

Abstract
Tracking a bug’s life: We describe the first direct and universal approach for labeling peptidoglycan (PG) of diverse bacteria by exploiting the surprising tolerance of cells for incorporating unnatural D-amino acids of various sizes and functionalities. These non-toxic D-amino acids preferably label the sites of active PG synthesis, enabling fine spatiotemporal tracking of cell wall dynamics in phylogenetically and morphologically diverse bacteria.
Keywords: bacteria, biosensors, D-amino acids, fluorescent probes, peptidoglycan
Bacterial growth is controlled by the domain-specific peptidoglycan (PG) cell wall, a rigid and essential structure composed of glycan strands cross-linked by D-amino acid (DAA)-containing short peptides, whose biosynthesis machinery is a high-value target for antibiotics.[1] Despite the incredible importance of this megapolymer, our knowledge of PG dynamics has been severely hampered by the lack of a global strategy for direct imaging of sites of PG synthesis in live cells. Significant limitations of current labeling methods, such as toxic effects and poor membrane permeability of the probes, have limited their applicability to only a small set of bacterial species.[2] Commonly, these methods are labor-intensive and their sensitivity suffers from their indirect and multiple-step nature. To overcome these limitations, we sought a chemical biology approach that would enable the rapid and covalent incorporation and detection of a fluorescently derivatized PG component during cell wall synthesis in real time, in a wide range of bacterial species and in live cells.
Given the recently established role of PG biosynthetic machinery in incorporating various natural DAAs in the PG of diverse bacteria, [3] we hypothesized that the mechanisms[3b] for DAA incorporation in PG are common to the bacterial domain, but still highly selective for the D-center, and that molecules with various functionalities could be incorporated into the sites of new PG synthesis if attached to a DAA backbone. To test this hypothesis, we attached the relatively small fluorophores, 7-hydroxycoumarin 3-carboxylic acid (HCC-OH) and 4-chloro-7-nitrobenzofurazan (NBD-Cl), to a D-amino acid backbone, 3-amino-D-alanine, via a modular and simple synthetic scheme, generating HADA (emission maximum 450 nm, blue) and NADA (emission maximum 538 nm, green) (Scheme 1 and Figure S2).
Scheme 1.

Modular structures of the fluorescent D-amino acids HADA, NADA, FDL and TDL and the ‘clickable’ D-amino acids EDA & ADA, color coded by the color of the fluorophores (blue, green and red).
Growth of the phylogenetically diverse model species Escherichia coli, Agrobacterium tumefaciens and Bacillus subtilis in the presence of these fluorescent D-amino acids (FDAAs) for as little as one generation (Figure S1a) resulted in strong peripheral and septal labeling of entire cell populations (Figures 1 and S3a, upper panels) without affecting growth rate (Figure S3b). Neither of the fluorescent L-amino acids (FLAAs, prepared from 3-amino-L-alanine, Figure S2c) resulted in significant labeling (Figure S3c), indicating that labeling is specific to the D-enantiomers. The labeling was exclusive to viable cells treated with the FDAAs and was not the result of non-specific interaction of FDAAs with the PG (Figure S4). Additionally, incorporation did not occur into teichoic acids for B. subtilis as indicated by identical labeling of wild-type and a ΔdltA mutant that does not D-alanylate its teichoic acids (Figure S5a–b).
Figure 1.
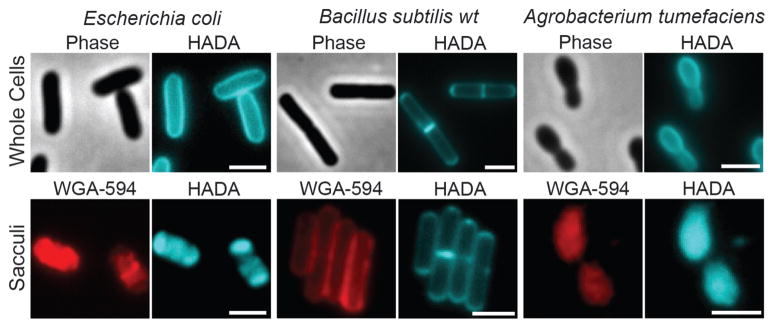
Long labeling pulses with HADA uniformly label PG in live cells of E. coli, B. subtilis and A. tumefaciens. The FDAA fluorescence is retained in isolated sacculi, which are also stained with the N-acetylglucosamine-specific WGA lectin conjugated to Alexa Fluor 594 (red). Scale bars, 2 μm.
Retained fluorescence on the purified sacculi (Figures 1 and S3a, lower panels) demonstrated that the labeling of PG by the FDAAs was covalent. HPLC analyses of muropeptides isolated from labeled cells (Figures 2a–c, S6 and S7) revealed that 0.2% – 2.8% of total muropeptides were modified (Figure 2c), which is sufficient for detection in various experiments while avoiding possible toxicity issues that could result from abundant incorporation. Significantly, the FDAA-specific peaks, which were absent in samples treated with FLAAs, could be distinguished from unlabeled muropeptides at FDAA-specific absorption wavelengths (Figure 2b). MS/MS analyses of FDAA-modified muropeptides in B. subtilis indicated that FDAAs were exclusively incorporated in the fifth position of the stem peptide (Figures 2d, S7a, d and Table S1). Interestingly, in a ΔdacA mutant of B. subtilis, the fraction of labeled muropeptides and the fluorescent signal were substantially higher than in wild-type B. subtilis, which is likely due to the D,D-carboxypeptidase activity of DacA (Figure S5c–d). In contrast, the detectable incorporation was solely at the fourth position in E. coli and A. tumefaciens (Figures 2d and S7). These results are in agreement with the known sites of incorporation of various natural DAAs in these species and suggest that, similar to DAAs, FDAAs incorporate mainly through periplasmic exchange reactions with the muropeptides catalyzed either by D,D-transpeptidases, e.g. in B. subtilis, or by L,D-transpeptidases, e.g. in E. coli and A. tumefaciens.[3b, 4] This DAA-like behavior together with the ease of fluorescent detection make FDAAs a strong alternative to radioactive probes for studying in vitro[3c] and in vivo[5] activities of PG synthesis enzymes.
Figure 2.

FDAA incorporation into the stem peptide of the PG subunit. a) Schematic representing the muramylpentapeptide precursor unit. b) HPLC detection of modified muropeptides in E. coli incubated with HADA or HALA and NADA or NALA. Samples were monitored using a dual wavelength UV monitor set for general muropeptide detection and for FDAA specific wavelengths. Peaks HEC-1 and NEC-1 correspond to the HADA or NADA modified muropeptides in E. coli that were further characterized by electrospray ionization MS/MS (ESI-MS/MS). c) Percentage of FDAA incorporation into the total muropeptides varies among different bacteria as revealed by HPLC analysis. d) MS/MS analyses show that FDAAs exclusively incorporate into the 4th position of muropeptides in E. coli and A. tumefaciens and the 5th position in B. subtilis. See also Figures S6–7 and Table S1.
In contrast to current approaches, [2b, 2f] pulse-chase experiments with HADA allowed the real-time tracking of new PG incorporation during growth via time-lapse microscopy. In E. coli and B. subtilis ΔdacA (Figure 3a and Supplementary Movies 1–2), the polar caps retained the HADA signal, but the signal from the lateral walls dispersed as the cells grew, in agreement with previous reports of cell wall growth along the length of the lateral walls. [2f, 5]
Figure 3.

FDAAs label diverse bacterial growth patterns. a) Time-lapse microscopy of HADA labeled E. coli and B. subtilis ΔdacA cells imaged during growth on LB agarose pads. Superresolution microscopy of b) E. coli and c) A. tumefaciens after short pulses with HADA. d) Superresolution microscopy of S. aureus cells after a short pulse with HADA. Autofluorescence is shown in red. e) Triple labeling of A. tumefaciens with HADA (blue), EDA (clicked with red sulfo-Cy3-azide), and NADA (green). f) Triple labeling of S. venezuelae with NADA (green), TDL (red), and HADA (blue). Arrows in the triple labeling panels indicate the sequence of labeling. White scale bars, 2 μm; red scale bars, 1 μm.
Strikingly, short labeling times (2% – 8% of doubling time) using E. coli and B. subtilis ΔdacA with HADA resulted in preferential localization of the signal at the septal plane of predivisional cells and in punctate patterns on the lateral walls of elongating cells (Figure 4). Super-resolution microscopy of E. coli revealed reticulated hoop-like patterns of HADA labeling around the lateral wall (Figure 3b & Supplementary Movie 3), supportive of bursts of PG incorporation in the side-walls.[6] This ability of FDAAs to resolve insertion of new PG provides the first direct detection of zones of PG synthesis in a structured rather than a random pattern in E. coli, consistent with recent results following the movement of the cell wall elongation machinery.[7] Short labeling times with A. tumefaciens, whose growth occurs predominantly from a single pole and the site of cell division while the mother cell remains inert, [4] resulted in polar and septal labeling. Super-resolution fluorescence microscopy of labeled cells further enhanced the spatial resolution of the site of active PG synthesis (Figure 3c & Supplementary Movie 4).
Figure 4.

Short pulses of HADA label distinct modes of growth in diverse bacteria. Strains were labeled for ~2–8% of the doubling time: E. coli (30 s), A. tumefaciens (2 min), B. subtilis ΔdacA (30 s), S. aureus (2 min), L. lactis (2 min), S. pneumoniae (4 min), C. crescentus (5 min), Synechocystis sp. PCC 6803 (1 h), S. venezuelae (2 min), B. conglomeratum (8 min), B. phytofirmans (20 min), V. Spinosum (10 min). Scale bars, 2 μm.
PG labeling in three evolutionarily distant species suggested that FDAAs could specifically label the active site of PG synthesis across the entire bacterial domain. When species representing diverse phyla and modes of growth were briefly incubated with FDAAs, we observed strong labeling at the sites of cell division in actively dividing cells (Figure 4 and Figure S1b). This septal probe incorporation was the sole mode in Synechocystis sp. PCC 6803, Lactococcus lactis, and Staphylococcus aureus as previously described.[8] Super-resolution microscopy of Staphylococcus aureus further highlighted the different stages of these constricting septal rings (Figure 3d and Supplementary Movie 5). Labeling of Streptococcus pneumoniae occurred in single or split equatorial rings depending on the length of the cell, with peripheral labeling between the split rings as described.[8] Labeling of Streptomyces venezuelae was predominantly apical as reported, [9] with some weak labeling of vegetative septa and lateral walls suggestive of a low but continuous lateral PG synthesis. In Caulobacter crescentus, labeling occurred at the sites of septal elongation, lateral elongation, and stalk synthesis, as reported elsewhere.[10]
Bacteria exhibit a myriad of growth patterns that provide selective advantages in the environment.[11] The strong correlation between FDAA labeling and previously inferred regions of new PG synthesis in diverse model species has a number of important implications. First, FDAA labeling marks the sites of active PG synthesis and therefore provides a long sought broadly applicable tool to study the spatial dynamics of PG synthesis. Second, these results establish for the first time that the enzymes responsible for DAA incorporation in the PG are associated with active growth sites and that not all the surface of the sacculus is equivalent in terms of accessibility to these enzymes. Finally, FDAAs can be used to discover the growth modes of previously uncharacterized taxa. For example, our results show that that Burkholderia phytofirmans exhibits polar and midcell PG synthesis, that Brachybacterium conglomeratum exhibits prominent peripheral PG synthesis in addition to seemingly alternating perpendicular division planes, and that Verrucomicrobium spinosum exhibits strong peripheral PG synthesis and asymmetric septal labeling (Figure 4).
The efficient label incorporation in all the bacteria studied thus far also suggests that FDAA, therefore DAA, incorporation is common to the bacterial domain and FDAAs can thus be used to analyze natural bacterial populations, providing a convenient and quick standard to measure bacterial activity and to probe the diversity of growth modes in complex microbiomes.[11] Indeed, labeling times with FDAAs as short as 2 hours revealed diverse modes of growth in saliva and freshwater samples in situ, but did not label dead cells as suggested by the strong correlation with Live-Dead staining (Figures S4b and S8).
Encouraged by the efficiency of FDAAs, we sought to increase our toolkit for PG detection and modification using differently functionalized unnatural D-amino acids. Following a similar approach, we derivatized the brighter and more versatile core fluorophore, fluorescein (emission maximum ~515 nm, green), and its analogue, carboxytetramethylrhodamine (TAMRA, emission maximum ~565 nm, red), [12] with D-lysine in order to separate the bulky fluorophore from the DAA backbone, generating FDL and TDL (Scheme 1 and Figure S2a). Incubation of both E. coli and B. subtilis cells with FDL (536 Da) showed patterns similar to NADA, although labeling of B. subtilis was stronger than E. coli, presumably due to reduced permeability of the E. coli outer-membrane to molecules larger than ~500 Da[13] (Figure S9a–c). Indeed, the larger TDL (560 Da) did not label E. coli cells, but labeling of B. subtilis was prominent and showed patterns similar to other FDAAs (Figure S9a–b). In order to expand the toolkit further, we used ‘clickable’ D-amino acids, namely ethynyl-D-alanine (EDA) or azido-D-alanine (ADA) (Scheme 1) that can be specifically captured by any molecule carrying the conjugate functional group via click-chemistry.[14] Similar to FDAAs, these bioorthogonal DAAs, but not the L-enantiomer control ELA, labeled both E. coli and B. subtilis cells when captured by commercially available azido/alkyne fluorophores (Figures S2c–d and S9d–e).
Furthermore, custom D-amino acids containing different colored fluorophores can be used sequentially to enable “virtual time-lapse microscopy.” Since addition of each new probe indicates the location and extent of PG synthetic activity during the respective labeling periods, this approach provides a chronological account of shifts in PG synthesis of individual cells over time. This powerful method is exemplified here through click chemistry in gram-negative A. tumefaciens (Figures S1a and 3e), or by the use of TDL in gram-positive S. venezuelae (Figure 3f).
In contrast to the bacterial protein-making machinery’s inherent bias against unnatural amino acids, [15] here we have shown that taxonomically diverse bacteria display a remarkable specific tolerance for incorporating D-amino acids with different sizes and functionalities into PG. Exploiting this tolerance provides a rapid, non-toxic, and universal method for real time tracking of PG at sites of active synthesis for the first time. Moreover, the combined utilization of two or more probes permits a temporal resolution never before achieved in cell wall growth studies. We expect that in combination with fluorescent protein fusions, mutational analysis, and chemical perturbations, this methodology will allow a comprehensive analysis of the regulation and coordination of bacterial growth. Furthermore, the tolerance for DAAs with substantial sizes or with bio-orthogonal handles will enable selective and specific modification of bacterial cell surfaces with various functionalities paving the way for development of DAA-based bacteria-specific diagnostic or therapeutic probes. Finally, when cells labeled with FDAAs are hybridized with fluorescent in situ hybridization (FISH) probes, this approach will allow the culture-independent and concurrent measurement of growth (with FDAAs) and taxonomy (with FISH) and the response of bacteria to varying conditions in medical or environmental microbiomes.
Supplementary Material
Footnotes
We thank David Kysela for discussions and critical reading of the manuscript, Bill Turner and Alvin Kalinda for their help in simplifying the synthesis of FDAAs and Professor Patricia Foster and the members of her laboratory for their support during the initial stages of the project. MSV was funded by National Institutes of Health (NIH) grant AI 059327, YVB by NIH grant GM051986, MAP by Ministry of Education and Science (Spain) grant BFU2006-04574 and by the Fundación Ramón Areces, FC by MICINN (Spain) grant RYC-2010-06241 and P.J.B. by NIH National Research Service Award AI072992.
Supporting information for this article is available on the WWW under http://www.angewandte.org.
Contributor Information
Mr. Erkin Kuru, Indiana University, Bloomington, IN 47405 (USA)
Ms. H. Velocity Hughes, Indiana University, Bloomington, IN 47405 (USA)
Dr. Pamela J. Brown, Indiana University, Bloomington, IN 47405 (USA)
Mr. Edward Hall, Indiana University, Bloomington, IN 47405 (USA)
Mr. Srinivas Tekkam, Indiana University, Bloomington, IN 47405 (USA)
Prof. Felipe Cava, Universidad Autonoma de Madrid, Campus de Cantoblanco Madrid, 28049 (Spain)
Prof. Miguel A. de Pedro, Universidad Autonoma de Madrid, Campus de Cantoblanco Madrid, 28049 (Spain)
Prof. Yves V. Brun, Email: ybrun@indiana.edu, Indiana University, Bloomington, IN 47405 (USA)
Prof. Michael S. VanNieuwenhze, Email: mvannieu@indiana.edu, Indiana University, Bloomington, IN 47405 (USA)
References
- 1.Typas A, Banzhaf M, Gross CA, Vollmer W. Nat Rev Microbiol. 2012;10:123–136. doi: 10.1038/nrmicro2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.a) van Dam V, Olrichs N, Breukink E. Chembiochem. 2009;10:617–624. doi: 10.1002/cbic.200800678. [DOI] [PubMed] [Google Scholar]; b) Daniel RA, Errington J. Cell. 2003;113:767–776. doi: 10.1016/s0092-8674(03)00421-5. [DOI] [PubMed] [Google Scholar]; c) Tiyanont K, Doan T, Lazarus MB, Fang X, Rudner DZ, Walker S. Proc Natl Acad Sci U S A. 2006;103:11033–11038. doi: 10.1073/pnas.0600829103. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Olrichs NK, Aarsman MEG, Verheul J, Arnusch CJ, Martin NI, Herve M, Vollmer W, de Kruijff B, Breukink E, den Blaauwen T. Chembiochem. 2011;12:1124–1133. doi: 10.1002/cbic.201000552. [DOI] [PubMed] [Google Scholar]; e) Sadamoto R, Niikura K, Ueda T, Monde K, Fukuhara N, Nishimura SI. J Am Chem Soc. 2004;126:3755–3761. doi: 10.1021/ja039391i. [DOI] [PubMed] [Google Scholar]; f) de Pedro MA, Quintela JC, Höltje JV, Schwarz H. J Bacteriol. 1997;179:2823–2834. doi: 10.1128/jb.179.9.2823-2834.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.a) Lam H, Oh DC, Cava F, Takacs CN, Clardy J, de Pedro MA, Waldor MK. Science. 2009;325:1552–1555. doi: 10.1126/science.1178123. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Cava F, de Pedro MA, Lam H, Davis BM, Waldor MK. EMBO J. 2011;30:3442–3453. doi: 10.1038/emboj.2011.246. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Lupoli TJ, Tsukamoto H, Doud EH, Wang TSA, Walker S, Kahne D. J Am Chem Soc. 2011;133:10748–10751. doi: 10.1021/ja2040656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brown PJB, de Pedro MA, Kysela DT, Van der Henst C, Kim J, De Bolle X, Fuqua C, Brun YV. Proc Natl Acad Sci U S A. 2012;109:1697–1701. doi: 10.1073/pnas.1114476109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mobley HL, Koch AL, Doyle RJ, Streips UN. J Bacteriol. 1984;158:169–179. doi: 10.1128/jb.158.1.169-179.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Furchtgott L, Wingreen NS, Huang KC. Mol Microbiol. 2011;81:340–353. doi: 10.1111/j.1365-2958.2011.07616.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.a) Domínguez-Escobar J, Chastanet A, Crevenna AH, Fromion V, Wedlich-Söldner R, Carballido-López R. Science. 2011;333:225–228. doi: 10.1126/science.1203466. [DOI] [PubMed] [Google Scholar]; b) Garner EC, Bernard R, Wang WQ, Zhuang XW, Rudner DZ, Mitchison T. Science. 2011;333:222–225. doi: 10.1126/science.1203285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.a) Rippka R, Herdman M. Ann Inst Pasteur/Microbiol. 1985;136:33–39. doi: 10.1016/s0769-2609(85)80018-1. [DOI] [PubMed] [Google Scholar]; b) Zapun A, Vernet T, Pinho MG. FEMS Microbiol Rev. 2008;32:345–360. doi: 10.1111/j.1574-6976.2007.00098.x. [DOI] [PubMed] [Google Scholar]
- 9.Flärdh K. Curr Opin Microbiol. 2003;6:564–571. doi: 10.1016/j.mib.2003.10.011. [DOI] [PubMed] [Google Scholar]
- 10.Aaron M, Charbon G, Lam H, Schwarz H, Vollmer W, Jacobs-Wagner C. Mol Microbiol. 2007;64:938–952. doi: 10.1111/j.1365-2958.2007.05720.x. [DOI] [PubMed] [Google Scholar]
- 11.Young KD. Microbiol Mol Biol Rev. 2006;70:660–703. doi: 10.1128/MMBR.00001-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lavis LD, Raines RT. ACS Chem Biol. 2008;3:142–155. doi: 10.1021/cb700248m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Decad GM, Nikaido H. J Bacteriol. 1976;128:325–336. doi: 10.1128/jb.128.1.325-336.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Prescher JA, Bertozzi CR. Nat Chem Biol. 2005;1:13–21. doi: 10.1038/nchembio0605-13. [DOI] [PubMed] [Google Scholar]
- 15.a) Neumann H, Wang K, Davis L, Garcia-Alai M, Chin JW. Nature. 2010;464:441–444. doi: 10.1038/nature08817. [DOI] [PubMed] [Google Scholar]; b) Wang L, Brock A, Herberich B, Schultz PG. Science. 2001;292:498–500. doi: 10.1126/science.1060077. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.