

Abstract
Low-grade brain tumors (pilocytic astrocytomas) arising in the neurofibromatosis type 1 (NF1) inherited cancer predisposition syndrome are hypothesized to result from a combination of germline and acquired somatic NF1 tumor suppressor gene mutations. However, genetically engineered mice (GEM) in which mono-allelic germline Nf1 gene loss is coupled with bi-allelic somatic (glial progenitor cell) Nf1 gene inactivation develop brain tumors that do not fully recapitulate the neuropathological features of the human condition. These observations raise the intriguing possibility that, while loss of neurofibromin function is necessary for NF1-associated low-grade astrocytoma development, additional genetic changes may be required for full penetrance of the human brain tumor phenotype. To identify these potential cooperating genetic mutations, we performed whole-genome sequencing (WGS) analysis of three NF1-associated pilocytic astrocytoma (PA) tumors. We found that the mechanism of somatic NF1 loss was different in each tumor (frameshift mutation, loss of heterozygosity, and methylation). In addition, tumor purity analysis revealed that these tumors had a high proportion of stromal cells, such that only 50%–60% of cells in the tumor mass exhibited somatic NF1 loss. Importantly, we identified no additional recurrent pathogenic somatic mutations, supporting a model in which neuroglial progenitor cell NF1 loss is likely sufficient for PA formation in cooperation with a proper stromal environment.
NF1 is one of the most common autosomal dominant tumor predisposition syndromes in which affected individuals develop brain tumors. In this regard, 15%–20% of children with NF1 develop World Health Organization (WHO) grade I pilocytic astrocytomas (PAs) (Listernick et al. 1994). These low-grade glial neoplasms typically arise in children <7 yr of age and most commonly occur in the optic pathway (optic nerves, chiasm, and tracts) or in the brainstem (Listernick et al. 1997). Similar to other tumor predisposition syndromes, children with NF1 are born with one mutated, nonfunctional copy of the NF1 gene, such that loss of the remaining allele in appropriate progenitor cells enables tumorigenesis. Consistent with this “two hit” hypothesis, previous studies of NF1-associated PA (NF1-PA) have demonstrated loss of heterozygosity at the DNA level (Kluwe et al. 2001; Gutmann et al. 2003) and loss of NF1 protein (neurofibromin) expression (Gutmann et al. 2000).
While NF1 loss in neuroglial progenitors is necessary for PA formation in individuals with NF1, genetically engineered mice (GEM) with conditional loss of Nf1 gene expression in neuroglial progenitor cells fail to develop brain tumors (Bajenaru et al. 2002; Zhu et al. 2005). This failure to develop gliomas reflects the need for cooperating Nf1+/− non-neoplastic cells in the tumor microenvironment. In this regard, similar to children with NF1 harboring one nonfunctional and one functional NF1 allele in every cell of their bodies, Nf1+/− mice (one functional and one inactivated Nf1 allele) with absent neuroglial progenitor Nf1 expression develop optic gliomas with nearly 100% penetrance (Bajenaru et al. 2003). Similar to their human counterparts, these murine brain tumors exhibit low levels of proliferation, increased numbers of endothelial cells and microglia, and nuclear pleomorphism (Bajenaru et al. 2005; Kim et al. 2010).
Interestingly, these mouse optic gliomas do not fully recapitulate the classic histopathological features of the human tumors, in that they lack Rosenthal fibers and eosinophilic granular bodies (Louis et al. 2007), raising the intriguing possibility that other genetic alterations exist in human NF1-PA. The purpose of the current study was to employ advanced whole-genome sequencing technologies to establish the genomic landscape of human NF1-PA to facilitate the development of improved approaches to the diagnosis and management of these brain tumors.
Results
Whole-genome sequencing (WGS) was performed on three PA-matched normal blood (PBMC) cases from individuals with NF1 established using NIH Consensus Development Conference diagnostic criteria (Stumpf et al. 1988; Gutmann et al. 1997). These tumors are difficult to obtain, as they are rarely surgically resected and, when removed, typically provide very limited amounts of frozen tissue for comprehensive analysis. Based on our previous studies (Leonard et al. 2006), we specifically selected three PA tumors that were atypical by virtue of patient age (>7 yr of age at the time of glioma discovery) and tumor location (outside of the optic pathway) to maximize our ability to detect additional causative mutations (Table 1). All of these tumors contained >90% tumor (and not normal surrounding brain) by routine hematoxylin and eosin histopathologic review, exhibited the classic histopathologic features of PA (eosinophilic granule bodies, Rosenthal fibers), had low proliferative indices, and lacked necrotic areas or mitotic figures. Informed consent was obtained for all patients on an active protocol approved by the Washington University School of Medicine Institutional Review Board. We obtained paired end sequencing data to a minimum average depth of >32-fold haploid depth per genome. An overall concordance of >99% was obtained when comparing WGS-derived SNP genotypes with SNP array data at identical genome positions (Supplemental Table 1).
Table 1.
Clinical attributes and NF1 mutations in the three NF1-PA tumors
In all three patients, the germline NF1 mutations were identified based on their presence in both tumor and blood DNA (Table 1; Fig. 1): T01 had a point mutation (c.2002-14) in intron 17 (NM_001042492.2) previously demonstrated to create a novel splice variant (Spurlock et al. 2010); T02 had a germline splice site mutation (c.205-2), while T03 had a germline nonsense mutation (p.W1662*). Despite the use of both PCR and capillary sequencing as well as targeted capture-based sequencing from validation, we only identified a somatic NF1 gene mutation in T02 (frameshift mutation; p.C2426fs), suggesting that additional second hit mechanisms were responsible for low-grade astrocytoma formation in the other two tumors. Moreover, no mutations were identified in the putative promoter region encompassing 50 kb 5′ of the initiation codon.
Figure 1.

NF1 truncation mutations in three whole-genome-sequenced NF1-PA tumors. The locations of all truncation mutations are designated with a circle mapped to transcript NM_001042492. Truncating mutations were initially identified in WGS data and further validated with targeted approaches. Predicted functional effects are based on transcript NM_001042492. In patient T03, a somatic, copy-neutral LOH of 57.3 Mbp resulted in duplication of the p.W1662* germline variant.
To exclude the possibility that T01 and T03 retained NF1 gene expression, we performed immunohistochemistry to demonstrate loss of neurofibromin protein expression in all three NF1-PA tumors (Fig. 2A), a result consistent with complete NF1 inactivation in NF1-PA tumors. We next sought to define the mechanism(s) underlying loss of the second allele in T01 and T03. Loss of heterozygosity (LOH) analyses were performed using VarScan-2 (Koboldt et al. 2012), requiring a variant allele frequency at heterozygous germline SNPs of ≥10% in the normal sample and of either ≤5% or ≥95% in the tumor (only high-confidence calls were considered) to identify LOH regions. Using this method, we identified a 57.3 Mbp copy-neutral LOH on the q arm of chromosome 17 from position 23.9 Mbp to the telomere, encompassing the NF1 gene, in T03 (Fig. 2B). Further examination of supporting variant and reference allele frequencies indicated bi-allelic inactivation of the NF1 gene in this tumor occurred through duplication of the p.W1662* nonsense germline mutation and loss of the wild-type NF1 allele.
Figure 2.

Mechanism of somatic NF1 inactivation. (A) Immunohistochemistry using rabbit polyclonal neurofibromin antibodies demonstrates positive immunoreactivity in the normal human brain (HBr). (Inset) Single neuron with neurofibromin expression. In each of the three NF1-PA specimens, no neurofibromin expression was detected. Scale bars, 100 μm. Magnification, 400×. Inset, 600×. (B) In T03, a somatic copy-neutral LOH of the q arm of chromosome 17 from position 23.9 Mbp to the telomere results in duplication of the NF1 W1662* germline variant. Shown are the median variant allele frequencies of high-confidence germline heterozygous SNPs identified in the normal DNA in 100,000-bp bins across chromosome 17. The position of the NF1 gene is indicated. (C) Methylation changes in T01 relative to T02 and T03 from Illumina Infinium HumanMethylation450 arrays for 42 probes spanning the NF1 genomic region. T01 exhibits increased methylation (>2 standard deviations) at NCBI 36 chr17:26648522 (probe cg13759778). This location is within 200 bp of the transcription start site of OMG, one of three antisense nested genes within intron 36 of NF1 mRNA NM_001042492.2.
Since T01 had neither an identified somatic NF1 mutation nor clear evidence of LOH based on the VarScan analysis of 81 heterozygous sites within the NF1 gene (mean VAFs: 49.2% in the normal DNA and 55.6% in the tumor DNA), we examined methylation patterns. Using the Illumina Infinium HumanMethylation450 BeadChip, we assayed the three whole-genome-sequenced NF1-PA samples and two additional sporadic PA tumors. Genome-wide analysis comparing T01 with T02 and T03 identified 118 probes clustered in 105 regions that exhibited significantly different methylation (Supplemental Table 2). Upon further examination, no methylation alterations affected genes previously reported to be mutated in sporadic PA tumors (Wimmer et al. 2002b; Tada et al. 2003; Sharma et al. 2005; Deshmukh et al. 2008; Jones et al. 2008; Pfister et al. 2008). For most of the 42 probes localized within the NF1 gene region, we found that methylation patterns were similar in all five PA tumor samples analyzed. However, as a group, the NF1-PA tumors exhibited increased methylation (greater than two standard deviations) at probe cg21850879 (NCBI 36 chr17:26646069) compared with the sporadic counterparts. Interestingly, in T01 (the tumor lacking a second NF1 somatic mutation) there was increased methylation (greater than two standard deviations) at probe cg13759778 (NCBI 36 chr17:26648522) compared with T02 and T03. Probe cg13759778 is within 200 bp of the transcription start site of OMG, one of three antisense nested genes within intron 27b of the NF1 gene (intron 36 of NM_001042492.2) (Fig. 2C). While this methylation event occurs in intronic sequences within the NF1 locus and cannot be definitively demonstrated to cause NF1 gene silencing, the absence of neurofibromin expression in this tumor is consistent with loss of NF1 gene expression from this allele. Together, these data support the existence of multiple distinct mechanisms (mutation, LOH, and methylation) underlying somatic NF1 inactivation in NF1-PA tumors.
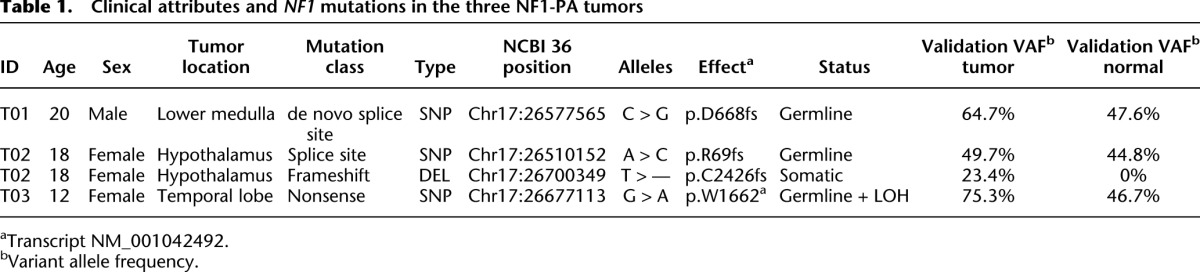
Using WGS, we were also able to accurately quantify the NF1 mutant allele frequencies and determine the relative clonality and cellular heterogeneity of these low-grade glial cell neoplasms (Ding et al. 2012). For clonality estimates, we evaluated data for somatic SNVs in nonrepetitive regions consisting of validated nonsynonymous coding variants, and high-confidence, manually reviewed events in potential regulatory regions, as well as the remaining nonrepetitive portion of the genome, representing a total of 133, 181, and 94 variants for tumors T01, T02, and T03, respectively (Supplemental Tables 3–5). Single clusters identified by kernel density estimation (Fig. 3A) indicated that each tumor consisted primarily of a single founding clone having no subclones.
Figure 3.
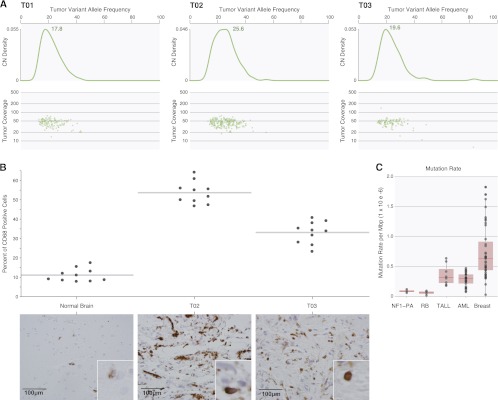
NF1-PA tumor clonality, cellular heterogeneity, and mutation rate. (A) Somatic mutation clusters were defined based on allele-specific supporting read counts in whole-genome data for mutations identified in nonrepetitive genomic with diploid copy number. Kernel density estimation indicates a monoclonal origin for each tumor and reveals a substantial proportion of stromal cells in the tumor biopsy with estimated tumor purities of 35%, 51%, and 39% for T01, T02, and T03, respectively. (B) CD68 immunostaining confirms the marked cellular heterogeneity of NF1-PA tumors, with 35%–55% of the cells in these tumors representing microglia/macrophages. Representative photomicrographs are included for each NF1-PA tumor. Scale bar, 100 μm. Magnification, 400×. Insets, 600×. (C) BMRs are based on the total sum of somatic mutations identified in all nonrepetitive genomic regions from WGS of three NF1-PA tumors, and compared with pediatric retinoblastoma (N = 4) (Zhang et al. 2012a), pediatric TALL (N = 8) (Zhang et al. 2012b), the M1 and M3 subtypes of adult AML (N = 25) (Welch et al. 2012), and luminal breast cancer (N = 46).
We used these analyses to calculate tumor purity, and found that these tumors were composed of 49%–64% non-neoplastic cells. Based on the combination of tumor purity and the coverage obtained from WGS data, the estimated sensitivity of our somatic mutation detection process is 70%. The large contribution of NF1+/− non-neoplastic cells to the tumor cellularity is consistent with parallel microglia (CD68+ cells) immunostaining results (Fig. 3B), where 35%–55% of the cells in these PA tumors were microglia/macrophages. This level of stromal contamination with microglia/macrophages is similar to that observed in both sporadic low-grade and high-grade gliomas (Simmons et al. 2011).
Next, we evaluated that possibility of large-scale somatic rearrangements in other genes potentially cooperate with NF1 loss during PA formation or growth by performing copy number alteration analysis of WGS read data using the cnvHMM algorithm (Chen et al. 2012). Importantly, read-count analysis showed no evidence of copy number aberrations (CNA) in the NF1 gene regions in any tumor (Supplemental Fig. 1). We then employed BreakDancer (Chen et al. 2009) and SquareDancer (derived from CREST [Wang et al. 2011]) to predict structural variants (SVs), Based on the detection of aberrant mate-pair read mapping distance or orientation, BreakDancer predicted 921, 825, and 449 putative somatic SVs in samples T01, T02, and T03, respectively. None of the predicted somatic SVs were supported by reads containing a physical breakpoint identified by SquareDancer, no large deletions were accompanied by corresponding drops in coverage, and no SV events were validated by custom capture or PCR-based validation sequencing. In this analysis, no CNV or SV were confirmed in the NF1-PA tumors, thus excluding CNA and SV from possibly altering the NF1 locus or its partners.
Predicted nonsynonymous somatic mutations that affect coding regions were validated using PCR followed by capillary sequencing (Supplemental Table 3). In contrast to previous reports on PA, we found no copy number changes or mutations in the BRAF, RAS (including KRAS, NRAS, and HRAS), MATN2, HIPK2, PTEN, TP53, CDKN2A, or EGFR genes (Tada et al. 2003; Fishbein et al. 2005; Sharma et al. 2005; Deshmukh et al. 2008; Jones et al. 2008; Pfister et al. 2008). However, nonsynonymous somatic mutations were identified in DST and SVIL in patient T01, OR13C3 in T02, and GPATCH2, FYTTD1, ACTBL2, PIK3R2, and ZNF257 in T03. To address the possibility that these somatically mutated genes could be involved in NF1-PA tumorigenesis, we used custom hybrid capture product sequencing to screen the coding exons of ACTBL2, DST, GPATCH2, OR13C3, PIK3R2, SVIL, FYTTD1, and ZNF257 in PA tumor samples from three additional NF1 patients. No additional somatic mutations were identified in these additional samples. Due to the low background mutation rate (BMR) in NF1-PA tumors compared with other solid tumor types, mutations identified in noncoding regions were not included in the recurrent screening efforts. Previous attempts to identify recurrent noncoding mutations in larger cohorts of solid tumors with higher BMRs have only identified a small number of recurrent mutations outside of coding regions (Ellis et al. 2012).
While these somatic mutations are novel, it is unlikely that they are pathogenic. First, the majority of the mutations identified (OR13C3, ACTBL2, FYTTD1, PIK3R2, and ZNF257) were revealed to be neutral (conservative changes in the protein sequence by in silico evaluation with Polyphen2 [Adzhubei et al. 2010], SIFT [Ng and Henikoff 2003], and Condel [Gonzalez-Perez and Lopez-Bigas 2011]) using pre-computed values from the Ensembl Variant Effects Predictor (Supplemental Table 3). Second, of the mutations in DST, SVIL, and GPATCH2 predicted to be deleterious, none occurred in an annotated functional protein domain related to carcinogenesis (Sigrist et al. 2010). Third, none of these eight mutations were identified in more than a single tumor, and thus were not recurrent in the limited number of these uncommonly banked tumors. Fourth, none of these genes exhibited bi-allelic inactivation either by mutation or methylation.
Finally, it had been previously postulated that the mutation frequency was high in individuals with NF1, with estimates ranging between 1.4 × 10−4 (Crowe et al.1956) and 6.5 × 10−5 gamete/generation (Samuelsson and Axelsson 1981; Huson et al. 1989; Clementi et al. 1990). Here, in contrast, using WGS data, we find that the mutation frequency in NF1-PA is relatively low, similar to that of other pediatric solid tumors where carcinogenesis often results from antecedent germline mutations. In T01, T02, and T03, the BMR, excluding noncoding repetitive sequence, was 9.0 × 10−8, 1.2 × 10−7, and 6.0 × 10−8 mutations per sequenced base, respectively, comparable with the BMRs of several pediatric retinoblastoma genomes (Zhang et al. 2012a) (2.0 × 10−8, 5.0 × 10−8, 8.0 × 10−8, and 9.0 × 10−8). These mutation frequencies differ slightly from hematological malignancies, such as pediatric TALL and the M1 and M3 subtypes of adult AML (2.0 × 10−8 to 4.1 × 10−8 mutations per base) (Mardis et al. 2009; Welch et al. 2012; Zhang et al. 2012b), and differ markedly from adult solid tumors, such as luminal breast cancer, that often have rates that are orders of magnitude higher (Fig. 3C; Mardis et al. 2009).
Discussion
This study represents the first WGS analysis of low-grade glioma and the first such examination of such tumors arising in the context of a common inherited brain tumor predisposition syndrome. Moreover, these data provide important information about the genomic landscape of NF1-PA, the clonal origins of these tumors, the various mechanisms underlying somatic NF1 tumor suppressor gene inactivation, and the extent of tumor cellular heterogeneity.
First, we employ WGS to show that bi-allelic NF1 gene inactivation is the primary genomic mutation in two of the three NF1-associated PA tumors. While we identified other genetic changes in each of the NF1-PA specimens, none of these mutations were found in more than one tumor, none exhibited bi-allelic inactivation, and none were located within a predicted functional domain of the encoded protein. Consistent with our previous studies (Li et al. 2001), no changes in the TP53, CDKN2A, RB1, EGFR, or CDK4 genes were observed. Together, these data favor the time-honored hypothesis that NF1 loss in the neoplastic cellular compartment is the key mutational driver of NF1-PA tumorigenesis.
Second, we demonstrate that NF1-PA have a monoclonal origin, similar to their sporadic counterparts (Payton et al. 2011). While the majority of studies that examined clonal origins of glioma using single X-chromosome loci, fluorescent in situ hybridization, microsatellite markers, or single substitute mutation methods demonstrated a monoclonal pattern (Kattar et al. 1997; Zhu et al. 1997; Mueller et al. 2001), there are reports of gliomas exhibiting a multi-clonal origin (Dong et al. 2002; Gomori et al. 2002; Vital et al. 2010). Since the few brain tumors with presumed polyclonal origins were high-grade gliomas, we favor a model of low-grade pediatric astrocytoma development in which tumors arise from a single population of glial progenitors during fetal or early postnatal life.
Third, we show that there exist multiple possible mechanisms of somatic NF1 gene silencing, including LOH, mutation, and methylation. While LOH and mutation have been reported in numerous NF1-associated tumor types, prior studies on another NF1-associated tumor (peripheral nerve sheath tumor; neurofibroma) revealed that NF1 promoter hypermethylation is a rare “second hit” event (Horan et al. 2000; Wimmer et al. 2002a; Harder et al. 2004; Fishbein et al. 2005). While we have not demonstrated a direct cause-and-effect relationship between NF1 locus hypermethylation and reduced NF1 gene expression, the finding that absent neurofibromin (immunohistochemistry) expression was observed in T01 is consistent with this conclusion. Our observations coupled with results on another childhood nervous system tumor, retinoblastoma (Zhang et al. 2012a), suggest that somatic tumor suppressor gene inactivation by methylation may represent another mode to achieve bi-allelic inactivation. However, it is possible that other mechanisms are operative, such as epigenetic silencing by microRNA.
Last, Nf1 GEM optic glioma models have been particularly instructive in elucidating the requirements for tumorigenesis. Collectively, these studies have revealed that the unique pattern of low-grade astrocytoma development (predominantly in the optic pathway/brainstem of children) reflects the coupling of at least two requirements: (1) Nf1 loss in specific neuroglial progenitor cells (third ventricle or brainstem) during a defined temporal window (embryonic development) (Lee et al. 2010, 2012) and (2) a supportive local microenvironment composed of critical molecular signals (Warrington et al. 2007, 2010) and non-neoplastic cell types (Daginakatte and Gutmann 2007; Daginakatte et al. 2008). The current study emphasizes the significant cellular heterogeneity in human NF1-PA, consistent with previous studies on glioma in which a large proportion of the cellular composition is non-neoplastic cells. One of these stromal cell types is microglia, an immune-like cell, analogous to macrophages outside of the central nervous system (Graeber et al. 2002; Watters et al. 2005). The importance of these stromal cells to NF1 glioma growth is further supported by several studies using Nf1 GEM strains. In these experiments, pharmacologic or genetic silencing of microglia reduces glioma growth and proliferation (Daginakatte and Gutmann 2007; Daginakatte et al. 2008; Simmons et al. 2011). The mechanism underlying their glioma-promoting properties has not been fully elucidated, but likely involves the generation of growth factors and chemokines (Daginakatte and Gutmann 2007; Warrington et al. 2007; Wesolowska et al. 2008; Zhai et al. 2011). This potentially instructive contribution of microglia has recently prompted us to apply RNA sequencing methods to more comprehensively define the role of microglia in sporadic PA formation and continued growth.
Methods
Patients
Blood and tumor specimens were obtained from three patients diagnosed with NF1 according to established criteria (Stumpf et al. 1988) treated at Washington University School of Medicine or the University of Toronto under active Human Studies Protocols approved by the Institutional Review Board at the Washington University School of Medicine and the University of Toronto. WGS was performed for tumor and normal DNA from three children with pilocytic astrocytomas. Tumor samples were obtained from banked resected tumors characterized by pathologic examination. Matched nontumor samples were obtained from PBMCs by phlebotomy.
Custom capture hybridization and sequencing
Sequencing libraries were hybridized to biotinylated 120-mer oligonucleotide probes designed to cover all coding exons of target genes. Illumina library quantification was completed using the KAPA SYBR FAST qPCR Kit (KAPA Biosystems).
Capture alignment, de-duplication, and BAM file generation
Illumina reads were aligned individually to the Ensembl release 45 version of Human NCBI Build 36 using BWA 2 v0.5.7 with soft trimming (-q 5) as previously reported (Mardis et al. 2009). For each sample, individual lane alignments in BAM format were merged together using SAMtools 3 r544. Duplicates were marked in the merged BAM files by the MarkDuplicates class of Picard 4 v1.17. Reads with mapping quality of zero, or that were marked as duplicates by Picard, were excluded from further analysis.
SNV and small indel detection
Candidate point mutations were predicted using SomaticSniper, previously referred to as glfSomatic 1.5. Putative SNVs with somatic score ≥40 and average mapping quality ≥40 were considered high-confidence (HC); all others were deemed low-confidence (LC). Small indels were predicted using a combination of GATK 6 IndelGenotyper, Pindel 7, and a modified version of SAMtools 3. Both SNVs and indels were annotated using gene structure, conservation information, and regulatory potential, then binned as previously described (Mardis et al. 2009). High-confidence noncoding mutations in nonrepetitive genomic regions were visualized to filter false positives, while nonsynonymous coding mutations were selected for additional PCR- and custom-capture-based validation sequencing.
Copy number variation (CNV) analysis
Copy number calls from WGS data were generated using the cnvHMM algorithm (http://genome.wustl.edu/software/cnvhmm). First, cnvHMM divided the genome into nonoverlapping windows 10 kbp in size. For each window, the raw copy number was estimated as the number of reads confidently mapped (mapping quality >35) divided by the median of such value across all windows in the genome, then multiplied by two (to reflect diploid genomes). A hidden Markov model (HMM) was then used to perform the segmentation. Each state in the HMM corresponds to a discrete copy number value. The emission probability was modeled as a single Gaussian probably density function. A standard Viterbi algorithm was used to estimate the HMM parameters and determined the segmentation. We derived copy number altered regions (CNARs) by comparing the segmentation of the tumor genome versus that of the normal genome. A log likelihood ratio (LLR) score was computed for each CNAR to quantify the possibility of being a true CNAR from being the null (copy number neutral) hypothesis. Calls with LLR score <20 were dropped from further consideration.
Structural variation (SV) analysis
Structural variants (SVs) were predicted by BreakDancer 9 with TIGRA_SV assembly 10, SquareDancer (Wang et al. 2011), and Pindel (Ye et al. 2009). Copy number differences with regions of SV were determined by BreakDancer (Chen et al. 2009). Putative SVs from BreakDancer were chosen for localized, de novo assembly if they (1) had at least two supporting discordant read pairs in the tumor, but none in the normal, (2) were supported by a copy number change of >0.5 in the SV region, (3) affected a coding exon, or (4) had one or more breakpoints in any part of a known cancer gene. SV regions were defined as ±500 bp from the predicted breakpoints. The depth-weighted average contig length was used as a quality measure of the assembly:
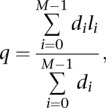 |
where li and di are the length and the average depth of contig i, respectively, and M is the total number of contigs in the TIGRA assembly. Short contigs (<30 bp) that are assembled from the homopolymer reads are excluded in the calculation. We found this statistic properly summarizes both the size and the coverage and therefore is more informative to measure the assembly quality than the N50 size. Based on our manual examination of the alignment results, we found that the high-quality assemblies that contain valid sequence frequently have q > 100 bp. Once assembled, contigs were aligned to their respective local reference sequences (SV region ±700 bp; NCBI Build b36) using cross_match v1.080721 with the parameters “–bandwidth 20 –minmatch 20 –minscore 25 –penalty –10 –discrep_lists –tags –gap_init –10 –gap_ext –1”. Contigs whose alignments had more than two segments or were significantly different from the reference (one segment with substitution >0.006 or indel rate of >0.002; or two segments with substitution rate >0.005, indel rate of >0.001, or more than five inserted/deleted bases) were removed. A complementary in-house algorithm, SquareDancer, was used to identify candidate SVs by utilizing soft-clipping information from the BWA aligner. Large deletions and moderately sized insertions were also predicted using a modified version of the Pindel 7 tool. Pindel calls with read support in the tumor but not the normal, as well as calls supported by copy number evidence from cnvHMM (copy number >0.5) were identified. Candidate SVs from BreakDancer, SquareDancer, and Pindel SV regions from different methods that overlapped >75% (allowing a buffer of ±50 bp) were merged into a single event.
PCR-based validation and screening
Using Primer3, universal-tailed PCR primers were designed to amplify all coding exons of NF1 in addition to the localized region surrounding all predicted nonsynonymous coding variants. Tumor DNA was PCR-amplified using AmpliTaq Gold PCR Master mix and sequenced using BigDye 3.1 Sequencing Kits and loaded on ABI 3730xl capillary sequencers. Data were assembled using Consed and putative mutations identified using Polyscan and Polyphred. All putative variants were visually reviewed to eliminate artifacts.
Hybridization validation data analysis
To validate predicted nonsynonymous coding SNVs and small (<3 bp) indels, we used VarScan 2.2.11 with the following parameters: –mincoverage 30 –min-var-freq 0.08 –P-value 0.10 –somatic-P-value 0.01 –validation 1. To validate low-frequency (2%–8%) SNVs, we reran VarScan with adjusted parameters: –min-coverage 100 –min-var-freq 0.02 –P-value 0.10 –somatic-P-value 0.01 –validation 1. In validation mode, VarScan reads data from tumor and normal samples simultaneously, performing pairwise comparisons at every position covered in both samples. Each position is classified as Reference (wild type), Germline, LOH, or Somatic, based upon a comparison of the consensus genotypes and supporting read counts (Fisher's exact test). Positions called Somatic are further subjected to our internally developed false-positive filter that removes sequencing- and alignment-related artifacts using several criteria (read count, mapping quality, average read position, strand representation, homopolymer-like sequence context, mismatch quality sum difference, trimmed read length, Q2 distance).
Mutation rate and spectrum analysis
Mutation rate analyses utilized validated nonsynonymous coding SNVs, high-confidence SNVs in potential regulatory regions, and SNVs in nonrepetitive regions that passed visual inspection. For the mutation rate calculation, the number of SNVs in each bin was compared to the overall size of the exome, noncoding genome with regulatory potential, and remaining nonrepetitive genome, which on NCBI Build 36, consisted of 43.89 Mbp, 248.21 Mbp, and 1198.99 Mbp, respectively. The individual mutation rates (mutations per megabase) were calculated independently then combined for all three bins to determine the overall mutation rate.
Methylation analysis
Methylation state was determined using the Infinium HumanMethylation450 BeadChip Kit (Gu et al. 2010; Bibikova et al. 2011; Sandoval et al. 2011). The HumanMethylation450 chip arrays >485,000 methylation sites per sample at single-nucleotide resolution. Aside from covering 96% of CpG islands, sites are also associated with regions of biological interest—e.g., miRNA promoter regions, differentially methylated sites identified in tumor versus normal (multiple forms of cancer) and across several tissue types—for epigenome-wide association studies (EWAS). Of the >485,000 sites on the Infinium array, 42 of the probes are located within the NF1 gene footprint (including upstream promoter region). The NF1 probes include UCSC-defined CpG island annotation (i.e., island, shore, and shelf designations). The 42 probes culled from the NF1 gene footprint have 13 probes associated with genes on the opposite strand of NF1: six for OMG, two for EVI2B, and five for EVI2A. These 42 probes were reviewed for methylation state across three different pilocytic astrocytomas from patients with known NF1 defects and two spontaneous pilocytic astrocytomas without NF1 defects. Infinium chip Beta-values (the ratio of the methylated probe intensity and the overall intensity: the sum of methylated and unmethylated probe intensities) were converted to M-values16 (log2 ratio of the intensities of methylated probe versus unmethylated probe) using the MethyLumi package from Bioconductor19 (Davis et al. 2012). Illumina's raw IDAT files were fed to the MethyLumi package for value conversion. Resulting M-values were tracked/interrogated across our samples by converting to YAML format, with subsequent parsing mediated by Perl scripts developed in-house. In this manner, M-values related to the 42 NF1-specific probes were collected and contrasted for our samples. Values were imported and manually reviewed as custom tracks in the UCSC Genome Browser (Kent et al. 2002) as methylation probe tracks, as well as M-value heat map blocks across our tumor and sporadic samples. Differences in M-values (Δ M-values) were calculated across our samples, in a pairwise round-robin fashion, with standard deviation used to assess significance in differential change. As values were assessed localized to the 42 probe NF1 gene region, a follow-up global assessment was undertaken for all 485,000+ probes across all samples, to review global significance of change, as Δ M-values, between samples. Our five tumor samples were evaluated in an all-versus-all fashion with 4,855,770 comparison values sampled in total. The mean Δ M-value was 0.52, standard deviation being 0.58. Any change in M-value >2.24 would be 3 standard deviations. For our purposes, we scaled back to an absolute Δ M-value of ≥2.0 in value being “significant”. This provided 137,628 comparisons matching the criteria (or only 2.83% of the total 4,855,770 cross-comparisons being relatively significant). Accepting these values as being representative of the sensitivity for the technology (i.e., not “error”), any Δ M-value ≥2.0 within NF1 would be considered significant. In addition, we performed a t-test to identify regions genome-wide exhibiting significantly differential methylation per probe. We clustered together probes 200 bp or closer with P ≤ 0.05 and determined 118 such probes in 105 clusters that exhibited significantly differential methylation. These regions were further annotated by identifying all genes occurring within 10 kbp (Supplemental Table 2).
Immunohistochemistry and qRT-PCR
Immunohistochemistry and real-time quantitative RNA RT-PCR were performed as previously reported (Gutmann et al. 2000; Sharma et al. 2007).
Data access
All sequence data have been submitted to the database of Genotypes and Phenotypes (dbGaP) (http://www.ncbi.nlm.nih.gov/gap) under accession number phs000563.v1.p1.
Acknowledgments
We thank Mr. Ryan Emnett for expert technical assistance and Sonika Dahiya for neuropathological consultation. We acknowledge the efforts of the following people and groups at The Genome Institute for their contributions to this manuscript: the Analysis Pipeline group for developing the automated analysis pipelines that generated alignments and somatic variants; the LIMS group for developing tools to manage validation array ordering, capture, and sequencing, Joelle Veizer and Heather Schmidt for manual review of predicted mutations; and Joshua F. McMichael and Charles Lu for their assistance preparing the tables and figures. This work was supported by a Doris Duke Fellowship (to I.H.) and core facilities in the Washington University CTSA (UL1-RR024992). We dedicate this work in memory of our dear colleague and friend, Abhijit Guha, who died during the final preparation of this manuscript.
Author contributions: D.H.G., E.R.M., and L.D. designed the experiments, L.L.F. and R.S.F. supervised data production and validation processes, L.D., M.D.M, and J.W.W. led the data analysis, V.M., R.D., C.A.M., C.K., T.W., and M.D.M. performed data analysis, L.D., J.W.W., T.W., and M.D.M prepared figures and tables, A.G. and J.R.L. provided samples and clinical data, T.W., R.D., and V.M. performed laboratory experiments, D.H.G. and E.R.M. developed the project concept, and I.H., J.W.W., M.D.M., L.D., E.R.M., and D.H.G. wrote the manuscript.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.142604.112.
References
- Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR 2010. A method and server for predicting damaging missense mutations. Nat Methods 7: 248–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajenaru ML, Zhu Y, Hedrick NM, Donahoe J, Parada LF, Gutmann DH 2002. Astrocyte-specific inactivation of the neurofibromatosis 1 gene (NF1) is insufficient for astrocytoma formation. Mol Cell Biol 22: 5100–5113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajenaru ML, Hernandez MR, Perry A, Zhu Y, Parada LF, Garbow JR, Gutmann DH 2003. Optic nerve glioma in mice requires astrocyte Nf1 gene inactivation and Nf1 brain heterozygosity. Cancer Res 63: 8573–8577 [PubMed] [Google Scholar]
- Bajenaru ML, Garbow JR, Perry A, Hernandez MR, Gutmann DH 2005. Natural history of neurofibromatosis 1-associated optic nerve glioma in mice. Ann Neurol 57: 119–127 [DOI] [PubMed] [Google Scholar]
- Bibikova M, Barnes B, Tsan C, Ho V, Klotzle B, Le JM, Delano D, Zhang L, Schroth GP, Gunderson KL, et al. 2011. High density DNA methylation array with single CpG site resolution. Genomics 98: 288–295 [DOI] [PubMed] [Google Scholar]
- Chen K, Wallis JW, McLellan MD, Larson DE, Kalicki JM, Pohl CS, McGrath SD, Wendl MC, Zhang Q, Locke DP, et al. 2009. BreakDancer: An algorithm for high-resolution mapping of genomic structural variation. Nat Methods 6: 677–681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K, Wallis JW, Kandoth C, Kalicki-Veizer JM, Mungall KL, Mungall AJ, Jones SJ, Marra MA, Ley TJ, Mardis ER, et al. 2012. BreakFusion: Targeted assembly-based identification of gene fusions in whole transcriptome paired-end sequencing data. Bioinformatics 28: 1923–1924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clementi M, Barbujani G, Turolla L, Tenconi R 1990. Neurofibromatosis-1: A maximum likelihood estimation of mutation rate. Hum Genet 84: 116–118 [DOI] [PubMed] [Google Scholar]
- Crowe FW, Schull WJ, Neel JV 1956. A clinical pathological and genetic study of multiple neurofibromatosis. Thomas, Springfield, IL [Google Scholar]
- Daginakatte GC, Gutmann DH 2007. Neurofibromatosis-1 (Nf1) heterozygous brain microglia elaborate paracrine factors that promote Nf1-deficient astrocyte and glioma growth. Hum Mol Genet 16: 1098–1112 [DOI] [PubMed] [Google Scholar]
- Daginakatte GC, Gianino SM, Zhao NW, Parsadanian AS, Gutmann DH 2008. Increased c-Jun-NH2-kinase signaling in neurofibromatosis-1 heterozygous microglia drives microglia activation and promotes optic glioma proliferation. Cancer Res 68: 10358–10366 [DOI] [PubMed] [Google Scholar]
- Davis S, Du P, Bilke S, Triche T Jr, Bootwalla M 2012. methylumi: Handle Illumina methylation data. Bioconductor R package version 1.9.9. http://www.bioconductor.org/packages/release/bioc/html/methylumi.html
- Deshmukh H, Yeh TH, Yu J, Sharma MK, Perry A, Leonard JR, Watson MA, Gutmann DH, Nagarajan R 2008. High-resolution, dual-platform aCGH analysis reveals frequent HIPK2 amplification and increased expression in pilocytic astrocytomas. Oncogene 27: 4745–4751 [DOI] [PubMed] [Google Scholar]
- Ding L, Ley TJ, Larson DE, Miller CA, Koboldt DC, Welch JS, Ritchey JK, Young MA, Lamprecht T, McLellan MD, et al. 2012. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature 481: 506–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong ZQ, Pang JC, Tong CY, Zhou LF, Ng HK 2002. Clonality of oligoastrocytomas. Hum Pathol 33: 528–535 [DOI] [PubMed] [Google Scholar]
- Ellis MJ, Ding L, Shen D, Luo J, Suman VJ, Wallis JW, Van Tine BA, Hoog J, Goiffon RJ, Goldstein TC, et al. 2012. Whole-genome analysis informs breast cancer response to aromatase inhibition. Nature 486: 353–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fishbein L, Eady B, Sanek N, Muir D, Wallace MR 2005. Analysis of somatic NF1 promoter methylation in plexiform neurofibromas and Schwann cells. Cancer Genet Cytogenet 157: 181–186 [DOI] [PubMed] [Google Scholar]
- Gomori E, Fulop Z, Meszaros I, Doczi T, Matolcsy A 2002. Microsatellite analysis of primary and recurrent glial tumors suggests different modalities of clonal evolution of tumor cells. J Neuropathol Exp Neurol 61: 396–402 [DOI] [PubMed] [Google Scholar]
- Gonzalez-Perez A, Lopez-Bigas N 2011. Improving the assessment of the outcome of nonsynonymous SNVs with a consensus deleteriousness score, Condel. Am J Hum Genet 88: 440–449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graeber MB, Scheithauer BW, Kreutzberg GW 2002. Microglia in brain tumors. Glia 40: 252–259 [DOI] [PubMed] [Google Scholar]
- Gu H, Bock C, Mikkelsen TS, Jager N, Smith ZD, Tomazou E, Gnirke A, Lander ES, Meissner A 2010. Genome-scale DNA methylation mapping of clinical samples at single-nucleotide resolution. Nat Methods 7: 133–136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutmann DH, Aylsworth A, Carey JC, Korf B, Marks J, Pyeritz RE, Rubenstein A, Viskochil D 1997. The diagnostic evaluation and multidisciplinary management of neurofibromatosis 1 and neurofibromatosis 2. JAMA 278: 51–57 [PubMed] [Google Scholar]
- Gutmann DH, Donahoe J, Brown T, James CD, Perry A 2000. Loss of neurofibromatosis 1 (NF1) gene expression in NF1-associated pilocytic astrocytomas. Neuropathol Appl Neurobiol 26: 361–367 [DOI] [PubMed] [Google Scholar]
- Gutmann DH, James CD, Poyhonen M, Louis DN, Ferner R, Guha A, Hariharan S, Viskochil D, Perry A 2003. Molecular analysis of astrocytomas presenting after age 10 in individuals with NF1. Neurology 61: 1397–1400 [DOI] [PubMed] [Google Scholar]
- Harder A, Rosche M, Reuss DE, Holtkamp N, Uhlmann K, Friedrich R, Mautner VF, von Deimling A 2004. Methylation analysis of the neurofibromatosis type 1 (NF1) promoter in peripheral nerve sheath tumours. Eur J Cancer 40: 2820–2828 [DOI] [PubMed] [Google Scholar]
- Horan MP, Cooper DN, Upadhyaya M 2000. Hypermethylation of the neurofibromatosis type 1 (NF1) gene promoter is not a common event in the inactivation of the NF1 gene in NF1-specific tumours. Hum Genet 107: 33–39 [DOI] [PubMed] [Google Scholar]
- Huson SM, Compston DA, Harper PS 1989. A genetic study of von Recklinghausen neurofibromatosis in south east Wales. II. Guidelines for genetic counselling. J Med Genet 26: 712–721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones DT, Kocialkowski S, Liu L, Pearson DM, Backlund LM, Ichimura K, Collins VP 2008. Tandem duplication producing a novel oncogenic BRAF fusion gene defines the majority of pilocytic astrocytomas. Cancer Res 68: 8673–8677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kattar MM, Kupsky WJ, Shimoyama RK, Vo TD, Olson MW, Bargar GR, Sarkar FH 1997. Clonal analysis of gliomas. Hum Pathol 28: 1166–1179 [DOI] [PubMed] [Google Scholar]
- Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D 2002. The human genome browser at UCSC. Genome Res 12: 996–1006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KY, Ju WK, Hegedus B, Gutmann DH, Ellisman MH 2010. Ultrastructural characterization of the optic pathway in a mouse model of neurofibromatosis-1 optic glioma. Neuroscience 170: 178–188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kluwe L, Hagel C, Tatagiba M, Thomas S, Stavrou D, Ostertag H, von Deimling A, Mautner VF 2001. Loss of NF1 alleles distinguish sporadic from NF1-associated pilocytic astrocytomas. J Neuropathol Exp Neurol 60: 917–920 [DOI] [PubMed] [Google Scholar]
- Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, Miller CA, Mardis ER, Ding L, Wilson RK 2012. VarScan 2: Somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res 22: 568–576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee DY, Yeh TH, Emnett RJ, White CR, Gutmann DH 2010. Neurofibromatosis-1 regulates neuroglial progenitor proliferation and glial differentiation in a brain region-specific manner. Genes Dev 24: 2317–2329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee DY, Gianino SM, Gutmann DH 2012. Innate neural stem cell heterogeneity determines the patterning of glioma formation in children. Cancer Cell 22: 131–138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard JR, Perry A, Rubin JB, King AA, Chicoine MR, Gutmann DH 2006. The role of surgical biopsy in the diagnosis of glioma in individuals with neurofibromatosis-1. Neurology 67: 1509–1512 [DOI] [PubMed] [Google Scholar]
- Li J, Perry A, James CD, Gutmann DH 2001. Cancer-related gene expression profiles in NF1-associated pilocytic astrocytomas. Neurology 56: 885–890 [DOI] [PubMed] [Google Scholar]
- Listernick R, Charrow J, Greenwald M, Mets M 1994. Natural history of optic pathway tumors in children with neurofibromatosis type 1: A longitudinal study. J Pediatr 125: 63–66 [DOI] [PubMed] [Google Scholar]
- Listernick R, Louis DN, Packer RJ, Gutmann DH 1997. Optic pathway gliomas in children with neurofibromatosis 1: Consensus statement from the NF1 Optic Pathway Glioma Task Force. Ann Neurol 41: 143–149 [DOI] [PubMed] [Google Scholar]
- Louis DN, Ohgaki H, Wiestler OD, Cavenee WK 2007. The WHO classification of tumours of the central nervous system. IARC, Lyon [Google Scholar]
- Mardis ER, Ding L, Dooling DJ, Larson DE, McLellan MD, Chen K, Koboldt DC, Fulton RS, Delehaunty KD, McGrath SD, et al. 2009. Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med 361: 1058–1066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller W, Lass U, Herms J, Kuchelmeister K, Bergmann M, von Deimling A 2001. Clonal analysis in glioblastoma with epithelial differentiation. Brain Pathol 11: 39–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng PC, Henikoff S 2003. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res 31: 3812–3814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payton JE, Schmidt J, Yu J, Lusis EA, Watson MA, Gutmann DH 2011. Genome-wide polymorphism analysis demonstrates a monoclonal origin of pilocytic astrocytoma. Neuropathol Appl Neurobiol 37: 321–325 [DOI] [PubMed] [Google Scholar]
- Pfister S, Janzarik WG, Remke M, Ernst A, Werft W, Becker N, Toedt G, Wittmann A, Kratz C, Olbrich H, et al. 2008. BRAF gene duplication constitutes a mechanism of MAPK pathway activation in low-grade astrocytomas. J Clin Invest 118: 1739–1749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuelsson B, Axelsson R 1981. Neurofibromatosis. A clinical and genetic study of 96 cases in Gothenburg, Sweden. Acta Derm Venereol Suppl (Stockh) 95: 67–71 [PubMed] [Google Scholar]
- Sandoval J, Heyn H, Moran S, Serra-Musach J, Pujana MA, Bibikova M, Esteller M 2011. Validation of a DNA methylation microarray for 450,000 CpG sites in the human genome. Epigenetics 6: 692–702 [DOI] [PubMed] [Google Scholar]
- Sharma MK, Zehnbauer BA, Watson MA, Gutmann DH 2005. RAS pathway activation and an oncogenic RAS mutation in sporadic pilocytic astrocytoma. Neurology 65: 1335–1336 [DOI] [PubMed] [Google Scholar]
- Sharma MK, Mansur DB, Reifenberger G, Perry A, Leonard JR, Aldape KD, Albin MG, Emnett RJ, Loeser S, Watson MA, et al. 2007. Distinct genetic signatures among pilocytic astrocytomas relate to their brain region origin. Cancer Res 67: 890–900 [DOI] [PubMed] [Google Scholar]
- Sigrist CJ, Cerutti L, de Castro E, Langendijk-Genevaux PS, Bulliard V, Bairoch A, Hulo N 2010. PROSITE, a protein domain database for functional characterization and annotation. Nucleic Acids Res 38: D161–D166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmons GW, Pong WW, Emnett RJ, White CR, Gianino SM, Rodriguez FJ, Gutmann DH 2011. Neurofibromatosis-1 heterozygosity increases microglia in a spatially and temporally restricted pattern relevant to mouse optic glioma formation and growth. J Neuropathol Exp Neurol 70: 51–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spurlock G, Knight SJ, Thomas N, Kiehl TR, Guha A, Upadhyaya M 2010. Molecular evolution of a neurofibroma to malignant peripheral nerve sheath tumor (MPNST) in an NF1 patient: Correlation between histopathological, clinical and molecular findings. J Cancer Res Clin Oncol 136: 1869–1880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stumpf DA, Alksne JF, Annegers JF 1988. Neurofibromatosis. Conference statement. National Institutes of Health Consensus Development Conference. Arch Neurol 45: 575–578 [PubMed] [Google Scholar]
- Tada K, Kochi M, Saya H, Kuratsu J, Shiraishi S, Kamiryo T, Shinojima N, Ushio Y 2003. Preliminary observations on genetic alterations in pilocytic astrocytomas associated with neurofibromatosis 1. Neuro Oncol 5: 228–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vital AL, Tabernero MD, Crespo I, Rebelo O, Tao H, Gomes F, Lopes MC, Orfao A 2010. Intratumoral patterns of clonal evolution in gliomas. Neurogenetics 11: 227–239 [DOI] [PubMed] [Google Scholar]
- Wang J, Mullighan CG, Easton J, Roberts S, Heatley SL, Ma J, Rusch MC, Chen K, Harris CC, Ding L, et al. 2011. CREST maps somatic structural variation in cancer genomes with base-pair resolution. Nat Methods 8: 652–654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warrington NM, Woerner BM, Daginakatte GC, Dasgupta B, Perry A, Gutmann DH, Rubin JB 2007. Spatiotemporal differences in CXCL12 expression and cyclic AMP underlie the unique pattern of optic glioma growth in neurofibromatosis type 1. Cancer Res 67: 8588–8595 [DOI] [PubMed] [Google Scholar]
- Warrington NM, Gianino SM, Jackson E, Goldhoff P, Garbow JR, Piwnica-Worms D, Gutmann DH, Rubin JB 2010. Cyclic AMP suppression is sufficient to induce gliomagenesis in a mouse model of neurofibromatosis-1. Cancer Res 70: 5717–5727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watters JJ, Schartner JM, Badie B 2005. Microglia function in brain tumors. J Neurosci Res 81: 447–455 [DOI] [PubMed] [Google Scholar]
- Welch JS, Ley TJ, Link DC, Miller CA, Larson DE, Koboldt DC, Wartman LD, Lamprecht TL, Liu F, Xia J, et al. 2012. The origin and evolution of mutations in acute myeloid leukemia. Cell 150: 264–278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wesolowska A, Kwiatkowska A, Slomnicki L, Dembinski M, Master A, Sliwa M, Franciszkiewicz K, Chouaib S, Kaminska B 2008. Microglia-derived TGF-β as an important regulator of glioblastoma invasion—an inhibition of TGF-β-dependent effects by shRNA against human TGF-β type II receptor. Oncogene 27: 918–930 [DOI] [PubMed] [Google Scholar]
- Wimmer K, Eckart M, Meyer-Puttlitz B, Fonatsch C, Pietsch T 2002a. Mutational and expression analysis of the NF1 gene argues against a role as tumor suppressor in sporadic pilocytic astrocytomas. J Neuropathol Exp Neurol 61: 896–902 [DOI] [PubMed] [Google Scholar]
- Wimmer K, Zhu XX, Rouillard JM, Ambros PF, Lamb BJ, Kuick R, Eckart M, Weinhausl A, Fonatsch C, Hanash SM 2002b. Combined restriction landmark genomic scanning and virtual genome scans identify a novel human homeobox gene, ALX3, that is hypermethylated in neuroblastoma. Genes Chromosomes Cancer 33: 285–294 [DOI] [PubMed] [Google Scholar]
- Ye K, Schulz MH, Long Q, Apweiler R, Ning Z 2009. Pindel: A pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics 25: 2865–2871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhai H, Heppner FL, Tsirka SE 2011. Microglia/macrophages promote glioma progression. Glia 59: 472–485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Benavente CA, McEvoy J, Flores-Otero J, Ding L, Chen X, Ulyanov A, Wu G, Wilson M, Wang J, et al. 2012a. A novel retinoblastoma therapy from genomic and epigenetic analyses. Nature 481: 329–334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Ding L, Holmfeldt L, Wu G, Heatley SL, Payne-Turner D, Easton J, Chen X, Wang J, Rusch M, et al. 2012b. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature 481: 157–163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu JJ, Leon SP, Folkerth RD, Guo SZ, Wu JK, Black PM 1997. Evidence for clonal origin of neoplastic neuronal and glial cells in gangliogliomas. Am J Pathol 151: 565–571 [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Harada T, Liu L, Lush ME, Guignard F, Harada C, Burns DK, Bajenaru ML, Gutmann DH, Parada LF 2005. Inactivation of NF1 in CNS causes increased glial progenitor proliferation and optic glioma formation. Development 132: 5577–5588 [DOI] [PMC free article] [PubMed] [Google Scholar]