

Abstract
Large-scale analysis of balanced chromosomal translocation breakpoints has shown nonhomologous end joining and microhomology-mediated repair to be the main drivers of interchromosomal structural aberrations. Breakpoint sequences of de novo unbalanced translocations have not yet been investigated systematically. We analyzed 12 de novo unbalanced translocations and mapped the breakpoints in nine. Surprisingly, in contrast to balanced translocations, we identify nonallelic homologous recombination (NAHR) between (retro)transposable elements and especially long interspersed elements (LINEs) as the main mutational mechanism. This finding shows yet another involvement of (retro)transposons in genomic rearrangements and exposes a profoundly different mutational mechanism compared with balanced chromosomal translocations. Furthermore, we show the existence of compound maternal/paternal derivative chromosomes, reinforcing the hypothesis that human cleavage stage embryogenesis is a cradle of chromosomal rearrangements.
With the advent of genomic technologies, our understanding of the mechanisms underlying human chromosomal rearrangements has expanded rapidly in recent years. Analysis of the breakpoints of copy number variants (CNVs) has revealed that “nonrecurrent CNVs” are mainly generated by microhomology-mediated end joining (MMEJ) (Liang et al. 1996; McVey and Lee 2008), nonhomologous end joining (NHEJ) (Vissers et al. 2009; Conrad et al. 2010), replication fork stalling and template switching (FoSTeS), or microhomology-mediated break-induced replication (MMBIR) (Lee et al. 2007; Bauters et al. 2008; Hastings et al. 2009a). “Recurrent CNVs” are caused by NAHR between low copy repeats (LCRs) (Sharp et al. 2006). In addition to LCRs, NAHR between Alu repetitive elements has also emerged as a major driver for CNV formation (Lehrman et al. 1985; Shaw and Lupski 2005; Luo et al. 2011). The mechanisms underlying chromosomal translocations have been less intensively studied. Recurrent balanced translocations can result from NAHR between paralogous low copy repeats (Giglio et al. 2002; Ou et al. 2011) or from rearrangements between palindromic AT-rich repeats (Edelmann et al. 2001; Kurahashi and Emanuel 2001). Two recent studies reported the first systematic high-throughput sequence-based studies of nonrecurrent balanced chromosomal translocations (Higgins et al. 2008; Chiang et al. 2012). In 30.5% of 141 de novo breakpoints, microhomology was detected, suggesting MMEJ or MMBIR, whereas the remaining breakpoints showed <2 bp of homology implying NHEJ, with or without prior processing of the exposed DNA ends (Lieber 2008). In contrast to balanced translocations, the breakpoint sequences of de novo unbalanced translocations have barely been investigated.
Unbalanced translocations, defined as derivative chromosomes comprising a terminal deletion and a duplication of a terminal segment of another chromosome, are found in 0.7%–1.1% of individuals with developmental disabilities (Ravnan et al. 2006; Ballif et al. 2007; Shao et al. 2008) and are generally assumed to result from the unbalanced transmission of a derivative chromosome from a balanced translocation carrier. During our systematic screen of patients with developmental disabilities, we noticed that a substantial number, namely, 0.23% of the patients and comprising 30% of all unbalanced translocations, actually arise de novo. To determine when those de novo unbalanced translocations originate and to learn about the mutational mechanisms leading to those rearrangements, we performed a systematic analysis.
Results
We collected 12 de novo translocations, ascertained either by conventional karyotyping or by array analysis. The translocations were present in all cells, except for case 8, where conventional karyotyping observed the translocation to be present in only eight out of 10 cells. The karyotypes describing the chromosomal rearrangements and the size of the affected segments are presented in Table 1. All parental karyotypes were normal. To fine-map the translocation breakpoints and identify the parental origin of both segments of the derivative chromosomes, DNA from all patients and their parents was analyzed by Affymetrix 250K SNP arrays. The resulting copy number calls confirmed the deleted and duplicated segments previously found on lower-resolution arrays. To determine the parental origin of the aberrant regions, we used a parent-of-origin algorithm that specifies the allelic origin of (aberrant) loci by identifying and visualizing SNPs with a Mendelian error in a parent-specific manner at the genome-wide level (see Methods) (Voet et al. 2011). The results concurred with the A and B allele ratios in SNP cluster plots of individual SNPs. In 11/12 samples, the parental origin could be determined, and SNP analysis confirmed paternity in all. The SNP plots of homozygous but different SNPs in both parents (e.g., AA in mother and BB in father) demonstrate either that both the deletion and duplication are of a single parent or are derived of both parents. Two translocations combine a paternal deletion with a maternal duplication and are therefore of postzygotic origin (Fig. 1; Supplemental Fig. 3). The other derivative chromosomes were maternal in seven and paternal in two cases. The results are presented in Table 1. Unbalanced translocations with involvement of both a maternal and a paternal segment originated postzygotically. To discriminate a premeiotic or meiosis I origin from a meiosis II or postzygotic origin, SNPs heterozygous in the parent of origin of the duplication and homozygous in the other parent (e.g., AB in the mother and BB in the father) were compared with the SNP call in the child. An ABB can only result from a premeiotic or meiosis I error because both alleles of the mother are present (Fig. 2A; Supplemental Fig. 4). Six translocations were of meiotic I origin, five maternal and one paternal. In three patients, the genotypes are consistent with either a mitotic origin or a meiosis II error with segregation of the translocated and normal sister chromatids to the same gamete (Table 1).
Table 1.
Origin and breakpoints of de novo unbalanced translocations
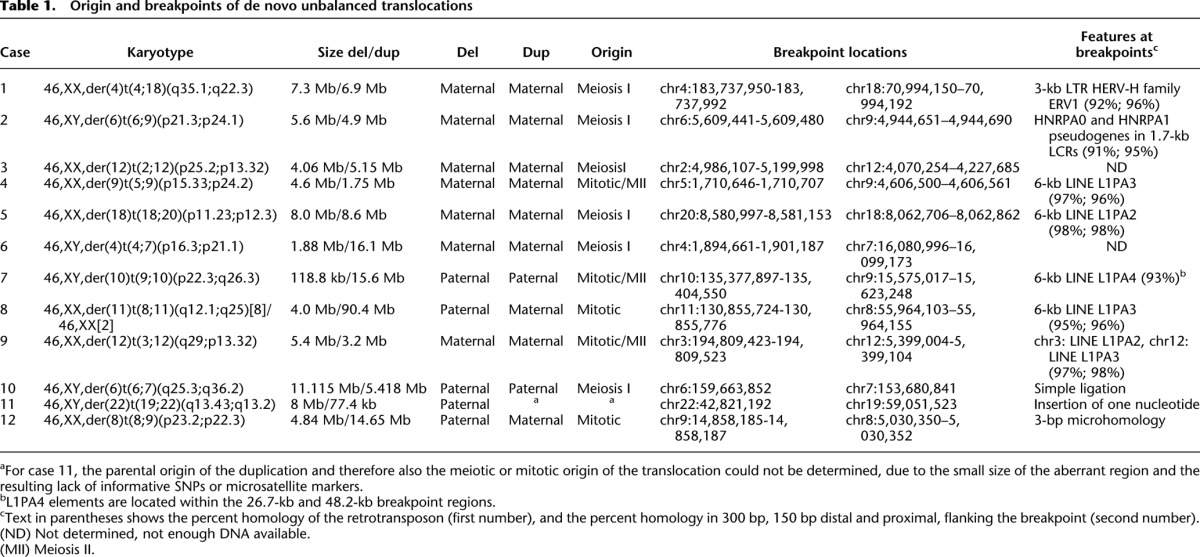
Figure 1.

Determination of the origin and breakpoint sequence of the translocation in case 8. (A) SNP array intensity ratio plots. Log2 intensity ratio of test over reference of the affected chromosomes is plotted on the y-axis against the position from pter to qter on the x-axis. (Gray line) The average test over reference ratio per 10 SNP values. (B) SNP cluster plots of individual SNPs in case 8. Green, red, and black dots represent controls with a BB, AB, and AA genotype, respectively. The pink and blue dots represent the genotypes of the mother and father and the longer arrows point to them. The yellow dots indicate the genotype call of the index patient and are indicated by the shorter arrow. The SNP plots of parental homozygous SNPs demonstrate that the duplication (left two panels) is of maternal origin, while the deletion (right panel) is of paternal origin. (C) The graphs indicate the percentage (left) and total number (right) of informative SNPs found in the deleted and duplicated region of case 8. (D) The breakpoint junction of case 8 is located in 6-kb LINE L1PA3 elements of the same orientation (black bars) with 95% DNA sequence identity in the chromosome regions 8q12.1 (top) and 11q25 (bottom). (Red arrows) The location of the breakpoint junction determined by sequence analysis. (E) DNA sequence alignment of the PCR-amplified translocation junction fragment of case 8 (middle sequence). The breakpoint site was narrowed to a 53-bp segment (gray box) with 100% DNA sequence identity between chromosomes 11 (top) and 8 (bottom). (Purple nucleotides) Alignment with the chromosome 11 sequence; (green nucleotides) alignment with the chromosome 8 sequence; (yellow highlighted nucleotides) trans-morphic mismatches.
Figure 2.

Resolving meiotic origin of de novo unbalanced translocations. (A) A schematic illustration of different segregants following a meiotic I or II de novo unbalanced translocation event. (Rose rectangles) Maternal gametes; (blue rectangles) paternal gametes; (gray rectangles) the zygote. In this figure, the translocation event occurs in the maternal gamete. Within those rectangles, the black and red colors represent maternal chromosomes, and dark gray and dark pink colors represent paternal chromosomes. To discriminate a meiosis I from a meiosis II event, maternal heterozygous SNPs and paternal homozygous SNPs are considered. The presence of ABB SNP-calls (underlined with yellow color) present in the duplicated region of the translocated chromosomes can only be the consequence of a premeiotic or a meiotic I event. (B) The SNP plots of parental homozygous SNPs demonstrate that both the deletion and duplication (left two panels) are of maternal origin, while the analysis of the maternal heterozygous and paternal homozygous SNPs demonstrates a premeiotic or meiosis I origin (right panel). (C) The SNP plots of parental homozygous SNPs demonstrate that both the deletion and duplication (left two panels) are of paternal origin, while the analysis of the paternal heterozygous and maternal homozygous SNPs demonstrates the premeiotic or meiosis I origin (right panel). Green, red, black, and gray dots represent controls with a BB, AB, AA, and NoCall genotype, respectively. The pink and blue dots represent the genotypes of the mother and father and the longer arrows point to them. The yellow dots indicate the genotype call of the index patient and are indicated by the shorter arrow. The SNP plots of parental homozygous SNPs demonstrate that the duplication (left two panels) is of maternal origin, while the deletion (right panel) is of paternal origin.
Mate pair or paired-end whole-genome sequencing enabled mapping of the translocation breakpoints in three cases (samples 10–12) (Fig. 1; Supplemental Fig. 1; Table 1). In the other cases, microarray and qPCR allowed fine-mapping of the breakpoint regions, enabling the generation of breakpoint-spanning long-range PCR fragments and subsequent breakpoint junction sequencing of those long-range fragments by Sanger or massive parallel sequencing. Overall, we identified 6/9 translocations with breakpoints localized in long stretches of homology: four in 6-kb LINEs, one in a 3-kb LTR, and one in a pseudogene inside a 1.7-kb low copy repeat (LCR), the LCR being unusual in that it is a sequence with paralogs at 54 genomic locations on 19 different chromosomes. Each of these repeats is >90% identical, and DNA sequencing identified the crossover sites within the repeat elements within a 40- to 157-bp interval with 100% identical reference sequence (Supplemental Fig. 1). The 3-kb LTR is an HERV-H transposable element. This element has been implicated in the formation of a de novo der(18)t(4;18)(q35.1;q22.3), complementary to the der(4)t(4;18)(q35.1;q22.3) identified here (Gunn et al. 2003; Moncla et al. 2004; Horbinski et al. 2008; Hermetz et al. 2012). In addition, in case 7, the breakpoint junction could not be sequenced, but the breakpoint region on both chromosomes harbors an intact 6-kb L1PA4 element, suggesting that NAHR occurred within. Hence, based on these findings, NAHR between homologous interspersed transposable elements appears to be the major mechanism in the formation of de novo unbalanced translocations.
Discussion
Alu repetitive elements are known to act as substrates for NAHR leading to CNVs (Lehrman et al. 1985; Shaw and Lupski 2005; Luo et al. 2011). These elements tend to lie close together, promoting recombination events that result in small, often viable, deletions and duplications. LINEs constitute ∼20% of the human genome (Lander et al. 2001) and are distributed throughout the genome. In addition, they are spaced farther apart and comprise significantly longer tracts of sequence homology than Alu elements and would thus be better suited for NAHR-mediated translocations. Hence, it has been hypothesized that LINEs would be a mediator of chromosomal rearrangements (Gilbert et al. 2005; Hedges and Deininger 2007). NAHR between LINEs has, indeed, been instrumental in shaping the structure of the human genome (Han et al. 2008). However, thus far, NAHR between LINEs has rarely been detected in large-scale studies analyzing the breakpoints of pathogenic CNVs and balanced translocations (Burwinkel and Kilimann 1998; Segal et al. 1999; Gribble et al. 2005, 2007; Higgins et al. 2008; Chiang et al. 2012), nor have they been observed in the many case reports mapping balanced translocation breakpoints (McMullan et al. 2002; Gajecka et al. 2008). In a recent systematic mapping study of chromosomal subtelomeric rearrangements, the breakpoints of four unbalanced translocation breakpoints were sequenced (Luo et al. 2011). All but one originated via NHEJ, while the fourth resulted from NAHR between L1PA2 elements in direct orientation. It is tempting to speculate that the latter arose as a de novo unbalanced translocation, whereas the others were the consequence of the unbalanced transmission of a balanced rearrangement in one of the parents.
That NAHR between LINEs is observed much more frequently in unbalanced compared with balanced translocations is surprising, but likely implies the involvement of a nonconservative homologous repair pathway. Homologous recombination (HR) is the basis of several mechanisms of accurate DNA repair that use identical sequence to repair damaged sequences (Hastings et al. 2009b). Conservative homologous recombination repair (HRR) pathways such as double-strand break repair and synthesis-dependent strand annealing, which demonstrate a high degree of preservation of sequence information, underlie NAHR that generates recurrent copy number changes in human and are also the cause of recurrent balanced translocations. HR is used, not only to repair two-ended double-stranded breaks, but also to repair collapsed or broken replication forks in a process called break-induced replication (BIR, a semiconservative form of HR). In BIR-induced unbalanced translocations, the broken chromosome end would invade another chromosome and is replicated to the chromosomal end. In addition single-strand annealing (SSA) a nonconservative HR could underlie this process and happens when neither end at a two-ended double-strand break invades homologous sequence. The random breaks are “chewed back,” allowing homologous sequences adjacent to the exposed ends to associate and ligate. As in the case of NHEJ, neither BIR nor SSA requires a sister chromatid and thus remains available throughout the cell cycle. Possibly, unbalanced translocations emerge during G1 of the cell cycle.
Although chromosomal breakage could originate anywhere in the genome, it is tempting to speculate that LINE sequences themselves may be involved in break formation. In mice, transposable elements are epigenetically silenced throughout most of life (Maksakova et al. 2008; Sasaki and Matsui 2008). This methylation prevents the involvement of retrotransposons in transposition events and chromosomal rearrangements (Yoder et al. 1997). Following fertilization, the zygote genome undergoes a short demethylation phase, meaning that LINEs can be activated during early embryogenesis (Maksakova et al. 2008; Hancks and Kazazian Jr. 2012). The LINEs identified at the breakpoints are all full length, but do not belong to the family L1H of active LINEs. HERV-H is an ancient transposable element that contains major deletions (Belshaw et al. 2005). Nevertheless, all LINEs identified at the translocation breakpoints are intact and accumulated only a limited number of mutations rendering them inactive (Supplemental Fig. 2). It seems plausible that transcription does occur at those elements inducing transcriptional breaks (Branzei and Foiani 2010).
A trivial reason for the discovery of LINEs here as compared with other studies of translocation breakpoints could lie in the difficulty of mapping breakpoints within repeated elements (e.g., Luo et al. 2011). If so, the de novo unbalanced translocations could also be the result of germline or low-level somatic mosaicisms for balanced translocations in one of the parents. Here, we amplified DNA molecules spanning >6 kb to map the breakpoints in repeated elements by applying different (massive parallel) sequencing strategies. An argument against this hypothesis is that unbiased sequencing of breakpoints of balanced translocations following chromosome flow sorting did not reveal NAHR (Gribble et al. 2007).
In addition to the breakpoints, we identified the parental origin of both deletions and duplications. Based on our observation that chromosomal deletions and duplications are very frequent in human cleavage-stage embryos following in vitro fertilization (Vanneste et al. 2009; Voet et al. 2011), we hypothesized that the detected unbalanced translocations might originate during cleavage of the zygote, which is in addition a time frame of active retrotransposon transcription (Maksakova et al. 2008; Vanneste et al. 2009; Hancks and Kazazian Jr. 2012). In this case, some unbalanced translocations originating during early embryogenesis would recombine both maternal and paternal chromosomes. In accordance with this prediction, we show the presence of two derivative chromosomes of combined parental origin. A similar number of rearrangements during cleavage stage would involve uniquely maternal or uniquely paternal chromosomes. The presence of three derivatives of either mitotic or meiosis II origin supports this thesis. Based on the limited number of cases studied here, up to 30%–40% of all de novo unbalanced translocations could originate postzygotically. It seems likely that a similar percentage of other chromosomal disorders also arises during early embryonic development.
Methods
Cytogenetic, FISH, and array analyses
Blood samples were obtained from 11 patients and parents referred for cytogenetic investigation due to the presence of congenital abnormalities and dysmorphic features, and one sample was obtained via amniocentesis (case 6). GTG-banded chromosome analysis was performed on metaphase chromosomes from PHA-stimulated peripheral blood lymphocytes or amniocytes using standard culture and chromosome preparation protocols. Deletions and duplications were confirmed by FISH using BAC clones as described (Vermeesch et al. 1995). Conventional karyotyping of postnatal case 8 revealed a mosaic karyotype with a derivative chromosome 11 present in 80% of white blood cells. G-banding did not reveal any abnormalities in postnatal cases 1–5 and 9–11 and was not performed in case 12.
Samples from cases 3, 9, and 11 were analyzed on a 1-Mb BAC array (Vermeesch et al. 2005), cases 4, 6, and 10 on the CytoSure Syndrome Plus v2 oligonucleotide array (Oxford Gene Technology), and the remaining cases on a CytoSure 180K Custom Microarray composed of probes from the CytoSure Syndrome Plus v2 array supplemented with probes from the CytoSure ISCA v2 60K array (Srisupundit et al. 2010). DNA from all cases and from the parents was analyzed on an Affymetrix GeneChip Human Mapping 250K Nsp Array (Affymetrix, Inc.). DNA digestion, labeling, and hybridization were performed according to the manufacturer’s protocol. After hybridization, arrays were washed and stained on the Affymetrix GeneChip fluidics station 450 and scanned using the Affymetrix GeneChip Scanner 3000 7G. SNP copy number analysis was performed using “Copy Number Analyzer for Genechip (CNAG) version 2.0” (Nannya et al. 2005) or the “Copy Number Analysis Tool” (CNAT4.0.1; Affymetrix) and the Genotyping Console Software (Affymetrix, Inc.).
SNP-based parent-of-origin determination
SNP probe intensities were analyzed by GTYPE 4.1 (Affymetrix) using the dynamic model (Di et al. 2005) with stringency P0.12. The parent-of-origin algorithm determined the allelic origin of (aberrant) loci in the patient by identifying and visualizing, in a parent-specific manner, SNPs that demonstrated a Mendelian error (Voet et al. 2011). SNPs were scored as either +1 or +0.5 (maternal) or −1 or −0.5 (paternal), and true chromosomal aberrations were identified by consecutive SNPs that were assigned to either a maternal or paternal origin. This analysis also confirms paternity.
The results of the parent-of-origin algorithm were supplemented with SNP cluster plots of individual SNPs. To determine the parental origin of the DNA copy number aberrations by a SNP-cluster strategy, normalized SNP A- and B-allele intensities as well as SNP genotype calls were computed using Affymetrix power tools (APT-1.10.1) in combination with the Birdseed command (Korn et al. 2008). Besides the described trio data sets, in-house-produced Affymetrix 250K SNP NspI data from 102 additional DNA samples were coprocessed for accurate SNP-cluster and genotype determination. Subsequently, the probe intensities and genotype calls for SNPs within the regions of interest were retrieved for all DNA samples and interpreted by custom R-code (http://www.r-project.org/) in which patient and parental SNP-probe intensities are visualized in Birdseed SNP clusters from the 102 individuals.
Q-PCR analysis
qPCR primers were designed using Primer 3 Software (http://frodo.wi.mit.edu/primer3). Primers were chosen free from any repeats using RepeatMasker provided by the UCSC Genome Browser (http://genome.ucsc.edu/). qPCR was performed using the Lightcycler 480 instrument (Roche Applied Science) working in a total volume of 15 μL including 7.5 μL of SYBR Green I mastermix (Roche Applied Science), 2.5 μL of primer mix (2.5 μM), and 50 ng of template DNA. The following amplification conditions were used: 5 min at 95°C, and 40 cycles of 10 sec at 95°C and 20 sec at 60°C. After the amplification protocol, a melting curve was obtained for 30 sec at 95°C, 30 sec at 60°C, and 95°C for a continuous mode (five acquisitions per °C), and finally cooling down for 30 sec to 40°C. Next, data were analyzed with Excel (Microsoft) according to the comparative ddCt method (Sequence Detection System bulletin 2; Applied Biosystems). The breakpoint regions were narrowed down to ∼10 kb.
Long-range PCR, amplicon paired-end sequencing, and nested junction PCR
Long-range PCR primers for cases 1, 2, 4, 5, 8, and 9 were designed flanking or inside the qPCR or array CGH-determined breakpoint regions. The primers were designed using Primer 3 Software (http://frodo.wi.mit.edu/primer3) with the following specifications: primer size 27–32–35 bp, primer Tm 65°C–67°C–70°C, and primer GC% 45%–50%–80%. Interspersed repeat sequences were blocked using RepeatMasker provided by the UCSC Genome Browser (http://genome.ucsc.edu/). Amplification of fragments was performed using the Expand Long Template PCR System (Roche Applied Science), following the manufacturer’s protocol. Briefly, we used 50-μL reaction mixtures containing 150–300 ng of genomic DNA, 0.5 mM dNTP (each), 0.3 μM primers (each), and 3.75 units of Expand Long Template Enzyme Mix. The PCR conditions were 2 min at 94°C, 10 cycles of 10 sec at 94°C, 30 sec at 65°C (−1°C/cycle), and 2 min at 68°C, followed by 25 cycles of 15 sec at 94°C, 30 sec at 55°C, and 6 min at 68°Cn (+20 sec/cycle), with a final extension of 7 min at 68°C.
The PCR products (7–12 kb) of cases 1, 2, and 4 were used to construct a paired-end library using the TruSeq DNA Sample Prep Kit v2 (Illumina), following the manufacturer’s protocol. The library, containing fragments of ∼450 bp, was loaded onto 1/10 of an Illumina flow cell at a concentration of 11 pM using the Illumina Cluster Station and Paired-End Cluster Kit v3. The flow cell was sequenced as a 76 × 2 paired-end read on an Illumina HiSeq 2000, followed by forced mapping of the sequences to the putative breakpoint region. To further define the exact breakpoint junction, the 0.5- to 1.5-kb center region was amplified by nested-PCR. PCR primers were designed as described above. Amplification of fragments was performed using the Taq DNA Polymerase PCR System (Roche Applied Science). Briefly, we used 25-μL reaction mixtures containing 1 μL of PCR product, 0.2 mM dNTP (each), 0.2 μM primers (each), 2.5 μL of 10× PCR buffer, and 1.5 units of Taq DNA Polymerase. The PCR conditions were 2 min at 94°C, followed by 10 cycles of 15 sec at 94°C, 30 sec at 65°C (−1°C/cycle), and 1 min at 68°C, with a final extension of 5 min at 68°C. PCR products were treated with ExoSAP-IT (USB) and bidirectionally sequenced on an ABI 3130xl automated capillary DNA sequencer (Applied Biosystems) using the BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems). DNA sequences were visualized using SeqMan v. 4.00 (DNASTAR, Inc.).
The DNA sequence of cases 5, 8, and 9 was determined by Sanger sequencing of the long-range PCR products (7–10 kb), as described above.
Genomic mate pair and paired-end sequencing
Genome-wide mate pair sequencing and paired-end sequencing were used to narrow the breakpoint region in cases 10, 11, and 12. Mate pair libraries for samples 11 and 12 were constructed from 10 μg of genomic DNA using the Mate Pair Library Prep Kit v2 (Illumina), following the manufacturer’s instructions. Genomic DNA fragments of ∼3 kb were selected. The final library composed of 350- to 650-bp fragments was loaded onto 1/4 lane of an Illumina flow cell at a concentration of 11 pM using the Illumina Cluster Station and Paired End Cluster Kit v2. The flow cell was sequenced as a 76 × 2 paired-end read on an Illumina HiSeq 2000 Sequencing System using the SBS Sequencing Kit v2. A paired-end library for case 10 was constructed from 1.5 μg of genomic DNA using the TruSeq DNA Sample Prep Kit v2 (Illumina), following the manufacturer’s instructions. Fragments of 780–870 bp in size were loaded onto 1/4 lane of an Illumina flow cell at a concentration of 9 pM using the Illumina Cluster Station and Paired End Cluster Kit v3. The flow cell was sequenced as a 100 × 2 paired-end read on an Illumina HiSeq 2000 Sequencing System using the SBS Sequencing Kit v3.
Analysis of the translocation breakpoint region revealed one, three, and five pairs that mapped across the breakpoint junction for cases 10, 12, and 11, respectively. PCR primers flanking the mate pair or paired-end clones that mapped across the translocation junction were designed as described above. Amplification of fragments was performed using the Expand Long Template PCR System (Roche Applied Science), as described above, but with 0.35 mM dNTPs and 2 min of extension time instead of 6 min. The PCR products of ∼2 kb were Sanger-sequenced and analyzed as described above.
Data filtering and mapping and breakpoint junction analysis
The standard Illumina HiSeq 2000 primary data analysis workflow was followed for base calling and quality scoring. The resulting raw reads were demultiplexed with CASAVA 1.8 and mapped to the reference human genome (Build GRCh37/hg19) with BWA (Li and Durbin 2009).
The obtained DNA sequences were mapped to the human genome by the BLAT tool provided by the UCSC Genome Browser (http://genome.ucsc.edu/) (Build GRCh37/hg19) to determine the exact location of the breakpoints. The RepeatMasker track of the UCSC Genome Browser (http://www.repeatmasker.org/) (Smit AFA, Hubley R, Green P. RepeatMasker Open-3.0) was used to identify highly repetitive sequences including long interspersed elements (LINEs), short interspersed elements (SINEs), long terminal repeats (LTRs), and simple tandem repeats (STRs). The sequence homology of both breakpoint regions involved in each translocation was evaluated by the Basic Local Alignment Search Tool (BLAST) (http://blast.ncbi.nlm.nih.gov/).
Data access
The Affymetrix SNP array CHP and CEL files have been submitted to the NCBI Gene Expression Omnibus (GEO) (http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE39303.
Acknowledgments
We thank G. Peeters for help with the HiSeq library preparation and L. Dehaspe for analysis of the HiSeq data. This work was made possible by grants from the IWT (SBO-60848), FWO grant G.0320.07, KUL PFV/10/016 SymBioSys, and GOA/12/015 to J.R.V. and T.V. B.A.N. is supported by a KOLUMB fellowship from the Foundation for Polish Science.
Author contributions: J.R.V. conceived the study and participated in its design and coordination. C.R. carried out the molecular genetic studies and analyzed the results. T.V. and M.Z.E. developed the parent-of-origin and SNP-clustering pipeline and performed the data analysis. B.A.N. participated collecting samples. C.R., T.V., B.A.N., M.Z.E., and J.R.V. drafted the manuscript, and all authors read and approved the final manuscript.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.145631.112.
References
- Ballif BC, Sulpizio SG, Lloyd RM, Minier SL, Theisen A, Bejjani BA, Shaffer LG 2007. The clinical utility of enhanced subtelomeric coverage in array CGH. Am J Med Genet A 143A: 1850–1857 [DOI] [PubMed] [Google Scholar]
- Bauters M, Van Esch H, Friez MJ, Boespflug-Tanguy O, Zenker M, Vianna-Morgante AM, Rosenberg C, Ignatius J, Raynaud M, Hollanders K, et al. 2008. Nonrecurrent MECP2 duplications mediated by genomic architecture-driven DNA breaks and break-induced replication repair. Genome Res 18: 847–858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belshaw R, Dawson AL, Woolven-Allen J, Redding J, Burt A, Tristem M 2005. Genomewide screening reveals high levels of insertional polymorphism in the human endogenous retrovirus family HERV-K(HML2): Implications for present-day activity. J Virol 79: 12507–12514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branzei D, Foiani M 2010. Maintaining genome stability at the replication fork. Nat Rev Mol Cell Biol 11: 208–219 [DOI] [PubMed] [Google Scholar]
- Burwinkel B, Kilimann MW 1998. Unequal homologous recombination between LINE-1 elements as a mutational mechanism in human genetic disease. J Mol Biol 277: 513–517 [DOI] [PubMed] [Google Scholar]
- Chiang C, Jacobsen JC, Ernst C, Hanscom C, Heilbut A, Blumenthal I, Mills RE, Kirby A, Lindgren AM, Rudiger SR, et al. 2012. Complex reorganization and predominant non-homologous repair following chromosomal breakage in karyotypically balanced germline rearrangements and transgenic integration. Nat Genet 44: 390–397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad DF, Bird C, Blackburne B, Lindsay S, Mamanova L, Lee C, Turner DJ, Hurles ME 2010. Mutation spectrum revealed by breakpoint sequencing of human germline CNVs. Nat Genet 42: 385–391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di X, Matsuzaki H, Webster TA, Hubbell E, Liu G, Dong S, Bartell D, Huang J, Chiles R, Yang G, et al. 2005. Dynamic model based algorithms for screening and genotyping over 100 K SNPs on oligonucleotide microarrays. Bioinformatics 21: 1958–1963 [DOI] [PubMed] [Google Scholar]
- Edelmann L, Spiteri E, Koren K, Pulijaal V, Bialer MG, Shanske A, Goldberg R, Morrow BE 2001. AT-rich palindromes mediate the constitutional t(11;22) translocation. Am J Hum Genet 68: 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gajecka M, Gentles AJ, Tsai A, Chitayat D, Mackay KL, Glotzbach CD, Lieber MR, Shaffer LG 2008. Unexpected complexity at breakpoint junctions in phenotypically normal individuals and mechanisms involved in generating balanced translocations t(1;22)(p36;q13). Genome Res 18: 1733–1742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giglio S, Calvari V, Gregato G, Gimelli G, Camanini S, Giorda R, Ragusa A, Guerneri S, Selicorni A, Stumm M, et al. 2002. Heterozygous submicroscopic inversions involving olfactory receptor-gene clusters mediate the recurrent t(4;8)(p16;p23) translocation. Am J Hum Genet 71: 276–285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert N, Lutz S, Morrish TA, Moran JV 2005. Multiple fates of L1 retrotransposition intermediates in cultured human cells. Mol Cell Biol 25: 7780–7795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gribble SM, Prigmore E, Burford DC, Porter KM, Ng BL, Douglas EJ, Fiegler H, Carr P, Kalaitzopoulos D, Clegg S, et al. 2005. The complex nature of constitutional de novo apparently balanced translocations in patients presenting with abnormal phenotypes. J Med Genet 42: 8–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gribble SM, Kalaitzopoulos D, Burford DC, Prigmore E, Selzer RR, Ng BL, Matthews NS, Porter KM, Curley R, Lindsay SJ, et al. 2007. Ultra-high resolution array painting facilitates breakpoint sequencing. J Med Genet 44: 51–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunn SR, Mohammed M, Reveles XT, Viskochil DH, Palumbos JC, Johnson-Pais TL, Hale DE, Lancaster JL, Hardies LJ, Boespflug-Tanguy O, et al. 2003. Molecular characterization of a patient with central nervous system dysmyelination and cryptic unbalanced translocation between chromosomes 4q and 18q. Am J Med Genet A 120A: 127–135 [DOI] [PubMed] [Google Scholar]
- Han K, Lee J, Meyer TJ, Remedios P, Goodwin L, Batzer MA 2008. L1 recombination-associated deletions generate human genomic variation. Proc Natl Acad Sci 105: 19366–19371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hancks DC, Kazazian HH Jr 2012. Active human retrotransposons: Variation and disease. Curr Opin Genet Dev 22: 191–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hastings PJ, Ira G, Lupski JR 2009a. A microhomology-mediated break-induced replication model for the origin of human copy number variation. PLoS Genet 5: e1000327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hastings PJ, Lupski JR, Rosenberg SM, Ira G 2009b. Mechanisms of change in gene copy number. Nat Rev Genet 10: 551–564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedges DJ, Deininger PL 2007. Inviting instability: Transposable elements, double-strand breaks, and the maintenance of genome integrity. Mutat Res 616: 46–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermetz KE, Surti U, Cody JD, Rudd MK 2012. A recurrent translocation is mediated by homologous recombination between HERV-H elements. Mol Cytogenet 5: 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgins AW, Alkuraya FS, Bosco AF, Brown KK, Bruns GA, Donovan DJ, Eisenman R, Fan Y, Farra CG, Ferguson HL, et al. 2008. Characterization of apparently balanced chromosomal rearrangements from the developmental genome anatomy project. Am J Hum Genet 82: 712–722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horbinski C, Carter EM, Heard PL, Sathanoori M, Hu J, Vockley J, Gunn S, Hale DE, Surti U, Cody JD 2008. Molecular and clinical characterization of a recurrent cryptic unbalanced t(4q;18q) resulting in an 18q deletion and 4q duplication. Am J Med Genet A 146: 2898–2904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korn JM, Kuruvilla FG, McCarroll SA, Wysoker A, Nemesh J, Cawley S, Hubbell E, Veitch J, Collins PJ, Darvishi K, et al. 2008. Integrated genotype calling and association analysis of SNPs, common copy number polymorphisms and rare CNVs. Nat Genet 40: 1253–1260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurahashi H, Emanuel BS 2001. Long AT-rich palindromes and the constitutional t(11;22) breakpoint. Hum Mol Genet 10: 2605–2617 [DOI] [PubMed] [Google Scholar]
- Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, et al. 2001. Initial sequencing and analysis of the human genome. Nature 409: 860–921 [DOI] [PubMed] [Google Scholar]
- Lee JA, Carvalho CM, Lupski JR 2007. A DNA replication mechanism for generating nonrecurrent rearrangements associated with genomic disorders. Cell 131: 1235–1247 [DOI] [PubMed] [Google Scholar]
- Lehrman MA, Schneider WJ, Sudhof TC, Brown MS, Goldstein JL, Russell DW 1985. Mutation in LDL receptor: Alu–Alu recombination deletes exons encoding transmembrane and cytoplasmic domains. Science 227: 140–146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25: 1754–1760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang F, Romanienko PJ, Weaver DT, Jeggo PA, Jasin M 1996. Chromosomal double-strand break repair in Ku80-deficient cells. Proc Natl Acad Sci 93: 8929–8933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieber MR 2008. The mechanism of human nonhomologous DNA end joining. J Biol Chem 283: 1–5 [DOI] [PubMed] [Google Scholar]
- Luo Y, Hermetz KE, Jackson JM, Mulle JG, Dodd A, Tsuchiya KD, Ballif BC, Shaffer LG, Cody JD, Ledbetter DH, et al. 2011. Diverse mutational mechanisms cause pathogenic subtelomeric rearrangements. Hum Mol Genet 20: 3769–3778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maksakova IA, Mager DL, Reiss D 2008. Keeping active endogenous retroviral-like elements in check: The epigenetic perspective. Cell Mol Life Sci 65: 3329–3347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMullan TW, Crolla JA, Gregory SG, Carter NP, Cooper RA, Howell GR, Robinson DO 2002. A candidate gene for congenital bilateral isolated ptosis identified by molecular analysis of a de novo balanced translocation. Hum Genet 110: 244–250 [DOI] [PubMed] [Google Scholar]
- McVey M, Lee SE 2008. MMEJ repair of double-strand breaks (director’s cut): Deleted sequences and alternative endings. Trends Genet 24: 529–538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moncla A, Missirian C, Philip N, Marlin S 2004. Another patient with cryptic unbalanced translocation between chromosomes 4q and 18q: Evidence by microarray CGH. Am J Med Genet A 131: 314–317 [DOI] [PubMed] [Google Scholar]
- Nannya Y, Sanada M, Nakazaki K, Hosoya N, Wang L, Hangaishi A, Kurokawa M, Chiba S, Bailey DK, Kennedy GC, et al. 2005. A robust algorithm for copy number detection using high-density oligonucleotide single nucleotide polymorphism genotyping arrays. Cancer Res 65: 6071–6079 [DOI] [PubMed] [Google Scholar]
- Ou Z, Stankiewicz P, Xia Z, Breman AM, Dawson B, Wiszniewska J, Szafranski P, Cooper ML, Rao M, Shao L, et al. 2011. Observation and prediction of recurrent human translocations mediated by NAHR between nonhomologous chromosomes. Genome Res 21: 33–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravnan JB, Tepperberg JH, Papenhausen P, Lamb AN, Hedrick J, Eash D, Ledbetter DH, Martin CL 2006. Subtelomere FISH analysis of 11 688 cases: An evaluation of the frequency and pattern of subtelomere rearrangements in individuals with developmental disabilities. J Med Genet 43: 478–489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki H, Matsui Y 2008. Epigenetic events in mammalian germ-cell development: Reprogramming and beyond. Nat Rev Genet 9: 129–140 [DOI] [PubMed] [Google Scholar]
- Segal Y, Peissel B, Renieri A, de Marchi M, Ballabio A, Pei Y, Zhou J 1999. LINE-1 elements at the sites of molecular rearrangements in Alport syndrome-diffuse leiomyomatosis. Am J Hum Genet 64: 62–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao L, Shaw CA, Lu XY, Sahoo T, Bacino CA, Lalani SR, Stankiewicz P, Yatsenko SA, Li Y, Neill S, et al. 2008. Identification of chromosome abnormalities in subtelomeric regions by microarray analysis: A study of 5,380 cases. Am J Med Genet A 146: 2242–2251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp AJ, Hansen S, Selzer RR, Cheng Z, Regan R, Hurst JA, Stewart H, Price SM, Blair E, Hennekam RC, et al. 2006. Discovery of previously unidentified genomic disorders from the duplication architecture of the human genome. Nat Genet 38: 1038–1042 [DOI] [PubMed] [Google Scholar]
- Shaw CJ, Lupski JR 2005. Non-recurrent 17p11.2 deletions are generated by homologous and non-homologous mechanisms. Hum Genet 116: 1–7 [DOI] [PubMed] [Google Scholar]
- Srisupundit K, Brady PD, Devriendt K, Fryns JP, Cruz-Martinez R, Gratacos E, Deprest JA, Vermeesch JR 2010. Targeted array comparative genomic hybridisation (array CGH) identifies genomic imbalances associated with isolated congenital diaphragmatic hernia (CDH). Prenat Diagn 30: 1198–1206 [DOI] [PubMed] [Google Scholar]
- Vanneste E, Voet T, Le CC, Ampe M, Konings P, Melotte C, Debrock S, Amyere M, Vikkula M, Schuit F, et al. 2009. Chromosome instability is common in human cleavage-stage embryos. Nat Med 15: 577–583 [DOI] [PubMed] [Google Scholar]
- Vermeesch JR, Mertens G, David G, Marynen P 1995. Assignment of the human glypican gene (GPC1) to 2q35-q37 by fluorescence in situ hybridization. Genomics 25: 327–329 [DOI] [PubMed] [Google Scholar]
- Vermeesch JR, Melotte C, Froyen G, Van VS, Dutta B, Maas N, Vermeulen S, Menten B, Speleman F, De MB, et al. 2005. Molecular karyotyping: Array CGH quality criteria for constitutional genetic diagnosis. J Histochem Cytochem 53: 413–422 [DOI] [PubMed] [Google Scholar]
- Vissers LE, Bhatt SS, Janssen IM, Xia Z, Lalani SR, Pfundt R, Derwinska K, de Vries BB, Gilissen C, Hoischen A, et al. 2009. Rare pathogenic microdeletions and tandem duplications are microhomology-mediated and stimulated by local genomic architecture. Hum Mol Genet 18: 3579–3593 [DOI] [PubMed] [Google Scholar]
- Voet T, Vanneste E, Van der Aa N, Melotte C, Jackmaert S, Vandendael T, Declercq M, Debrock S, Fryns JP, Moreau Y, et al. 2011. Breakage-fusion-bridge cycles leading to inv dup del occur in human cleavage stage embryos. Hum Mutat 32: 783–793 [DOI] [PubMed] [Google Scholar]
- Yoder JA, Walsh CP, Bestor TH 1997. Cytosine methylation and the ecology of intragenomic parasites. Trends Genet 13: 335–340 [DOI] [PubMed] [Google Scholar]