockerzi V. The TRP channels, a remarkably functional family. Cell. 2002;108:595–8. doi: 10.1016/S0092-8674(02)00670-0. [DOI] [PubMed] [Google Scholar]
- 69.Scharenberg AM. TRPM2 and TRPM7: channel/enzyme fusions to generate novel intracellular sensors. Pflugers Arch. 2005;451:220–7. doi: 10.1007/s00424-005-1444-0. [DOI] [PubMed] [Google Scholar]
- 70.Montell C. The TRP superfamily of cation channels. Sci STKE. 2005;2005:re3. doi: 10.1126/stke.2722005re3. [DOI] [PubMed] [Google Scholar]
- 71.Miller BA, Zhang W. TRP channels as mediators of oxidative stress. Adv Exp Med Biol. 2011;704:531–44. doi: 10.1007/978-94-007-0265-3_29. [DOI] [PubMed] [Google Scholar]
- 72.Pan Z, Yang H, Reinach PS. Transient receptor potential (TRP) gene superfamily encoding cation channels. Hum Genomics. 2011;5:108–16. doi: 10.1186/1479-7364-5-2-108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Nilius B, Owsianik G. The transient receptor potential family of ion channels. Genome Biol. 2011;12:218. doi: 10.1186/gb-2011-12-3-218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bae CY, Sun HS. TRPM7 in cerebral ischemia and potential target for drug development in stroke. Acta Pharmacol Sin. 2011;32:725–33. doi: 10.1038/aps.2011.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gaudet R. A primer on ankyrin repeat function in TRP channels and beyond. Mol Biosyst. 2008;4:372–9. doi: 10.1039/b801481g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Montell C. Mg2+ homeostasis: the Mg2+nificent TRPM chanzymes. Curr Biol. 2003;13:R799–801. doi: 10.1016/j.cub.2003.09.048. [DOI] [PubMed] [Google Scholar]
- 77.Runnels LW, Yue L, Clapham DE. TRP-PLIK, a bifunctional protein with kinase and ion channel activities. Science. 2001;291:1043–7. doi: 10.1126/science.1058519. [DOI] [PubMed] [Google Scholar]
- 78.Riazanova LV, Pavur KS, Petrov AN, Dorovkov MV, Riazanov AG. [Novel type of signaling molecules: protein kinases covalently linked to ion channels] Mol Biol (Mosk) 2001;35:321–32. [PubMed] [Google Scholar]
- 79.Nadler MJ, Hermosura MC, Inabe K, Perraud AL, Zhu Q, Stokes AJ, et al. LTRPC7 is a Mg.ATP-regulated divalent cation channel required for cell viability. Nature. 2001;411:590–5. doi: 10.1038/35079092. [DOI] [PubMed] [Google Scholar]
- 80.Hanano T, Hara Y, Shi J, Morita H, Umebayashi C, Mori E, et al. Involvement of TRPM7 in cell growth as a spontaneously activated Ca2+ entry pathway in human retinoblastoma cells. J Pharmacol Sci. 2004;95:403–19. doi: 10.1254/jphs.FP0040273. [DOI] [PubMed] [Google Scholar]
- 81.Jiang J, Li M-H, Inoue K, Chu X-P, Seeds J, Xiong Z-G. Transient receptor potential melastatin 7-like current in human head and neck carcinoma cells: role in cell proliferation. Cancer Res. 2007;67:10929–38. doi: 10.1158/0008-5472.CAN-07-1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kim BJ, Park EJ, Lee JH, Jeon J-H, Kim SJ, So I. Suppression of transient receptor potential melastatin 7 channel induces cell death in gastric cancer. Cancer Sci. 2008;99:2502–9. doi: 10.1111/j.1349-7006.2008.00982.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kim BJ, Kim S-Y, Lee S, Jeon J-H, Matsui H, Kwon YK, et al. The role of transient receptor potential channel blockers in human gastric cancer cell viability. Can J Physiol Pharmacol. 2012;90:175–86. doi: 10.1139/y11-114. [DOI] [PubMed] [Google Scholar]
- 84.Monteilh-Zoller MK, Hermosura MC, Nadler MJS, Scharenberg AM, Penner R, Fleig A. TRPM7 provides an ion channel mechanism for cellular entry of trace metal ions. J Gen Physiol. 2003;121:49–60. doi: 10.1085/jgp.20028740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hermosura MC, Monteilh-Zoller MK, Scharenberg AM, Penner R, Fleig A. Dissociation of the store-operated calcium current I(CRAC) and the Mg-nucleotide-regulated metal ion current MagNuM. J Physiol. 2002;539:445–58. doi: 10.1113/jphysiol.2001.013361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Murakami M, Xu F, Miyoshi I, Sato E, Ono K, Iijima T. Identification and characterization of the murine TRPM4 channel. Biochem Biophys Res Commun. 2003;307:522–8. doi: 10.1016/S0006-291X(03)01186-0. [DOI] [PubMed] [Google Scholar]
- 87.Zhang W, Chu X, Tong Q, Cheung JY, Conrad K, Masker K, et al. A novel TRPM2 isoform inhibits calcium influx and susceptibility to cell death. J Biol Chem. 2003;278:16222–9. doi: 10.1074/jbc.M300298200. [DOI] [PubMed] [Google Scholar]
- 88.Krapivinsky G, Mochida S, Krapivinsky L, Cibulsky SM, Clapham DE. The TRPM7 ion channel functions in cholinergic synaptic vesicles and affects transmitter release. Neuron. 2006;52:485–96. doi: 10.1016/j.neuron.2006.09.033. [DOI] [PubMed] [Google Scholar]
- 89.Brauchi S, Krapivinsky G, Krapivinsky L, Clapham DE. TRPM7 facilitates cholinergic vesicle fusion with the plasma membrane. Proc Natl Acad Sci USA. 2008;105:8304–8. doi: 10.1073/pnas.0800881105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ilardi JM, Mochida S, Sheng ZH. Snapin: a SNARE-associated protein implicated in synaptic transmission. Nat Neurosci. 1999;2:119–24. doi: 10.1038/5673. [DOI] [PubMed] [Google Scholar]
- 91.Rohács T, Lopes CMB, Michailidis I, Logothetis DE. PI(4,5)P2 regulates the activation and desensitization of TRPM8 channels through the TRP domain. Nat Neurosci. 2005;8:626–34. doi: 10.1038/nn1451. [DOI] [PubMed] [Google Scholar]
- 92.Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science. 2002;298:1912–34. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- 93.Drennan D, Ryazanov AG. Alpha-kinases: analysis of the family and comparison with conventional protein kinases. Prog Biophys Mol Biol. 2004;85:1–32. doi: 10.1016/S0079-6107(03)00060-9. [DOI] [PubMed] [Google Scholar]
- 94.Futey LM, Medley QG, Côté GP, Egelhoff TT. Structural analysis of myosin heavy chain kinase A from Dictyostelium. Evidence for a highly divergent protein kinase domain, an amino-terminal coiled-coil domain, and a domain homologous to the beta-subunit of heterotrimeric G proteins. J Biol Chem. 1995;270:523–9. doi: 10.1074/jbc.270.2.523. [DOI] [PubMed] [Google Scholar]
- 95.Côté GP, Luo X, Murphy MB, Egelhoff TT. Mapping of the novel protein kinase catalytic domain of Dictyostelium myosin II heavy chain kinase A. J Biol Chem. 1997;272:6846–9. doi: 10.1074/jbc.272.11.6846. [DOI] [PubMed] [Google Scholar]
- 96.Ryazanov AG, Ward MD, Mendola CE, Pavur KS, Dorovkov MV, Wiedmann M, et al. Identification of a new class of protein kinases represented by eukaryotic elongation factor-2 kinase. Proc Natl Acad Sci USA. 1997;94:4884–9. doi: 10.1073/pnas.94.10.4884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ryazanov AG, Pavur KS, Dorovkov MV. Alpha-kinases: a new class of protein kinases with a novel catalytic domain. Curr Biol. 1999;9:R43–5. doi: 10.1016/S0960-9822(99)80006-2. [DOI] [PubMed] [Google Scholar]
- 98.Pinna LA, Ruzzene M. How do protein kinases recognize their substrates? Biochim Biophys Acta. 1996;1314:191–225. doi: 10.1016/S0167-4889(96)00083-3. [DOI] [PubMed] [Google Scholar]
- 99.Middelbeek J, Clark K, Venselaar H, Huynen MA, van Leeuwen FN. The alpha-kinase family: an exceptional branch on the protein kinase tree. Cell Mol Life Sci. 2010;67:875–90. doi: 10.1007/s00018-009-0215-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Runnels LW, Yue L, Clapham DE. The TRPM7 channel is inactivated by PIP(2) hydrolysis. Nat Cell Biol. 2002;4:329–36. doi: 10.1038/ncb781. [DOI] [PubMed] [Google Scholar]
- 101.Clark K, Langeslag M, van Leeuwen B, Ran L, Ryazanov AG, Figdor CG, et al. TRPM7, a novel regulator of actomyosin contractility and cell adhesion. EMBO J. 2006;25:290–301. doi: 10.1038/sj.emboj.7600931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Clark K, Middelbeek J, Lasonder E, Dulyaninova NG, Morrice NA, Ryazanov AG, et al. TRPM7 regulates myosin IIA filament stability and protein localization by heavy chain phosphorylation. J Mol Biol. 2008;378:790–803. doi: 10.1016/j.jmb.2008.02.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Dorovkov MV, Ryazanov AG. Phosphorylation of annexin I by TRPM7 channel-kinase. J Biol Chem. 2004;279:50643–6. doi: 10.1074/jbc.C400441200. [DOI] [PubMed] [Google Scholar]
- 104.Perraud A-L, Zhao X, Ryazanov AG, Schmitz C. The channel-kinase TRPM7 regulates phosphorylation of the translational factor eEF2 via eEF2-k. Cell Signal. 2011;23:586–93. doi: 10.1016/j.cellsig.2010.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Schmitz C, Perraud A-L, Johnson CO, Inabe K, Smith MK, Penner R, et al. Regulation of vertebrate cellular Mg2+ homeostasis by TRPM7. Cell. 2003;114:191–200. doi: 10.1016/S0092-8674(03)00556-7. [DOI] [PubMed] [Google Scholar]
- 106.Ryazanova LV, Dorovkov MV, Ansari A, Ryazanov AG. Characterization of the protein kinase activity of TRPM7/ChaK1, a protein kinase fused to the transient receptor potential ion channel. J Biol Chem. 2004;279:3708–16. doi: 10.1074/jbc.M308820200. [DOI] [PubMed] [Google Scholar]
- 107.Matsushita M, Kozak JA, Shimizu Y, McLachlin DT, Yamaguchi H, Wei F-Y, et al. Channel function is dissociated from the intrinsic kinase activity and autophosphorylation of TRPM7/ChaK1. J Biol Chem. 2005;280:20793–803. doi: 10.1074/jbc.M413671200. [DOI] [PubMed] [Google Scholar]
- 108.Goldberg MP, Choi DW. Combined oxygen and glucose deprivation in cortical cell culture: calcium-dependent and calcium-independent mechanisms of neuronal injury. J Neurosci. 1993;13:3510–24. doi: 10.1523/JNEUROSCI.13-08-03510.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Aarts MM, Tymianski M. TRPM7 and ischemic CNS injury. Neuroscientist. 2005;11:116–23. doi: 10.1177/1073858404272966. [DOI] [PubMed] [Google Scholar]
- 110.Aarts MM, Tymianski M. TRPMs and neuronal cell death. Pflugers Arch. 2005;451:243–9. doi: 10.1007/s00424-005-1439-x. [DOI] [PubMed] [Google Scholar]
- 111.Kolman MF, Futey LM, Egelhoff TT. Dictyostelium myosin heavy chain kinase A regulates myosin localization during growth and development. J Cell Biol. 1996;132:101–9. doi: 10.1083/jcb.132.1.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Rico M, Egelhoff TT. Myosin heavy chain kinase B participates in the regulation of myosin assembly into the cytoskeleton. J Cell Biochem. 2003;88:521–32. doi: 10.1002/jcb.10361. [DOI] [PubMed] [Google Scholar]
- 113.Heid PJ, Wessels D, Daniels KJ, Gibson DP, Zhang H, Voss E, et al. The role of myosin heavy chain phosphorylation in Dictyostelium motility, chemotaxis and F-actin localization. J Cell Sci. 2004;117:4819–35. doi: 10.1242/jcs.01358. [DOI] [PubMed] [Google Scholar]
- 114.Murphy DA, Courtneidge SA. The ‘ins’ and ‘outs’ of podosomes and invadopodia: characteristics, formation and function. Nat Rev Mol Cell Biol. 2011;12:413–26. doi: 10.1038/nrm3141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Clark K, Middelbeek J, Morrice NA, Figdor CG, Lasonder E, van Leeuwen FN. Massive autophosphorylation of the Ser/Thr-rich domain controls protein kinase activity of TRPM6 and TRPM7. PLoS ONE. 2008;3:e1876. doi: 10.1371/journal.pone.0001876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Su L-T, Agapito MA, Li M, Simonson WTN, Huttenlocher A, Habas R, et al. TRPM7 regulates cell adhesion by controlling the calcium-dependent protease calpain. J Biol Chem. 2006;281:11260–70. doi: 10.1074/jbc.M512885200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Glading A, Chang P, Lauffenburger DA, Wells A. Epidermal growth factor receptor activation of calpain is required for fibroblast motility and occurs via an ERK/MAP kinase signaling pathway. J Biol Chem. 2000;275:2390–8. doi: 10.1074/jbc.275.4.2390. [DOI] [PubMed] [Google Scholar]
- 118.Su L-T, Chen H-C, González-Pagán O, Overton JD, Xie J, Yue L, et al. TRPM7 activates m-calpain by stress-dependent stimulation of p38 MAPK and c-Jun N-terminal kinase. J Mol Biol. 2010;396:858–69. doi: 10.1016/j.jmb.2010.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Su L-T, Liu W, Chen H-C, González-Pagán O, Habas R, Runnels LW. TRPM7 regulates polarized cell movements. Biochem J. 2011;434:513–21. doi: 10.1042/BJ20101678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Oancea E, Wolfe JT, Clapham DE. Functional TRPM7 channels accumulate at the plasma membrane in response to fluid flow. Circ Res. 2006;98:245–53. doi: 10.1161/01.RES.0000200179.29375.cc. [DOI] [PubMed] [Google Scholar]
- 121.Theriot JA. Accelerating on a treadmill: ADF/cofilin promotes rapid actin filament turnover in the dynamic cytoskeleton. J Cell Biol. 1997;136:1165–8. doi: 10.1083/jcb.136.6.1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Bamburg JR. Proteins of the ADF/cofilin family: essential regulators of actin dynamics. Annu Rev Cell Dev Biol. 1999;15:185–230. doi: 10.1146/annurev.cellbio.15.1.185. [DOI] [PubMed] [Google Scholar]
- 123.Maciver SK, Hussey PJ. The ADF/cofilin family: actin-remodeling proteins. Genome Biol. 2002;3:s3007. doi: 10.1186/gb-2002-3-5-reviews3007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Van Troys M, Huyck L, Leyman S, Dhaese S, Vandekerkhove J, Ampe C. Ins and outs of ADF/cofilin activity and regulation. Eur J Cell Biol. 2008;87:649–67. doi: 10.1016/j.ejcb.2008.04.001. [DOI] [PubMed] [Google Scholar]
- 125.Arber S, Barbayannis FA, Hanser H, Schneider C, Stanyon CA, Bernard O, et al. Regulation of actin dynamics through phosphorylation of cofilin by LIM-kinase. Nature. 1998;393:805–9. doi: 10.1038/31729. [DOI] [PubMed] [Google Scholar]
- 126.Meng Y, Zhang Y, Tregoubov V, Janus C, Cruz L, Jackson M, et al. Abnormal spine morphology and enhanced LTP in LIMK-1 knockout mice. Neuron. 2002;35:121–33. doi: 10.1016/S0896-6273(02)00758-4. [DOI] [PubMed] [Google Scholar]
- 127.Meng Y, Zhang Y, Tregoubov V, Falls DL, Jia Z. Regulation of spine morphology and synaptic function by LIMK and the actin cytoskeleton. Rev Neurosci. 2003;14:233–40. doi: 10.1515/REVNEURO.2003.14.3.233. [DOI] [PubMed] [Google Scholar]
- 128.Low SE, Amburgey K, Horstick E, Linsley J, Sprague SM, Cui WW, et al. TRPM7 is required within zebrafish sensory neurons for the activation of touch-evoked escape behaviors. J Neurosci. 2011;31:11633–44. doi: 10.1523/JNEUROSCI.4950-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Jin J, Desai BN, Navarro B, Donovan A, Andrews NC, Clapham DE. Deletion of Trpm7 disrupts embryonic development and thymopoiesis without altering Mg2+ homeostasis. Science. 2008;322:756–60. doi: 10.1126/science.1163493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Jin J, Wu L-J, Jun J, Cheng X, Xu H, Andrews NC, et al. The channel kinase, TRPM7, is required for early embryonic development. Proc Natl Acad Sci USA. 2012;109:E225–33. doi: 10.1073/pnas.1120033109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Lipton P. Ischemic cell death in brain neurons. Physiol Rev. 1999;79:1431–568. doi: 10.1152/physrev.1999.79.4.1431. [DOI] [PubMed] [Google Scholar]
- 132.Jiang J, Li M, Yue L. Potentiation of TRPM7 inward currents by protons. J Gen Physiol. 2005;126:137–50. doi: 10.1085/jgp.200409185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Kozak JA, Matsushita M, Nairn AC, Cahalan MD. Charge screening by internal pH and polyvalent cations as a mechanism for activation, inhibition, and rundown of TRPM7/MIC channels. J Gen Physiol. 2005;126:499–514. doi: 10.1085/jgp.200509324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Li M, Du J, Jiang J, Ratzan W, Su L-T, Runnels LW, et al. Molecular determinants of Mg2+ and Ca2+ permeability and pH sensitivity in TRPM6 and TRPM7. J Biol Chem. 2007;282:25817–30. doi: 10.1074/jbc.M608972200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Shichi K, Fujita-Hamabe W, Harada S, Mizoguchi H, Yamada K, Nabeshima T, et al. Involvement of matrix metalloproteinase-mediated proteolysis of neural cell adhesion molecule in the development of cerebral ischemic neuronal damage. J Pharmacol Exp Ther. 2011;338:701–10. doi: 10.1124/jpet.110.178079. [DOI] [PubMed] [Google Scholar]
- 136.Fujita-Hamabe W, Tokuyama S. The involvement of cleavage of neural cell adhesion molecule in neuronal death under oxidative stress conditions in cultured cortical neurons. Biol Pharm Bull. 2012;35:624–8. doi: 10.1248/bpb.35.624. [DOI] [PubMed] [Google Scholar]
- 137.Linder S, Aepfelbacher M. Podosomes: adhesion hot-spots of invasive cells. Trends Cell Biol. 2003;13:376–85. doi: 10.1016/S0962-8924(03)00128-4. [DOI] [PubMed] [Google Scholar]
- 138.Proszynski TJ, Gingras J, Valdez G, Krzewski K, Sanes JR. Podosomes are present in a postsynaptic apparatus and participate in its maturation. Proc Natl Acad Sci USA. 2009;106:18373–8. doi: 10.1073/pnas.0910391106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Asrar S, Meng Y, Zhou Z, Todorovski Z, Huang WW, Jia Z. Regulation of hippocampal long-term potentiation by p21-activated protein kinase 1 (PAK1) Neuropharmacology. 2009;56:73–80. doi: 10.1016/j.neuropharm.2008.06.055. [DOI] [PubMed] [Google Scholar]
- 140.Zhou Z, Meng Y, Asrar S, Todorovski Z, Jia Z. A critical role of Rho-kinase ROCK2 in the regulation of spine and synaptic function. Neuropharmacology. 2009;56:81–9. doi: 10.1016/j.neuropharm.2008.07.031. [DOI] [PubMed] [Google Scholar]
- 141.Langeslag M, Clark K, Moolenaar WH, van Leeuwen FN, Jalink K. Activation of TRPM7 channels by phospholipase C-coupled receptor agonists. J Biol Chem. 2007;282:232–9. doi: 10.1074/jbc.M605300200. [DOI] [PubMed] [Google Scholar]
- 142.Chen H-C, Xie J, Zhang Z, Su L-T, Yue L, Runnels LW. Blockade of TRPM7 channel activity and cell death by inhibitors of 5-lipoxygenase. PLoS ONE. 2010;5:e11161. doi: 10.1371/journal.pone.0011161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Chubanov V, Mederos Y. Schnitzler M, Meißner M, Schäfer S, Abstiens K, Hofmann T, et al. Natural and synthetic modulators of SK (K(ca)2) potassium channels inhibit magnesium-dependent activity of the kinase-coupled cation channel TRPM7. British journal of pharmacology [Internet] 2012 [cited 2012 May 14]; Available from: http://www.ncbi.nlm.nih.gov/pubmed/22242975 [DOI] [PMC free article] [PubMed]
- 144.Chen X, Numata T, Li M, Mori Y, Orser BA, Jackson MF, et al. The modulation of TRPM7 currents by nafamostat mesilate depends directly upon extracellular concentrations of divalent cations. Mol Brain. 2010;3:38. doi: 10.1186/1756-6606-3-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Castillo B, Pörzgen P, Penner R, Horgen FD, Fleig A. Development and optimization of a high-throughput bioassay for TRPM7 ion channel inhibitors. J Biomol Screen. 2010;15:498–507. doi: 10.1177/1087057110368294. [DOI] [PMC free article] [PubMed] [Google Scholar]