

Abstract
Objective:
To report long-term efficacy and safety data for everolimus for the treatment of subependymal giant cell astrocytoma (SEGA) in patients with tuberous sclerosis complex (TSC).
Methods:
This was an open-label extension phase of a prospective, phase 1–2 trial (NCT00411619) in patients ≥3 years of age with SEGA associated with TSC. Patients received oral everolimus starting at 3 mg/m2 per day and subsequently titrated, subject to tolerability, to attain whole blood trough concentrations of 5–15 ng/mL. Change in SEGA volume, seizures, and safety assessments were the main outcome measures.
Results:
Of 28 patients enrolled, 25 were still under treatment at the time of analysis. Median dose was 5.3 mg/m2/day and median treatment duration was 34.2 months (range 4.7–47.1). At all time points (18, 24, 30, and 36 months), primary SEGA volume was reduced by ≥30% from baseline (treatment response) in 65%–79% of patients. All patients reported ≥1 adverse event (AE), mostly grade 1/2 in severity, consistent with that previously reported, and none led to everolimus discontinuation. The most commonly reported drug-related AEs were upper respiratory infections (85.7%), stomatitis (85.7%), sinusitis (46.4%), and otitis media (35.7%). No drug-related grade 4 or 5 events occurred.
Conclusion:
Everolimus therapy is safe and effective for longer term (median exposure 34.2 months) treatment of patients with TSC with SEGA.
Classification of evidence:
This study provides Class III evidence that everolimus, titrated to trough serum levels of 5–15 ng/mL, was effective in reducing tumor size in patients with SEGA secondary to TSC for a median of 34 months.
Tuberous sclerosis complex (TSC) is an autosomal dominant, genetic disorder with an estimated prevalence of 1:6,000.1 TSC is caused by mutations in TSC1 or TSC2, which result in constitutive activation of the mammalian target of rapamycin complex 1 (mTORC1), causing benign tumors (hamartomas) most commonly in the brain, kidneys, lungs, and skin.2 In the brain, subependymal giant cell astrocytomas (SEGA) occur in 5%–20% of patients with TSC.3,4 SEGA are slow-growing tumors that typically develop near the foramen of Monro.1 Standard treatment has been surgical resection, although complications include intraventricular hemorrhage, hydrocephalus, cognitive impairment, and inevitable recurrence if gross total resection is not achieved.2,5–7
In a phase 1–2, open-label study in 28 patients with evidence of serial SEGA growth, the mTOR inhibitor everolimus (Afinitor, Novartis, East Hanover, NJ) was associated with a reduction in SEGA volume and improved quality of life.8 As a result of this trial, the US Food and Drug Administration (FDA) approved everolimus for patients with SEGA associated with TSC who are not candidates for curative surgical resection.9 The core treatment phase lasted for 6 months, after which patients were eligible to enter a long-term extension phase and continue receiving everolimus for up to 5 years as long as clinical benefit was apparent by investigator opinion. Long-term tolerability and efficacy of everolimus from this extension phase in patients with TSC with SEGA for up to 3 years are reported.
METHODS
Study participants.
Patients completing the 6-month, open-label study of everolimus who had a reduction in tumor volume were eligible to continue treatment in an extension phase.8 All patients were ≥3 years of age, had a definite TSC diagnosis,10 and had serial SEGA growth (defined by increase in size compared with baseline on serial MRI). All patients were from the Cincinnati Children's Hospital Medical Center Tuberous Sclerosis Clinic. Patients were not eligible if they had a serious intercurrent infection or other uncontrolled medical disease that would compromise participation in the study (excepting patients with uncontrolled epilepsy), had used an investigational drug within the past 30 days, or had evidence of impending herniation or focal neurologic deficit related to the patient's astrocytoma.
Study design.
This was an extension phase of a prospective, open-label, phase 1–2 trial of oral everolimus, starting at a dose of 3 mg/m2 per day and titrated to a serum concentration between 5 and 15 ng/mL. During the extension phase of the trial, patients were continued on their prior everolimus dose. In those who experienced troublesome side effects (i.e., grade 2 or 3), dosage was transiently withheld or reduced by increments of 25% until these resolved.
Classification of evidence.
This study provides Class III evidence that everolimus, titrated to trough serum levels of 5–15 ng/mL, was effective in reducing tumor size in patients ≥3 years old with SEGA secondary to TSC for a median of 34 months.
Standard protocol approval, registrations, and participant consents.
This trial is registered with clinicaltrials.gov, number NCT00411619. The protocol was approved by the institutional review board, and a data and safety monitoring board has reviewed progress of the trial twice a year. In accordance with the Declaration of Helsinki,11 all adult patients provided written informed consent and patients <18 years old provided verbal assent if able, with written informed consent obtained from a parent or guardian.
Efficacy evaluation.
The primary efficacy evaluation during the initial treatment phase was change in primary SEGA volume from baseline at 6 months based on MRI volumetric measurements. During the extension phase, physical examinations and SEGA volumes were assessed using MRI every 6 months. Both local assessment and independent central reviews (performed by VirtualScopics Inc., Rochester, NY) of SEGA volumes were conducted. The independent central review was used for the primary efficacy endpoint. SEGA volumes were calculated using a Vitrea 2 workstation based on reformatted images of 1-mm-thick coronal views acquired from MRI: either postcontrast sagittal, 3D, spoiled-gradient recalled acquisition in steady-state 1.5-T MRI (General Electric Medical Systems, Milwaukee, WI, and Siemens, Malvern, PA) or 3D, T1-weighted, gradient-echo sequence 3.0-T MRI (Siemens). Progression was defined as ≥25% increase from nadir in primary SEGA volume to a value greater than baseline. Angiofibroma and seizure frequency were 2 secondary endpoints while changes in tuber and subependymal nodule (SEN) volumes were exploratory endpoints. Seizure frequency was reported by all participants or their caregivers at each study visit. Tuber and SEN volumetric assessments were performed using brain MRI.
Safety evaluation.
Assessment of safety included monitoring of hematologic (complete blood count plus differential) and other laboratory parameters (i.e., liver and renal panels, blood sugar, fasting lipid profiles, urinalysis) every 3 months. Adverse events (AEs) were monitored during the study using the Common Terminology Criteria for Adverse Events (version 3.0).12 According to study protocol, all infections were to be considered related to study drug by the investigators. The exposure–safety relationship was explored by subgroup analysis of AEs by everolimus exposure range (Cmin <5 ng/mL, 5–10 ng/mL, and >10 ng/mL); patients were assigned to the Cmin group corresponding to their mean Cmin value during the study. Stomatitis and infections were evaluated further.
Statistical analyses.
It was determined that a sample size of 28 patients would provide a statistical power of at least 90% to detect a median reduction in the volume of a SEGA of at least 1 cm3 from the baseline volume (primary endpoint at 6 months), on the basis of a one-sided Wilcoxon signed-rank test with an α level of 0.025. All statistical analyses were performed with the use of SAS software (SAS Institute). The primary population for efficacy analyses (equivalent to an intention-to-treat population) included all patients receiving at least one dose of everolimus. The safety population consisted of all patients receiving at least one dose of everolimus and who had at least one postbaseline safety assessment.
RESULTS
Patient characteristics.
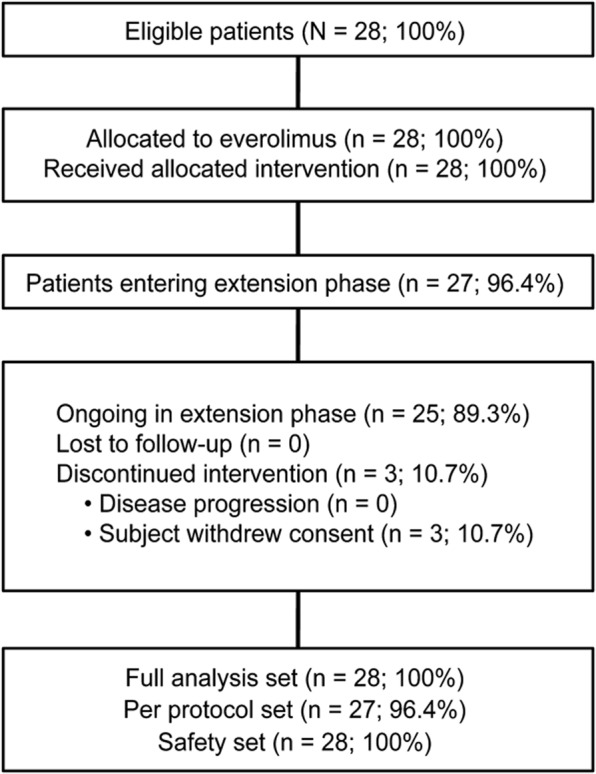
Of 28 patients enrolled in the trial, 27 (96.4%) patients entered the extension phase and 25 patients (89.3%) were still continuing treatment at the data cutoff of December 31, 2010 (figure 1). Three patients withdrew consent at 142, 532, and 653 days for noncompliance with antiepileptic drug, inability to maintain study visits, and withdrawal of parental consent. At baseline, the median age was 11 years (range 3 to 34 years), 16 were <12 years, and 6 patients were between 12 and 18 years old. Twelve patients (42.9%) had bilateral SEGA, 15 (53.6%) had 1 SEGA lesion, and 13 (46.4%) had 2 SEGA lesions. Six patients (21.4%) presented with hydrocephalus. Four patients (14.3%) had tumor recurrence after incomplete surgical resection of SEGA at outside centers before enrollment. The majority (25/28 = 89.3%) of patients had facial angiofibromas; 24 patients (82.1%) were receiving antiepileptic medications.
Figure 1. CONSORT diagram of patient disposition throughout the trial.
Efficacy.
Everolimus treatment resulted in a reduction in SEGA volume in the majority of patients, and this effect was maintained for up to 3 years (figures 2 and 3). Before everolimus treatment, the mean annual rate of change of primary SEGA volume was +0.54 cm3 (range −0.10 to 2.20). After everolimus treatment, the mean annual rate of change was −0.32 cm3 (range −2.63 to 0.60). Over time, the median primary SEGA volume decreased from 1.74 cm3 at baseline to 0.97 cm3 at 36 months (table 1). Primary SEGA volume was reduced by ≥30% from baseline in 79.2% (19/24), 64.7% (11/17), and 77.8% (7/9) of patients at 24, 30, and 36 months, respectively, and by ≥50% from baseline in 50.0% (12/24), 41.2% (7/17), and 55.6% (5/9) of patients, respectively. Nine patients had a ≥30% but <50% reduction in primary SEGA volume within 6 months. Six of these patients improved to a ≥50% reduction in primary SEGA volume at a later time point.
Figure 2. Effect of everolimus on primary subependymal giant cell astrocytoma volume over time.

Figure 3. Effect of everolimus on subependymal giant cell astrocytoma volume over time.

(A) Patient A: Note resolution of hydrocephalus and progressive decrease in bilateral subependymal giant cell astrocytoma (SEGA); subependymal nodule at left ventricular trigone appears to increase at 30 months, but this is artifactual due to slice variation. (B) Patient B: Infiltrating SEGA status after 2 failed resections and proton beam irradiation; note progressive reduction in enhancement and tumor size. (C) Patient C: SEGA volume at baseline and after everolimus for 4 years; patient's trough serum levels ranged from 1 to 3 ng/mL. (D) Patient D: SEGA volume at baseline and after everolimus for 4 years, although lesion approximates baseline volume note reduction in contrast enhancement.
Table 1.
Response of primary SEGA to everolimus therapy during the 6-month treatment phase and extension phase per independent central review
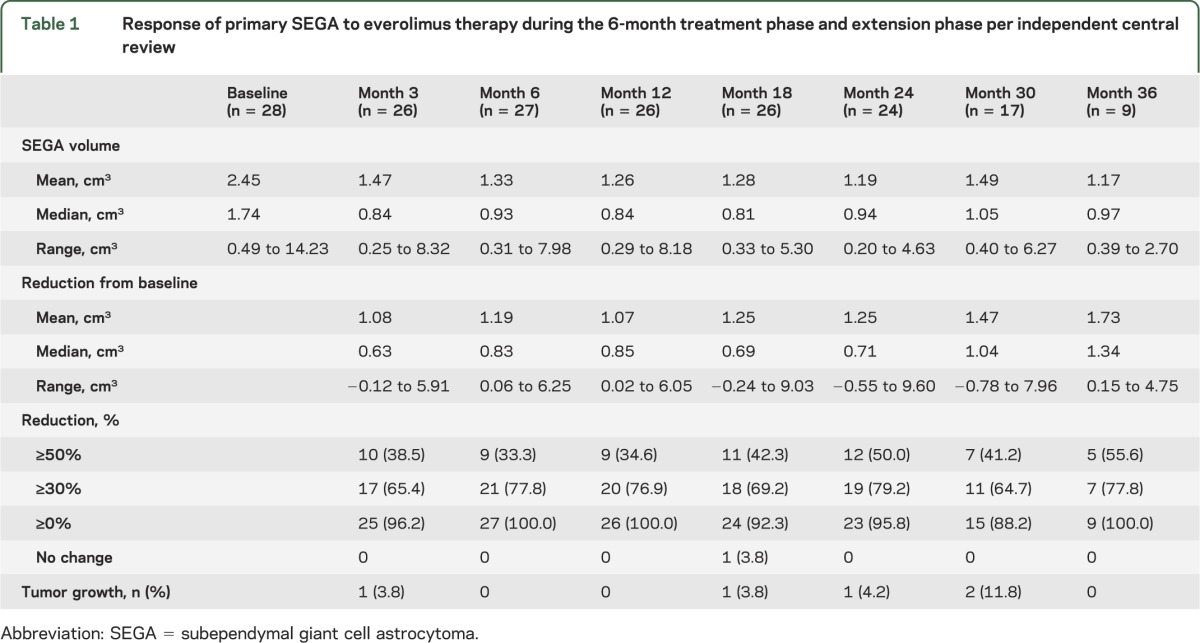
The ≥30% reduction in primary SEGA volume was maintained for a median duration of 23.8 months (range 0 to 39.4) up to progression or cutoff date. Overall, the median time to SEGA progression could not be estimated by the Kaplan-Meier method because too few progressions were observed. At the time of data cutoff, 3 patients had a progression, including 2 of 25 patients who had a ≥30% reduction in primary SEGA volume at 6 months. In 2 of these 3 patients, the primary lesion volume reverted back to volume at baseline, while in the other patient, an increase in volume of 51% compared with baseline was observed. These 3 patients continued on treatment due to investigators' judgment of overall clinical benefit. Despite this, no patient required surgery or additional therapy for their SEGA or for hydrocephalus.
Additionally, the median reduction in tuber (not tumor) volume from baseline to 24 months was 0.67 cm3 (range −14.06 to 49.69). At the same time points, median reduction in SEN from baseline was 0.15 cm3 (range −0.25 to 1.10). At 24 months (n = 9 with evaluation), an improvement in facial angiofibromas was observed in most patients (8/9 = 88.9%). However, comparisons were made relative to the previous visit rather than to baseline and were not always conducted by the same investigator. There was also an improvement in patient-reported seizure frequency over time. The percentage of patients who reported no seizures since their last visit (or more than 6 months since their last seizure from baseline) increased from 38.5% (10/26) at baseline to 65.2% (15/23) at 24 months. Further, the number of patients who reported experiencing at least 1 seizure per day decreased from 26.9% (7/26) at baseline to 13.0% (3/23) at 24 months.
Safety.
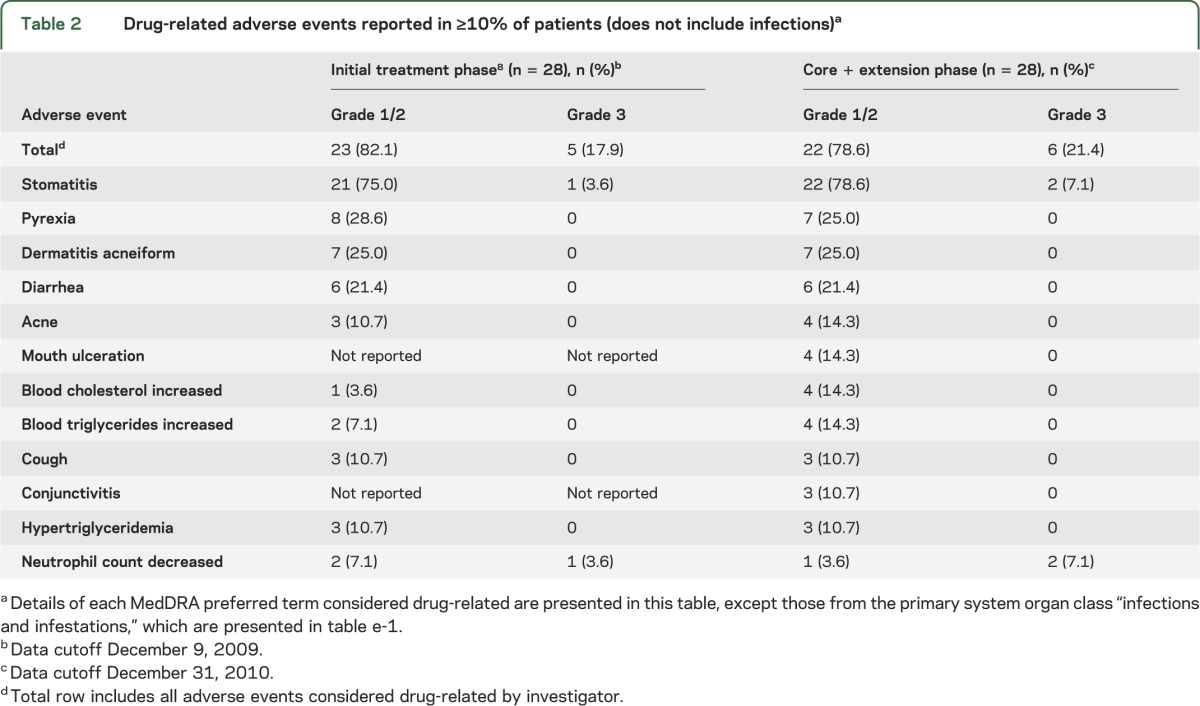
Overall, during both phases, the median daily dose of everolimus was 5.3 mg/m2 (range 2.1–12.3) and median duration of therapy was 34.2 months (range 4.7–47.1). All patients reported at least one drug-related AE, mostly mild (grade 1) to moderate (grade 2) in severity, which were managed through dose reduction, temporary interruption of therapy, or administration of concomitant drug therapy. No AEs resulted in discontinuation of everolimus therapy. The type, incidence, and severity of AEs reported by the new cutoff date were similar to those reported in the primary treatment phase (table 2 and table e-1 on the Neurology® Web site at www.neurology.org). The most frequently reported AEs were upper respiratory infections (24/28 = 85.7%), stomatitis (24/28 = 85.7%), sinusitis (13/28 = 46.4%), and otitis media (10/28 = 35.7%). The incidence of AEs and infections decreased over time (tables e-2 and e-3). Few grade 3 and no grade 4 or 5 AEs occurred over the core treatment and extension phase of the trial. Of 13 patients who experienced grade 3 AEs, 6 were considered to be related to everolimus therapy. The most frequently reported grade 3 AEs were stomatitis (2/28 = 7.1%) and neutropenia (2/28 = 7.1%). The only grade 4 AE to occur was a convulsion that was not believed to be related to everolimus therapy. No deaths were reported during the study.
Table 2.
Drug-related adverse events reported in ≥10% of patients (does not include infections)a
There were 6 patients who experienced 9 serious AEs (SAEs), 3 of which were suspected to be related to everolimus, 1 patient with pneumonia, 1 patient with viral bronchitis, and 1 patient with an abscess of the right leg (all grade 3 reactions). Management of these SAEs included hospitalization, concomitant drug therapy, and interruption of everolimus therapy or dose reduction. In each case, the SAE resolved without sequelae and everolimus treatment was continued. The 6 unrelated SAEs included 2 patients with seizures (prior history), gastroenteritis, leukocytosis, petechiae, and post–lumbar puncture syndrome.
Subgroup analysis of the exposure–safety relationship demonstrated that the incidence of various AEs was similar across the different everolimus trough concentrations (Cmin <5 ng/mL [n = 13], 5–10 ng/mL [n = 14], and >10 ng/mL [n = 1]). Further, no relationship was evident between the severity of infections or stomatitis (the 2 most frequently reported AEs) and everolimus trough levels.
DISCUSSION
This prospective, open-label extension study in patients with TSC provides further evidence of the safety demonstrated in the initial evaluation phase of this trial of everolimus.8 In patients who received everolimus for a median duration of almost 3 years, its effect in tumor burden reduction was maintained with an acceptable safety profile. The range of SEGA volume reduction was also not limited to 30%–50% as the proportion of patients experiencing a SEGA volume reduction of ≥50% at 24 months was greater than 50%. In fact, the percentage of patients with a SEGA response greater than 50% increased over time. The clinically relevant reduction in tumor burden did not diminish over the longer term extension phase demonstrating not only consistency in achieving SEGA volume reduction, but also that further SEGA growth was prevented. No increase in incidence of or previously unreported AEs associated with everolimus was observed. These data support everolimus use over the longer term, making it a viable alternative to surgery in patients unsuitable for surgical resection. Results of this study are similar to the phase 3, double-blind, placebo-controlled EXIST-1 (NCT00789828) trial in which the overall response rate (as defined by ≥50% reduction in primary SEGA volume from baseline) was 35% for everolimus- and 0% for placebo-treated patients (p < 0.0001).13 The median duration of therapy for everolimus-treated patients (n = 78) was 9.8 months (range 5.6–18.4), compared to the present study with median duration of therapy 34.2 months. As expected, the number of patients reporting AEs was lower in EXIST-1 compared to the present study given the shorter reporting period duration, but the pattern of AE severity (mostly grade 1 or 2) and types were nearly identical. The most commonly observed AEs for everolimus vs placebo, respectively, in EXIST-1, were mouth ulceration (32% vs 5%), stomatitis (31% vs 21%), convulsions (23% vs 26%), pyrexia (22% vs 15%), nasopharyngitis (18% vs 23%), vomiting (17% vs 13%), and upper respiratory infection (15% vs 18%).
Mechanistically, mTOR inhibition corrects the specific molecular defect involved in patients with TSC. Up to 85% of patients with TSC have identifiable mutations in TSC1 or TSC2, which normally limit mTORC1 activation.2 A deficiency in TSC1 or TSC2 leads to hyperactivation of mTORC1, resulting in increased protein synthesis, and abnormal cellular growth and proliferation.14,15 Clinical data not only support the use of mTOR inhibitors for the treatment of SEGA,16–19 but also renal angiomyolipomas,20–23 lymphangioleiomyomatosis,20,21,23–25 and facial angiofibromas.26 However, discontinuation of therapy has led to tumor regrowth or reappearance of lesions in other studies, suggesting that continuation of therapy is necessary for ongoing benefit.
In this trial, everolimus treatment resulted in rapid reduction in SEGA volume, and reduction of primary SEGA volume was sustained over time. Although only 9 patients received everolimus for 36 months by the time of data cutoff, the median SEGA volume was 0.97 cm3 and similar to earlier assessments. However, 4 patients in this study did not have ≥30% response during the evaluation phase, but responded after 12 and 24 months of therapy (possibly due to more slowly growing SEGA), indicating that benefit may still be observed with prolonged treatment. In 2 of 3 patients who had progression, a ≥30% response at 6 months was seen before progression; all 3 patients remained on therapy at data cutoff due to the investigators' judgment of overall clinical benefit. Two of the 3 patients with continued treatment demonstrated renewed tumor reduction at subsequent timepoints. These cases demonstrate that fluctuations in SEGA volume can occur with everolimus treatment; continued radiographic monitoring and titration to achieve target trough concentrations is useful in sustaining clinical benefit. Despite the effect of everolimus on SEGA volume reduction, SEGA did not completely disappear. This may suggest the potential for regrowth of lesions once therapy is discontinued. Most importantly, no patient developed signs of increased intracranial pressure or hydrocephalus, which in fact resolved in every patient who had evidence of it at baseline. No patients required surgery for hydrocephalus or SEGA.
Everolimus was safe and well-tolerated, with an AE profile similar to that reported in the 6-month core treatment phase of the trial. The majority of patients are still being treated with everolimus (25 of 28), and no AEs have led to treatment discontinuation. The incidence of AEs did not increase with higher everolimus trough concentrations, and there was no relationship between everolimus trough levels and the severity of specific AEs such as infections or stomatitis.
One concern may be the high rate of infections reported in this study (89.3% [25/28] in the 6-month treatment phase; 96.4% [27/28] in the core and extension phase). Given the open-label study design, all infections were considered drug-related by protocol. In comparison, infections were reported by 72% of patients who received everolimus and 67% of patients who received placebo in the EXIST-1 trial.13 At dosages typically employed in clinical practice, the rate of infections related to therapy is likely lower than that reported in this study. We believe that many of the reported infections would have occurred in the study population regardless of study medication. Nevertheless, mTOR inhibitors have immunosuppressant properties that must be taken into account as part of their clinical use.
The incidence of grade 3 drug-related AEs reported by the cutoff date remained low, and no grade 4 or 5 drug-related AEs were reported during the core or extension portion of the trial. Patients experiencing AEs associated with everolimus can be managed through dose reduction or interruption of therapy,27 without loss of efficacy.
Limitations of this study include its open-label, single-arm design, and small number of patients treated. The extension phase of this study is still ongoing, and efficacy and safety data continue to be monitored. In addition to SEGA, everolimus use for the treatment of renal angiomyolipomas in patients with TSC has been recently approved by the FDA based on a phase 3, double-blind, placebo-controlled trial (i.e., EXIST-2; NCT00790400). Trials of everolimus for the treatment of TSC-related epilepsy in a phase 2 open-label trial (NCT01070316) and on neurocognitive deficits in a phase 3, double-blind, placebo-controlled trial (NCT01289912) are underway.
In view of its effects on multiple aspects of TSC involving various organ systems, everolimus may represent a therapy not merely for SEGA or renal angiomyolipomas, but rather a disease-modifying treatment for the disorder. In this study, everolimus demonstrated a sustained effect on tumor burden and tolerability for a median duration of approximately 36 months of treatment.
Supplementary Material
ACKNOWLEDGMENT
The authors thank Jeremie Lebrec, PhD, for assistance in reviewing the statistical analysis of the study data.
GLOSSARY
- AE
adverse event
- FDA
US Food and Drug Administration
- mTORC1
mammalian target of rapamycin complex 1
- SAE
serious adverse event
- SEGA
subependymal giant cell astrocytoma
- SEN
subependymal nodules
- TSC
tuberous sclerosis complex
Footnotes
Supplemental data at www.neurology.org
AUTHOR CONTRIBUTIONS
D.A. Krueger: study conception and design, analysis and interpretation of data, drafting and revision of manuscript, acquisition of data, study supervision. M.M. Care: study conception and design, analysis and interpretation of data, acquisition of data. K. Agricola, C. Tudor: analysis and interpretation of data, acquisition of data, study coordination. M. Mays: acquisition of data, study coordination. D.N. Franz: study conception and design, analysis and interpretation of data, drafting and revision of manuscript, acquisition of data, study supervision, obtaining funding.
STUDY FUNDING
Novartis Pharmaceuticals, Inc. provided the study drug and funding for conduct of this research. Cincinnati Children's Hospital has received financial support from Novartis Pharmaceuticals for the conduct and monitoring of this study, and for consulting work done by Dr. Franz and Dr. Krueger.
DISCLOSURE
D.A. Krueger has received speaking fees from Novartis Pharmaceuticals and UCB Pharma. M.M. Care, K. Agricola, C. Tudor, and M. Mays report no disclosures. D.N. Franz has received speaking fees from Novartis Pharmaceuticals and UCB Pharma. ApotheCom provided assistance in editing and formatting the manuscript. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Baskin HJ., Jr The pathogenesis and imaging of the tuberous sclerosis complex. Pediatr Radiol 2008;38:936–952 [DOI] [PubMed] [Google Scholar]
- 2.Crino PB, Nathanson KL, Henske EP. The tuberous sclerosis complex. N Engl J Med 2006;355:1345–1356 [DOI] [PubMed] [Google Scholar]
- 3.Goh S, Butler W, Thiele EA. Subependymal giant cell tumors in tuberous sclerosis complex. Neurology 2004;63:1457–1461 [DOI] [PubMed] [Google Scholar]
- 4.Adriaensen ME, Schaefer-Prokop CM, Stijnen T, et al. Prevalence of subependymal giant cell tumors in patients with tuberous sclerosis and a review of the literature. Eur J Neurol 2009;16:691–696 [DOI] [PubMed] [Google Scholar]
- 5.Chu-Shore CJ, Major P, Camposano S, et al. The natural history of epilepsy in tuberous sclerosis complex. Epilepsia 2010;51:1236–1241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.de Ribaupierre S, Dorfmuller G, Bulteau C, et al. Subependymal giant-cell astrocytomas in pediatric tuberous sclerosis disease: when should we operate? Neurosurgery 2007;60:83–89 [DOI] [PubMed] [Google Scholar]
- 7.Moavero R, Pinci M, Bombardieri R, et al. The management of subependymal giant cell tumors in tuberous sclerosis: a clinician's perspective. Childs Nerv Syst 2011;27:1203–1210 [DOI] [PubMed] [Google Scholar]
- 8.Krueger DA, Care MM, Holland K, et al. Everolimus for subependymal giant-cell astrocytomas in tuberous sclerosis. N Engl J Med 2010;363:1801–1811 [DOI] [PubMed] [Google Scholar]
- 9.Afinitor (Everolimus) Tablets for Oral Administration [Prescribing Information].East Hanover, NJ:Novartis Pharmaceuticals Corporation;2012 [Google Scholar]
- 10.Roach ES, Gomez MR, Northrup H. Tuberous sclerosis complex consensus conference: revised clinical diagnostic criteria. J Child Neurol 1998;13:624–628 [DOI] [PubMed] [Google Scholar]
- 11.World Medical Association Declaration of Helsinki: ethical principles for medical research involving human subjects, 6th revision;2008Available at: www.wma.net/en/30publications/10policies/b3/index.html. Accessed April 23, 2012 [DOI] [PubMed]
- 12.Common Terminology Criteria for Adverse Events v3.0 (CTCAE).Bethesda, MD:National Cancer Institute, US National Institutes of Health;2006Available at: ctep.cancer.gov/protocoldevelopment/electronic_applications/docs/ctcaev3.pdf. Accessed April 23, 2012 [Google Scholar]
- 13.Franz DN, Belousova E, Curatolo P, et al. EXIST-1 trial: double-blind, placebo-controlled, phase 3 trial of everolimus in subependymal giant cell astrocytomas associated with tuberous sclerosis complex. Presented at the International TSC Research Conference, July 6–9, 2011, Washington, DC
- 14.Huang J, Manning BD. The TSC1-TSC2 complex: a molecular switchboard controlling cell growth. Biochem J 2008;412:179–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chan JA, Zang H. Pathogenesis of tuberous sclerosis subependymal giant cell astrocytomas: biallelic inactivation of TSC1 or TSC2 leads to mTOR activation. J Neuropathol Exp Neurol 2004;63:1236–1242 [DOI] [PubMed] [Google Scholar]
- 16.Franz DN, Leonard J, Tudor C, et al. Rapamycin causes regression of astrocytomas in tuberous sclerosis complex. Ann Neurol 2006;59:490–498 [DOI] [PubMed] [Google Scholar]
- 17.Koenig MK, Butler IJ, Northrup H. Regression of subependymal giant cell astrocytoma with rapamycin in tuberous sclerosis complex. J Child Neurol 2008;23:1238–1239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Birca A, Mercier C, Major P. Rapamycin as an alternative to surgical treatment of subependymal giant cell astrocytomas in a patient with tuberous sclerosis complex. J Neurosurg Pediatr 2010;6:381–384 [DOI] [PubMed] [Google Scholar]
- 19.Lam C, Bouffet E, Tabori U, et al. Rapamycin (sirolimus) in tuberous sclerosis associated pediatric central nervous system tumors. Pediatr Blood Cancer 2010;54:476–479 [DOI] [PubMed] [Google Scholar]
- 20.Wienecke R, Fackler I, Linsenmaier U, et al. Antitumoral activity of rapamycin in renal angiomyolipoma associated with tuberous sclerosis complex. Am J Kidney Dis 2006;48:e27–e29 [DOI] [PubMed] [Google Scholar]
- 21.Davies DM, Johnson SR, Tattersfield AE, et al. Sirolimus therapy in tuberous sclerosis or sporadic lymphangioleiomyomatosis. N Engl J Med 2008;358:200–203 [DOI] [PubMed] [Google Scholar]
- 22.Bissler JJ, McCormack FX, Young LR, et al. Sirolimus for angiomyolipoma in tuberous sclerosis complex or lymphangioleiomyomatosis. N Engl J Med 2008;358:140–151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Davies DM, de Vries PJ, Johnson SR, et al. Sirolimus therapy for angiomyolipoma in tuberous sclerosis and sporadic lymphangioleiomyomatosis: a phase 2 trial. Clin Cancer Res 2011;17:4071–4081 [DOI] [PubMed] [Google Scholar]
- 24.Taille C, Debray MP, Crestani B. Sirolimus treatment for pulmonary lymphangioleiomyomatosis. Ann Intern Med 2007;146:687–688 [DOI] [PubMed] [Google Scholar]
- 25.Morton JM, McLean C, Booth SS, et al. Regression of pulmonary lymphangioleiomyomatosis (PLAM)-associated retroperitoneal angiomyolipoma post-lung transplantation with rapamycin treatment. J Heart Lung Transpl 2008;27:462–465 [DOI] [PubMed] [Google Scholar]
- 26.Hofbauer GF, Marcollo-Pini A, Corsenca A, et al. The mTOR inhibitor rapamycin significantly improves facial angiofibroma lesions in a patient with tuberous sclerosis. Br J Dermatol 2008;159:473–475 [DOI] [PubMed] [Google Scholar]
- 27.Agricola K, Tudor C, Krueger DA, Care M, Holland K. First approved targeted medical therapy for subependymal giant-cell astrocytoma in patients with tuberous sclerosis complex: strategies to improve tolerability and patient outcomes by effective management of side effects. Presented at the 43rd annual educational meeting, American Association of Neuroscience Nurses, March 19–22, 2011, Kansas City, MO
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.