

Abstract
Objective:
To review current approaches for obtaining patient data in Duchenne muscular dystrophy (DMD) and consider how monitoring and comparing outcome measures across DMD clinics could facilitate standardized and improved patient care.
Methods:
We reviewed annual standardized data from cystic fibrosis (CF) clinics and DMD care guidelines and consensus statements; compared current approaches to obtain DMD patient data and outcome measures; and considered the best method for implementing public reporting of outcomes, to drive improvements in health care delivery.
Results:
Current methods to monitor DMD patient information (MD STARnet, DuchenneConnect, and TREAT-NMD) do not yet provide patients with comparative outcome data. The CF patient registry allows for reporting of standard outcomes across clinics and is associated with improved CF outcomes. A similar patient registry is under development for the Muscular Dystrophy Association (MDA) clinic network. Suggested metrics for quality care include molecular diagnosis, ambulatory status and age at loss of ambulation, age requiring ventilator support, and survival.
Conclusions:
CF longevity has increased by almost 33% from 1986 to 2010, in part due to a CF patient registry that has been stratified by individual care centers since 1999, and publically available since 2006. Implementation of outcome reporting for MDA clinics might promote a similar benefit to patients with DMD.
The quality of health care delivery is likened to a bell-shaped curve, where for any sample population, in this case physician and disease outcomes, there is a small percentage of the population with excellent outcomes and a small percentage with poor outcomes. However, the majority of outcomes is in the middle and defined as average. This is considered a normal distribution and in such a case, the curve appears in the shape of a bell. For the rare disease cystic fibrosis (CF), monitoring and reporting patient data has been associated with shifting this curve to the right, reflecting improved CF outcomes across clinics.1 There is no similar approach for monitoring outcomes in Duchenne muscular dystrophy (DMD). Current DMD care standards have been developed based on randomized clinical trials and expert consensus. We hypothesize that monitoring patient outcomes will promote translation of these standards into routine DMD care, provide information about their effectiveness, and improve patient outcomes.
DMD is an X-linked disorder affecting 1.3–1.8 per 10,000 males aged 5–24 years in the United States.2 Although no cure exists for the progressive muscle weakness of DMD, corticosteroids are established to be an effective treatment for up to 18 months, to improve muscle strength and function, and in combination with supportive medical care are associated with prolonged survival.3 However, despite recent consensus standards, there continues to be great heterogeneity in delivery of DMD care.4–7
Global rare disease registries advance translational research by connecting patients with researchers.8 However, many registries focus on obtaining genetic data for clinical trials, without measuring outcomes.9 In this article, we review the registry model developed by the CF Foundation and compare it to current DMD networks monitoring patient data. We propose that a standardized nationwide registry of patients within specialized neuromuscular centers would provide a way to monitor clinical outcomes and to promote more standardized care. Data, which could be publically reported, would identify opportunities for improvement in delivery of care and development of future care standards.
METHODS
We reviewed the development, utilization, and data published annually by the CF patient registry, through review of CF literature and information provided on the CF Web site, www.cff.org. This includes the foundation's yearly data report (most recent 2010) and publically reported data available on the CF Web site from 2005 to 2010, which compares individual CF clinic outcomes to national averages and achievable goals, based on outcomes at the highest performing centers.10–14
We reviewed published guidelines and consensus statements regarding the care of patients with DMD. Evidence-based guidelines are limited to the use of corticosteroids, published by the American Academy of Neurology and The Cochrane Collaboration.15,16 Consensus guidelines include the recently published guidelines in 2010, by Bushby et al.,4,5 which review previous consensus statements as well as present new opinion on DMD care from various subspecialties.
We reviewed 3 current networks obtaining DMD patient data for monitoring and registry use. These networks, identified through discussion with leaders in the DMD community and Web searches, include the Muscular Dystrophy Surveillance Tracking and Research Network (MD STARnet), Translational Research in Europe for the Assessment and Treatment of Neuromuscular Disease (TREAT-NMD), and DuchenneConnect.org. These 3 networks were compared with the CF patient registry. A nationwide DMD patient registry utilizing specialized neuromuscular clinics, similar to the CF model, is under development.
RESULTS
The Cystic Fibrosis Foundation patient registry.
The Cystic Fibrosis Foundation (CFF) began utilizing its specialized CF care centers as the foundation for a quality improvement initiative in 2002, combining their patient registry to track clinic outcomes with public reporting of data, while training the clinical workforce in quality improvement methodologies and dissemination of best practices.10 Through this initiative, the median age of survival for patients with CF has increased from 27 years in 1986 to 38.3 years in 2010. Additionally, greater than 90% median forced expiratory volume in 1 second percent predicted, a major outcome for CF, has been sustained in patients to age 15 in 2010, vs less than 6 years in 1990.14
The CFF registry was initiated in 1966, to monitor trends in national mortality. In 1999, the registry transitioned to stratifying outcomes by individual care center, which identified variation between clinics in practice patterns and outcomes.11 To accelerate improvements in care, center-specific performance indicators were made public in 2006, starting with reporting of the previous year's data, 2005.12 There are currently 26 evidence-based CF care guidelines, covering 6 treatment categories.17 The remaining guidelines, 84, are based on consensus review and have evolved by benchmarking clinics with top outcomes.18 Improvement in outcomes, through benchmarking, has been documented independent of treatment advancements by randomized clinical trials.10
The CF patient registry is optional, but the majority of patients agree to participate to improve quality of life. Trained professionals enter patient information at each clinic visit via an online portal, documenting over 300 variables.12,14,19 Multiple steps are in place throughout the data entry process to ensure valid data.10 The online portal also provides patients and providers with access to care guidelines, clinical reminders, and patient alerts. The same care guidelines are available on the CF Web site for patients to discuss with their providers.17 CF care centers are accredited with an onsite visit every 5 years by one of the peers serving on the CF Foundation's oversight committee (Center Committee), ensuring clinics are following CF standards and maintaining the presence of a multidisciplinary team.11 Care centers also receive an extensive guide to help implement new practices based on the Dartmouth Microsystems approach.20
Individual centers receive a thorough annual report of their practice patterns and outcomes as compared to other centers across the country and risk-adjusted center-level data are published yearly on the CF Web site. National data are available online and published in a CFF annual data report mailed to patients and providers.14 Hard data are accessible to researchers who have a valid scientific question through a peer review process. As of 2008, over 80 articles had been published utilizing registry data.13 Additionally, with over 85% of patients with CF (26,000) followed within the 113 CF care centers, the CFF has established a large cohort of patients for clinical trials and postmarketing research, a current initiative.14,21
Overview of current DMD guidelines and tracking/registry networks.
Evidence-based guidelines for the treatment of DMD are limited to corticosteroid treatment, which is recommended for up to 18 months.3,15,16 Two recent consensus articles, which review a multitude of recommendations across specialties, make specific recommendations concerning DMD care, including diagnostic, gastrointestinal and nutrition, rehabilitation, neuromuscular, psychosocial, cardiovascular, respiratory, orthopedic, and surgical aspects of DMD.4,5 However, there remains no quality standard to assess optimal delivery of care in neuromuscular clinics, nor are patients aware of what outcomes their provider should be meeting. There is evidence that improvements in care enable patients to live into their 30s vs a median age of survival of 14.4 years in the 1960s, but no systematic effort to measure and monitor improved outcomes has been implemented, to date.22,23 Three current efforts for obtaining and monitoring DMD data are reviewed: MD STARnet, TREAT-NMD, and DuchenneConnect.org. The table summarizes these networks and compares them to the CF registry and a Muscular Dystrophy Association (MDA) registry under development.
Table.
Comparison of registry/network data reporting
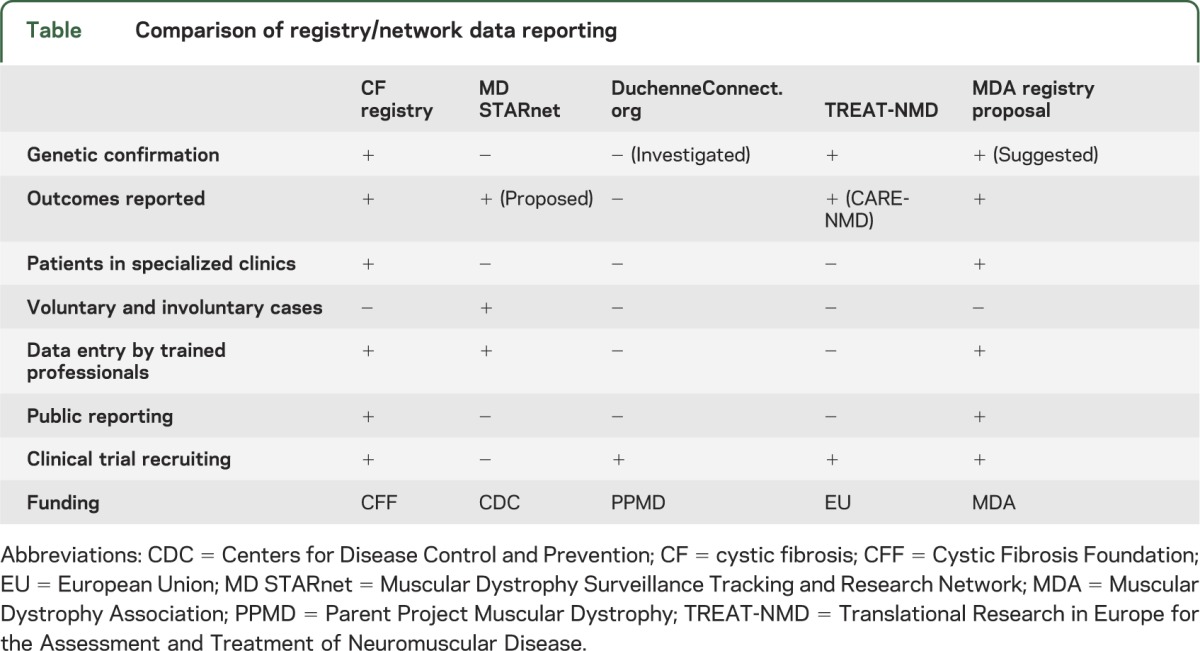
MD STARnet.
MD STARnet, founded in 2002, is a program established by the Centers for Disease Control and Prevention to identify all individuals with Duchenne and Becker muscular dystrophy (DBMD) in Iowa, Colorado, Arizona, Georgia, Hawaii, and 12 counties in western New York State. The network gathers information for all patients with DBMD born on or after January 1, 1982, in the defined areas. Data are used to determine prevalence and information about diagnosis, treatment, and disease course. Trained abstractors, who have a medical background, identify DBMD cases and abstract information from birth certificates, hospital medical records, neuromuscular and neurology clinics, and self-reports from families. Data are abstracted annually, and supplemented with patient or caregiver telephone interviews or mailed surveys. The MD STARnet Data Coordination Center at the University of Iowa maintains the data. All potential cases go through a clinical review committee, composed of neuromuscular experts from each participating site, to determine the confidence of DBMD diagnosis: definite, probable, possible, asymptomatic, or female. Data reviewed include age at first symptoms, serum creatine kinase, genetic mutation, muscle biopsy results, and family history. Only definite and probable patients are used in analysis, which may include patients without confirmatory genetic testing.24,25 Data are available for analysis to researchers within MD STARnet only, but public release is planned. Monitoring and reporting outcomes within the group is also a goal of the project. A strength, compared to other registries, is that MD STARnet information is collected through surveillance methodology rather than patient self-report. Data, however, are retrospective and limited to the defined geographic locations.
TREAT-NMD.
TREAT-NMD is a European initiative, founded in 2007, to connect patients to clinical trials for rare neuromuscular diseases, using a collaboration of international patient registries. An initial focus has been on DMD and spinal muscular atrophy.26 There are currently 37 national registries and over 9,000 registrants with DMD; however, the majority of registries are run by outside organizations and data are linked to a global TREAT-NMD patient database.26,27 Linked registries must adhere to TREAT-NMD standards and document a core dataset that includes the following: molecular data, diagnosis, motor function, steroid use, scoliosis surgery, cardiac medications, and enrollment in clinical trials. Additional information is suggested, but not required.27 Only patients with a confirmed or pending diagnosis of DMD by genetic testing are allowed in the registry, with all testing confirmed by a curator.27,28 There are no restrictions on who can enter data, but data must be updated yearly. Baseline demographic and background data are aggregated as an international cohort and graphed on the TREAT-NMD Web site.27 Third parties may use data for studies if approved by the institutional review board and the TREAT-NMD international oversight committee.26 CARE-NMD, a subset of TREAT-NMD, is a separate collaboration of 7 European countries, launched in 2008, to monitor implementation of DMD consensus care standards, measure patient quality of life, identify inequalities between the participating sites, and provide training workshops on dissemination of care standards. These data are submitted by patients online or through mailed surveys.29
DuchenneConnect.org.
DuchenneConnect, founded in 2007, is an initiative designed to connect patients to experts in the field of DBMD, utilizing an online registry, educational material, and access to clinical trials. The online patient registry is linked to the global TREAT-NMD registry. Registry participation is voluntary and open to males with DBMD and female carriers, who submit their information directly onto the DuchenneConnect Web site. Coordinators curate the data and assist with participation. The registry is open to patients outside of the United States and includes, as of June 2011, 1,756 registrants from 78 countries. A total of 78% (1,369) of the patients report a diagnosis of DMD, with 70% from the United States. A total of 7% have Becker muscular dystrophy and the remaining 15% are carriers, possible carriers, intermediate, or no identification. Data recorded include community affiliation, genetic mutation diagnosis, as well as pulmonary, cardiac, and ambulating status. A genetic profile is not required for participation, but registrants without genetic testing are not connected to the global TREAT-NMD registry. A total of 86% of patients in the registry report having genetic testing, but as of June 2011 only 47% have submitted a copy of the results. In addition to an online registry, registrants and clinics have the option of completing a survey about their specialized clinic. Only 3 clinics and 27 registrants have submitted surveys, to date. Study investigators or clinicians can search the database for potential clinical trial participants, but only affiliated providers may review registry data. Basic analyses of the cohort demographics are reported publicly on the Web site and published.30,31
A proposed MDA patient registry.
MDA is a nonprofit health agency founded in 1950, as a collaborative effort between adult patients, families, providers, and researchers. The association funds worldwide research, and also provides patients and families with comprehensive health care and support services, and advocacy and education programs. There are some 200 MDA-funded clinics in the United States and Puerto Rico, providing services for patients with 43 neuromuscular disorders. MDA has long promoted multidisciplinary care for individuals with neuromuscular diseases, and similar to CF clinics, many MDA clinics are staffed by teams of health experts, including physician specialists as well as genetic counselors and occupational, physical, speech, and respiratory therapists.32 Some MDA clinics, particularly smaller clinics in less populated areas, utilize a referral system for provision of specialty care. MDA also shares published clinical care guidelines with physicians and families via electronic and print communications and by posting on its Web site.33 However, it is not fully understood how variations in delivery of care—i.e., which experts are available in clinic, whether care guidelines are followed, what other interventions are provided, and qualitative aspects of care such as aggressiveness, consistency, and ingenuity—impact outcomes for those with DMD.
MDA maintains an internal, noncurated database of individuals registered with the association, which includes physician-provided diagnoses but no other clinical information. Currently there are more than 11,000 individuals with DBMD in the MDA database. Individual MDA clinics often track patient data or initiate retrospective studies, but no standard data collection or Web-based reporting currently exists across clinics. Transitioning to a DMD nationwide patient registry that utilizes MDA neuromuscular specialty clinics, similar to the CFF registry, will allow for collection of prospective, longitudinal data on large numbers of individuals with DMD in different geographic and clinical settings. A Web-based MDA registry, with reporting by trained professionals, can be used to track adherence to care guidelines and which interventions most impact outcomes, as well as identify eligibility for clinical trials. Public reporting of clinic and clinical outcomes, for physicians and patients to review, will allow for benchmarking of best practices and lead to overall improved outcomes for those with DBMD as well as provide validation for standardization of a multidisciplinary approach to care across MDA clinics. Simple metrics to monitor that would reflect quality of care include molecular diagnosis, ambulatory status and age at loss of ambulation, forced vital capacity and age requiring ventilator support, and survival. Other considerations include dose and age at initiation of corticosteroids, cardiac measures, and bone fractures. MDA is currently working with its Clinical Advisory Committee and Registry Advisory Board to define outcomes that will be measured in the neuromuscular registry. Additionally, recording zip code could permit linkage to socioeconomic data through area level census measures of socioeconomic status (SES). This could help delineate where there is a need for additional clinics and what effect SES has on patients receiving care and follow-up. MDA registry modules for amyotrophic lateral sclerosis and spinal muscular atrophy are also under development; eventually modules for all neuromuscular diseases within MDA's program will be implemented. A patient portal will examine issues such as quality of life, SES, and caregiver burden.
Limitations to this proposal.
The suggested proposal may be met with some resistance, as was initial public reporting of the CF registry outcomes. If significant variations in outcomes are found among clinics, a period of anonymity could provide clinics time to improve their individual outcomes against the national average. The 4 metrics to monitor were suggested because they are relatively easy to document. However, even these variables may require refinement. For example, the use of a wheelchair may provide increased mobility, but decrease the age of ambulation, so defining this variable as the complete loss of ambulation with wheelchair dependence may be required to allow for a transition period that many patients have when they are only partially wheelchair dependent. The CFF monitors over 300 variables, and it would be feasible to increase the suggested variables for the DMD registry. However, initially asking for extensive data entry could overburden clinic staff.
There are other potential limitations to this approach including funding, maintaining commitment from individual clinics, and staffing/standardizing both private facilities and larger institutions. The MDA is currently developing a Web-based system for clinicians to enter data into the MDA registry, beginning with a small subset of the clinics before branching out to all MDA clinics across the United States and Puerto Rico. In terms of the limitation of many small facilities vs academic institutions, this is seen also in the CF community, and the CFF has adapted their model to meet the needs of patients at private facilities and larger clinics. The CFF has 58 smaller satellite clinics; however, they have affiliated all of these clinics with their 113 larger institution clinics, which combined with the affiliated satellite clinic must provide adherence to the CFF accreditation process.14
Finally, arguments can be made that the CF registry has been successful because there is less racial and ethnic variability than in DMD. CF is primarily a Caucasian disease, 94.3%, compared to 53.4% of patients with DMD. This is important because in a retrospective study, it was shown that for black males with muscular dystrophies (DMD, BMD, limb girdle, and facioscapulohumeral MD, and other less common dystrophies), the median age at death was 10 years younger than in their white counterparts (23 years vs 33 years), with a greater increase in age at death over time in white males.14,34 The factors contributing to this difference, however, are not known and may be a combination of factors (type of MD, environmental factors, socioeconomic factors, access to care centers and treatment, or natural history/genetic factors). Despite differences in race and ethnicity between CF and DMD, there are large percentages of patients with both diseases who live below the poverty line and utilize Medicaid or other state health insurance programs. However, in a recent MD STARnet publication, poorer patients with DMD had earlier evaluations, a contradiction to a general belief regarding poverty gradients.35 Thus, more data will be useful to determine effect of race, ethnicity, and other socioeconomic factors on delivery of care and patient outcomes, which the described DMD registry could help delineate. With these data, the MDA and specialists can make adjustments to clinic locations, identify patients earlier, monitor for comorbidities, and minimize social barriers.
DISCUSSION
Patient registries and public reporting of outcomes is not limited to neuromuscular diseases. There is potential to use similar registries to improve patient outcomes for both common and rare diseases, whether or not there are well-developed evidence-based standards of care. However, for such registries to be effective, an infrastructure of specialized clinics such as with CFF or MDA should be in place, with documentation of variations in practice patterns. For those diseases, improving patient outcomes with this model is possible, as you can only begin to improve care when it is measured.
GLOSSARY
- CF
cystic fibrosis
- CFF
Cystic Fibrosis Foundation
- DBMD
Duchenne and Becker muscular dystrophy
- DMD
Duchenne muscular dystrophy
- MD STARnet
Muscular Dystrophy Surveillance Tracking and Research Network
- MDA
Muscular Dystrophy Association
- SES
socioeconomic status
- TREAT-NMD
Translational Research in Europe for the Assessment and Treatment of Neuromuscular Disease
AUTHOR CONTRIBUTIONS
M.A. Scully: drafting/revising the manuscript for content, including medical writing for content, acquisition of data, conceptualization of the study. V.A. Cwik: revising the manuscript for content, including medical writing for content, conceptualization of the study. B.C. Marshall: revising the manuscript for content, including medical writing for content, acquisition of data. E. Ciafaloni: revising the manuscript for content, including medical writing for content. J.M. Wolff: revising the manuscript for content, including medical writing for content. T. Getchius: revising the manuscript for content, including medical writing for content. R.C. Griggs: drafting/revising the manuscript for content, including medical writing for content, acquisition of data, conceptualization of the study.
STUDY FUNDING
No targeted funding reported.
DISCLOSURE
M.A. Scully reports no disclosures. V.A. Cwik is Interim President and Research and Medical Director for the Muscular Dystrophy Association (MDA). B.C. Marshall is the Vice President of clinical affairs for the Cystic Fibrosis Foundation. E. Ciafaloni is a clinical consultant for MD STARnet. J.M. Wolff reports no disclosures. T. Getchius is a full-time employee of the American Academy of Neurology (AAN) and is the Project Director of the CDC-RFA-DD10-1012 award. R.C. Griggs holds NIH funding for a multicenter trial of corticosteroids in DMD and for a multicenter clinical trial of dichlorphenamide in periodic paralysis. He served as a consultant for PTC Therapeutics (as Chair of the DSMB for a study of PTC 124 in DMD). His institution receives support from Taro Pharmaceuticals for a clinical trial in periodic paralysis. He chairs the Executive Committee of the Muscle Study Group, which receives support from pharmaceutical companies, the MDA, the NIH, and the US FDA. He receives research support from the MDA. He does not receive personal compensation from any of the foregoing grants. He is co-lead of the AAN and American Association of Neuromuscular & Electrodiagnostic Medicine effort to develop, disseminate, and implement the recommendations from 4 guidelines on muscular dystrophy. The Centers for Disease Control and Prevention funds this project under CDC-RFA-DD10-1012. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Gawande A. The bell curve. In: Better: A Surgeon's Notes on Performance. New York: Metropolitan Books; 2007:201–230. [Google Scholar]
- 2.Center for Disease Control and Prevention (CDC). Prevalence of Duchenne/Becker muscular dystrophy among males aged 5-24 years: four states, 2007. MMWR Morb Mortal Wkly Rep 2009;58:1119–1122. [PubMed] [Google Scholar]
- 3.Fenichel GM, Florence JM, Pestronk A, et al. Long-term benefit from prednisone therapy in Duchenne muscular dystrophy. Neurology 1991;41:1874–1877. [DOI] [PubMed] [Google Scholar]
- 4.Bushby K, Finkel R, Birnkrant DJ, et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and pharmacological and psychosocial management. Lancet Neurol 2010;9;77–93. [DOI] [PubMed] [Google Scholar]
- 5.Bushby K, Finkel R, Birnkrant DJ, et al. Diagnosis and management of Duchenne muscular dystrophy, part 2: implementation of multidisciplinary care. Lancet Neurol 2010;9:177–189. [DOI] [PubMed] [Google Scholar]
- 6.Aymé S, Rodwell C. 2011 Report on the state of the art of rare disease activities in Europe of the European Union committee of experts on rare diseases: part 1: overview of rare disease activities in Europe and key developments in 2010, July 2011. Available at: http://www.eucerd.eu/upload/file/Reports/2011ReportStateofArtRDActivities.pdf. Accessed February 7, 2012.
- 7.Matthews DJ, James KA, Miller LA, et al. Use of corticosteroids in a population based cohort of boys with Duchenne and Becker muscular dystrophy. J Child Neurol 2010;25:1319–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Forrest CB, Bartek RJ, Rubinstein Y, Groft SC. The case for a global rare-diseases registry. Lancet 2011;377:1057–1059. [DOI] [PubMed] [Google Scholar]
- 9.Hilbert JE, Kissel JT, Luebbe EA, et al. If you build a rare disease registry, will they enroll and will they use it? Methods and data from the National Registry of Myotonic Dystrophy (DM) and Facioscapulohumeral Muscular Dystrophy (FSHD). Contemp Clin Trials 2012;33:302–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schechter MS, Gutierrez HH. Improving the quality of care for patients with cystic fibrosis. Curr Opin Pediatr 2010;22:296–301. [DOI] [PubMed] [Google Scholar]
- 11.Schechter MS, Margolis P. Improving subspecialty healthcare: lessons from cystic fibrosis. J Pediatr 2005;147:295–301. [DOI] [PubMed] [Google Scholar]
- 12.Quon BS, Goss C. A story of success: continuous quality improvement in cystic fibrosis care in the USA. Thorax 2011;66:1106–1108. [DOI] [PubMed] [Google Scholar]
- 13.Schechter MS. Patient registry analyses: seize the data, but caveat lector. J Pediatr 2008;153:733–755. [DOI] [PubMed] [Google Scholar]
- 14.Cystic Fibrosis Foundation Patent Registry. Annual data report 2010. Available at: http://www.cff.org/UploadedFiles/LivingWithCF/CareCenterNetwork/PatientRegistry/2010-Patient-Registry-Report.pdf. Accessed February 7, 2012.
- 15.Manzur AY, Kuntzer T, Pike M, Swan A. Glucocorticoid corticosteroids for Duchenne muscular dystrophy. Cochrane Database Syst Rev 2008;1:CD003725. [DOI] [PubMed] [Google Scholar]
- 16.Moxley RT, III, Ashwal S, Pandya S, et al. Practice parameter: corticosteroid treatment of Duchenne dystrophy: report of the Quality Standards Subcommittee of the American Academy of Neurology and the Practice Committee of the Child Neurology Society. Neurology 2005;64:13–20. [DOI] [PubMed] [Google Scholar]
- 17.Robinson KA, Saldanha IJ, Mckoy NA. Identification of research gaps from evidence-based guidelines: a pilot study in cystic fibrosis. Int J Technol Asses 2011;27:247–252. [DOI] [PubMed] [Google Scholar]
- 18.Cystic Fibrosis Care Guidelines. Available at: http://www.cff.org/treatments/CFCareGuidelines/. Accessed February 7, 2012. [Google Scholar]
- 19.Quittner AL, Buu A, Messer MA, Modi AC, Watrous M. Development and validation of The Cystic Fibrosis Questionnaire in the United States: a health related quality-of-life measure for cystic fibrosis. Chest 2005;128:2347–2354. [DOI] [PubMed] [Google Scholar]
- 20.Action Guide for Accelerating Improvement in Cystic Fibrosis Care. Available at: http://www.clinicalmicrosystem.org/assets/materials/workbooks/cystic_fibrosis_action_guide.pdf. Accessed February 7, 2012. [Google Scholar]
- 21.Cystic Fibrosis Foundation. 2010 annual report. Available at: http://www.cff.org/UploadedFiles/aboutCFFoundation/AnnualReport/2010-Annual-Report.pdf. Accessed February 7, 2012.
- 22.Eagle M, Bourke J, Bullock R, et al. Managing Duchenne muscular dystrophy: the additive effect of spinal surgery and home nocturnal ventilation in improving survival. Neuromuscul Disord 2007;17:470–475. [DOI] [PubMed] [Google Scholar]
- 23.Eagle M, Baudouin SV, Chandler C, et al. Survival in Duchenne muscular dystrophy: improvements in life expectancy since 1967 and the impact of home nocturnal ventilation. Neuromuscul Disord 2002;12:926–929. [DOI] [PubMed] [Google Scholar]
- 24.Mathews KD, Cunniff C, Kantamneni JR, et al. Muscular Dystrophy Surveillance Tracking and Research Network (MD STARnet): case definitions in surveillance for childhood-onset Duchenne/Becker muscular dystrophy. J Child Neurol 2010;25:1089–1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Miller LA, Romitti PA, Cunniff C, et al. The Muscular Dystrophy Surveillance Tracking and Research Network (MD STARnet): surveillance methodology: birth defects research (part A). Clin Mol Technol 2006;76:793–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bushby K, Lynn S, Straub V. Collaborating to bring new therapies to the patient: the TREAT-NMD model. Acta Myo 2009;28:12–15. [PMC free article] [PubMed] [Google Scholar]
- 27.Charter for TREAT-NMD patient database/registry. Available at: http://www.treat-nmd.eu/downloads/file/registries_toolkit/charter_treat-nmd_global_database_approved.pdf. Accessed February 7, 2012.
- 28.Abbs S, Flanigan K, Tuffery-Giraud S. Mutation Entries in DMD Databases: A Handbook for National Curators. Available at: http://www.treat-nmd.eu/downloads/file/registries_toolkit/handbook_on_dmd_genetics_jan2009.pdf. Accessed February 7, 2012. [Google Scholar]
- 29.CARE-NMD. Available at: http://en.care-nmd.eu/. Accessed February 7, 2012.
- 30.DuchenneConnect. Available at: https://www.duchenneconnect.org/. Accessed February 7, 2012.
- 31.Rangel V, Martin AS, Peay HL. DuchenneConnect registry report [Internet]. Version 17. PLoS currents: Muscular Dystrophy. Available at: http://knol.google.com/k/vanessa-rangel/duchenneconnect-registry-report/3c8kwqirmpdbh/3. Accessed March 30, 2012. [DOI] [PMC free article] [PubMed]
- 32.Muscular Dystrophy Association. Available at: http://www.mdausa.org/. Accessed March 30, 2012.
- 33.Muscular Dystrophy Association Clinical Conference Information. Available at: http://www.mdausa.org/research/national-conferences/2012_clinical_conference/information.htm. Accessed on March 30, 2012.
- 34.Kenneson A, Vatave A, Finkel R. Widening gap in age at muscular dystrophy–associated death between blacks and whites, 1986–2005. Neurology 2010;75:982–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Holtzer C, Meaney FJ, Andrews J, et al. Disparities in the diagnostic process of Duchenne and Becker muscular dystrophy. Genet Med 2011;13:942–947. [DOI] [PubMed] [Google Scholar]