

Abstract
Transcriptional enhancement of X-linked genes to compensate for the sex chromosome monosomy in Drosophila males is brought about by a ribonucleoprotein assembly called Male-Specific-Lethal or Dosage Compensation Complex (MSL-DCC). This machinery is formed in male flies and specifically associates with active genes on the X chromosome. After assembly at dedicated high-affinity “entry” sites (HAS) on the X chromosome, the complex distributes to the nearby active chromatin. High-resolution, genome-wide mapping of the MSL-DCC subunits by chromatin immunoprecipitation (ChIP) on oligonucleotide tiling arrays suggests a rather homogenous spreading of the intact complex onto transcribed chromatin. Coupling ChIP to deep sequencing (ChIP-seq) promises to map the chromosomal interactions of the DCC with improved resolution. We present ChIP-seq binding profiles for all complex subunits, including the first description of the RNA helicase MLE binding pattern. Exploiting the preferential representation of direct chromatin contacts upon high-energy shearing, we report a surprising functional and topological separation of MSL protein contacts at three classes of chromosomal binding sites. Furthermore, precise determination of DNA fragment lengths by paired-end ChIP-seq allows decrypting of the local complex architecture. Primary contacts of MSL-2 and MLE define HAS for the DCC. In contrast, association of the DCC with actively transcribed gene bodies is mediated by MSL-3 binding to nucleosomes. We identify robust MSL-1/MOF binding at a fraction of active promoters genome-wide. Correlation analyses suggest that this association reflects a function outside dosage compensation. Our comprehensive analysis provides a new level of information on different interaction modes of a multiprotein complex at distinct regions within the genome.
Genes on the single X chromosome in Drosophila melanogaster males are subjected to transcriptional enhancement in order to meet the levels of expression product in females that carry two X chromosomes. This process is referred to as dosage compensation (DC). Even though similar compensatory processes can be observed in several unrelated heterogametic organisms, major principles and mechanisms differ substantially (Straub and Becker 2007; Mank 2009). In Drosophila, a ribonucleoprotein complex called Dosage Compensation Complex (DCC) or Male-Specific-Lethal (MSL) complex (MSL-DCC) constitutes specifically in males where it targets X-chromosomal genes (Larsson and Meller 2006; Gelbart and Kuroda 2009; Lucchesi 2009; Conrad and Akhtar 2011). Genetic screenings for male-specific lethality identified MSL-1, MSL-2, MSL-3, the histone acetyl transferase MOF, and the RNA/DNA helicase MLE as protein subunits. Two redundant noncoding RNAs—roX1 and roX2—complete the complex. MOF acetylates histone H4 at lysine 16 (H4K16ac), a modification that is expected to promote the unfolding of the chromatin fiber (Shogren-Knaak et al. 2006), boosting gene expression via enhanced transcriptional elongation (Larschan et al. 2011).
Correct targeting of the MSL-DCC poses a major challenge, as ∼1000 active genes on the X chromosome must be selectively identified. Based on a multitude of genetic and biochemical studies, a two-step model has been proposed (for reviews, see Gelbart and Kuroda 2009; Conrad and Akhtar 2011; Straub and Becker 2011): First, the dosage compensation machinery is attracted to ∼100 initiation sites along the X, termed high-affinity sites (HAS) or chromosomal entry sites (CES). In a second step the complex is disseminated to active target genes in the vicinity of these sites. Genetic analyses of the MSL genes point to a crucial role of MSL-2 and MSL-1 in the identification of HAS/CES as these two factors can bind these selected sites in the absence of all other dosage compensation components (Lyman et al. 1997). HAS targeting most likely involves specific DNA sequence motifs. A GA-rich motif is highly enriched in these regions and contributes to complex recruitment (Alekseyenko et al. 2008; Straub et al. 2008). Conceivably, a core complex consisting of MSL-2 and MSL-1 is involved in recognizing this sequence, since MSL-2 is a DNA binding protein (Fauth et al. 2010).
The distribution of the MSL-DCC to active gene targets requires the enzymatic activities of MLE and MOF (Gu et al. 2000; Morra et al. 2008), the presence of MSL-3, and at least one of the two roX RNAs (Kelley et al. 1999; Meller and Rattner 2002). It has been proposed that the contact with transcribed chromatin is established by recognition of H3 trimethylated on lysine 36 (H3K36me3) through MSL-3 (Larschan et al. 2007).
Complex assembly is triggered by male-specific expression of MSL-2. Importantly, all other MSL proteins are expressed in females, suggesting their involvement in processes outside the realm of dosage compensation. Given the male-specific lethal phenotype of loss-of-function mutations, however, these functions are probably not essential. MLE is required for the editing of a Na+-channel mRNA (Reenan et al. 2000). MOF is part of the so-called “Non-Specific-Lethal” (NSL) complex, which preferentially binds promoters of some housekeeping genes in both sexes, most likely serving a role in transcription initiation (Prestel et al. 2010; Raja et al. 2010; Feller et al. 2012). Functions for MSL-1 and MSL-3 outside of the dosage compensation system are not known even though both are expressed at low levels in females.
During recent years, genome-wide mapping studies have revealed in great details the global binding pattern of the MSL proteins and roX RNAs (Straub and Becker 2011). These studies confirm the overwhelming enrichment of the complex on the X chromosome in males. The MSL proteins studied so far (MSL-1, MSL-2, MSL-3, MOF) preferentially bind the bodies of active genes, in many cases with clear 3′ enrichment. Even though the binding patterns of the different MSLs show some variation, current models assume that all MSL proteins, in the context of a well-defined MSL-DCC, are involved in all steps of targeting and dissemination (Gelbart and Kuroda 2009; Conrad and Akhtar 2011; Straub and Becker 2011).
We present here the first comprehensive description of the MLE binding pattern. Comparing ChIP-chip with ChIP-seq profiles (in the former assay the ChIP material is used to probe DNA microarrays, whereas in the latter the recovered DNA is determined by deep sequencing) revealed striking differences. A systematic analysis of the phenomenon showed that the chromatin shearing protocol we employed allowed us to visualize the primary contacts of the MSL proteins at different chromatin targets. The data reveal different modes of MSL interactions at HAS and within genes, demonstrate an unexpected contribution of MLE to a novel HAS definition, and point to a novel function of MSL-1 and MOF outside the compensation process. Our experimental strategy allowed the assessment of the topology of large protein complexes at distinct classes of chromosomal interaction sites and it may be applied to other regulatory processes outside of the dosage compensation system.
Results
Comparative ChIP-chip and ChIP-seq mapping of all MSL proteins reveals striking differences
Global mapping of MSL-DCC subunits by ChIP-chip revealed a general co-localization of all tested components, mainly at transcribed gene sequences (for review, see Straub and Becker 2011). The RNA helicase MLE is thought to be more loosely associated with the other MSLs as it is easily lost upon purification of the complex or IP at slightly elevated stringency (Smith et al. 2000). A comparison between the chromosomal interactions of the RNA/DNA helicase MLE and the remainder of the MSLs was of interest. We generated ChIP-chip profiles for MLE and found the helicase broadly co-localizing with the remainder of the MSL proteins in male Drosophila S2 cells (Fig. 1A, see below).
Figure 1.

Systematic mapping of MSL proteins by ChIP-seq reveals novel binding qualities at high resolution. (A) ChIP-chip coverage profiles of MSL complex features on a representative X-chromosomal locus. (B) Corresponding ChIP-seq profiles. Genes above the x-scale are transcribed from left to right, genes below are transcribed from right to left. Exons are shown as boxes, introns as lines. The y-scale reflects the continuous unsmoothed average signal enrichment of the IP over the input samples. The x-scale reflects the chromosomal position in kilobases. (C) MOF and MSL-1 ChIP enrichment on the X-chromosomal Set2 locus as determined by ChIP-seq (cs) and ChIP-chip (cc). (D) qPCR quantitation of MOF enrichment using amplicons tiled along the Set2 locus. Chromatin was sheared to ∼180 bp using the Covaris S220 prior to IP. Amplicons correspond to the red boxes indicated in panel C ordered from left to right with the first serving as control (unbound) locus. Error bars reflect the standard error of the mean (SEM) of three independent biological replicates. (E) qPCR quantitation on a subset of amplicons comparing MSL-1-ChIP performed on weakly (800 bp, gray bars) and strongly (180 bp, white bars) sheared chromatin. Error bars reflect the standard error of the mean (SEM) of two independent biological replicates.
In order to increase the sensitivity and resolution of the mapping we applied more advanced ChIP-seq methodology to all MSL proteins, including MLE (Fig. 1B). Their interactions with S2 cell chromatin were mapped in at least two biological replicates. In an attempt to maximize the resolution and to obtain sequences from most of the immunoprecipitated DNA we subjected the chromatin preparations to extensive shearing. The most homogenous small size was achieved through Adaptive Focused Acoustics technology (Covaris) (Supplemental Fig. S1). The chromatin interaction profiles for MSL-3 and H4K16 acetylation were very similar in ChIP-chip and ChIP-seq data sets, demonstrating that, in principle, the two approaches are able to reveal the known extended binding qualities at transcribed sequences. Intriguingly, however, some striking differences between the profiles obtained by the microarray and sequencing strategies became obvious (Fig. 1, cf. A and B). The profiles of MSL-1 and MOF resembled each other but deviated qualitatively from the expected pattern. While the ChIP-chip mapping of these proteins shows the broad distribution over gene bodies, the ChIP-seq profiles mainly show sharp peaks. The chromosomal interactions of MSL-2 and MLE are again very similar, yet different from the others. They show a tendency toward well-defined peaks within the broad MSL-3 domains, but these are qualitatively different from the MSL-1/MOF signals. These results were highly provocative since we expected at least the “core” components MSL-1 and MSL-2 to co-localize on the chromosome.
We next validated the unexpected enrichments by quantitative PCR (qPCR). Figure 1C shows the analysis for a representative case: The ChIP-seq profile of MSL-1 and MOF on the X-chromosomal model gene Set2 shows two strong peaks and does not reproduce the broad enrichment on the transcription unit. The ChIP-seq peak at the 3′ end of Set2 coincides with an MSL-2 peak at a HAS (see below). The prominent signal at the promoter only appears in ChIP-seq studies. A series of qPCR amplicons were designed to interrogate different positions along the gene (red bars in Fig. 1C) taking care to precisely place amplicons #3 and #8 within the peak areas. The peak bases are usually <300 bp wide and a precise placement of primers is necessary to pick up the strongest enrichments. The qPCR analysis confirmed the strong enrichments at the promoter and the HAS as well as the low gene body binding (Fig. 1D). These results exclude a signal distortion due to the specifics of sample processing and subsequent data analysis related to the sequencing technology.
High-energy shearing as applied for ChIP-seq disrupts indirect chromatin interactions and exposes direct contacts
Discrepancies between global ChIP profiles in the literature can be related to differences in fragmentation of chromatin (Fan et al. 2008). In fact, MOF promoter peaks have been described before (Kind et al. 2008), when chromatin was sheared more intensely than in studies reporting rather distributed MOF binding (Straub et al. 2008). We therefore monitored the MSL-1 interactions at Set2 upon high- and low-energy shearing. Reducing the shear force converted the “peak”-type pattern into a broader profile that resembled the ChIP-chip pattern (Fig. 1, cf. E and C). Notably, the enrichment of the promoter signal (amplicon #3) over coding sequences (amplicons #5, #6) was completely leveled. Similar results were obtained for MOF (data not shown).
Although an increased length of input chromatin fragments reduces the resolution of the ChIP analysis, we do not think that this is the cause for the distinct binding profiles (Fig. 1A,B). First, the distances between amplicons #5 and #6 and the next peak is ∼3 kb, much larger than any average fragment size in any of our experiments. Second, the broad distribution observed on the gene bodies is asymmetric with reference to both, the promoter and 3′ peaks. A highly resolved peak would give rise to a symmetrically expanded area if the resolution were lowered. Third, the MSL-3 profiles, which are highly similar in both types of studies, are derived from the same chromatin preparations as all other profiles. Finally, similar changes in enrichment were detected on regions that are even further away from a HAS peak (>30 kb) (data not shown).
We furthermore observed that the MSL proteins get increasingly degraded by increased sonication levels (Supplemental Fig. S1G). Accordingly, we conclude that the key difference between high- and low-shear chromatin is that the MSL-DCC itself is fragmented and some binding qualities within the MSL complex are selectively lost. If the complex was disrupted during the fragmentation of the chromatin, the ChIP analysis should preferentially visualize those proteins that are directly cross-linked to DNA, i.e., the most direct chromatin interactions. More indirect chromatin associations mediated by protein–protein or protein–roX RNA cross-links within the complex would tend to be lost. In the context of reference ChIP-chip profiles derived from moderately sheared chromatin, the ChIP-seq signals obtained from highly fragmented chromatin provide an opportunity to uncover the chromatin binding modes of each individual MSL protein. The finding that the different MSL proteins show distinct binding patterns (Fig. 1B) suggests that the complex has a different topology at different chromatin locations. The following analysis is entirely consistent with this hypothesis and provides some striking new insights into the differential interactions of the dosage compensation machinery.
MSL-3 constitutes the interface of the MSL-DCC at transcribed chromatin marked with H3K36me3
The above argument states that increased shear force disrupts the MSL-DCC but not the underlying chromatin infrastructure. Indeed, ChIP-chip and ChIP-seq yielded very similar maps for nucleosomes carrying the H4K16 acetylation mark, a trace of the global action of the MSL-DCC on transcribed chromatin (Fig. 2A). The only MSL protein that likewise enriches on the bodies of active X-chromosomal genes in ChIP-seq is MSL-3 (Fig. 2A). Intriguingly, both types of ChIP analyses reveal that the enrichment of MSL-3 on transcribed sequences is frequently interrupted at positions of long introns (Fig. 2B). MSL-3 is supposed to tether the MSL-DCC to transcribed chromatin through recognition of the H3K36me3 mark with its chromo-barrel domain (Larschan et al. 2007). In agreement with this notion, the long MSL-3-free introns are also devoid of H3K36me3. Instead, these regions show a different chromatin signature, which is particularly enriched in histone acetylation and the histone H3.3 variant (Fig. 2B).
Figure 2.

MSL-3 associates with active chromatin marked by H3K36me3. (A) Average ChIP-seq enrichment of MSL-3 and H4K16ac along active genes on the X (red line, n = 1113) and the autosomes (dark blue line, n = 5341). Shaded areas above and below the solid lines describe the interquartile range of enrichment. (B) Chromatin features around a representative locus comprising a gene with a long intron. ChIP-seq (cs) and ChIP-chip (cc) derived maps for MSL-3 and selected ChIP-chip profiles derived from the modEncode project are shown. (C) Clustering of Drosophila genes based on their exon structure (active, non-nested, X-linked genes >1000 bp and <10000 bp in length, n = 592). The color scale indicates the intron density within a sampling bin. (D) Average MSL-3 ChIP-seq profiles on genes with (n = 201, top) and without (n = 391, bottom) large 3′ introns. Genes were scaled for length. Boxplots on the right show the distribution of the ratios of 3′ half to 5′ half of the signals. Signals on HAS have been masked for this analysis.
The example shown is representative of a major class of Drosophila genes that contain long introns. Clustering all genes based on their exon structure leads us to define two categories: Class 1 genes contain extended intronic regions, whereas class 2 genes have a low overall intron content. Interestingly, large introns in class 1 genes cluster at the 5′ end of genes (Fig. 2C). We observed a cumulative 3′ enrichment of MSL-3 on class 1 genes but not on class 2 genes (Fig. 2D). Our refined analysis suggests that this 3′ enrichment is not an intrinsic feature of MSL-DCC recruitment but rather due to a selective depletion of the MSL-3 interaction on long intronic DNA lacking the H3K36me3 mark that tends to be located toward the 5′ end of genes. Since no other MSL protein shows enrichment at transcribed chromatin similar to MSL-3, we conclude that the MSL-3–H3K36me3 interaction is the primary contact of the MSL-DCC with transcribed chromatin.
Co-localization of MLE with MSL-2 at high-affinity binding sites for the MSL-DCC
Following our reasoning, the narrow “peaks” of interactions observed for the other MSL proteins should reveal their primary contact sites. Until now, global MLE binding patterns have not been described. Using two different antibodies, we generated robust MLE profiles that complete the mapping of all dosage compensation protein components in Drosophila S2 cells (Fig. 3A). The ChIP-seq and ChIP-chip profiles of the MLE chromatin associations essentially look the same. Strikingly, the binding pattern of MLE on the X chromosome is very localized and matches very well the one of MSL-2 (Fig. 3A; Supplemental Fig. S3A). In fact, almost all MSL-2 peaks in the genome coincide with MLE peaks (Supplemental Fig. S3B). Furthermore, most of the combined MSL-2/MLE peak areas coincide with HAS for the MSL-DCC previously mapped using different strategies (Fig. 3B; Supplemental Fig. S3C; Alekseyenko et al. 2008; Straub et al. 2008). The increased resolution and signal-to-noise of our ChIP-seq profiles clearly suggests that the 241 sites that are characterized by co-localization of MSL-2 and MLE have a special quality and we consider that they may all belong to the HAS category. Conceivably, HAS may be defined as the composite MLE and MSL-2 binding sites. In agreement with our previous analysis (Straub et al. 2008), these 241 sites mainly map to noncoding parts of active genes, preferentially to their 3′ halves (Fig. 3C; Supplemental Fig. S3D,E). Roughly 200 of these sites contain a central (GA)8-motif, about half of them with multiple instances (Supplemental Fig. S3F,G).
Figure 3.

MLE binds with MSL-2 at high-affinity binding sites. (A) A representative 2 MB X-chromosomal region with ChIP-seq binding profiles for MSL-2 and MLE. In addition, a ChIP-chip profile for MLE (MLE cc) is shown. (B) MSL complex feature profiles at a known high-affinity binding site (HAS) in the last intron of the Tao-1 gene (Straub et al. 2008). (Red rectangle) The position and extent of the previous HAS definition. (C) Distribution of the newly defined 241 HAS on functional regions of the genome. (D) Average enrichment of MSL complex components along 2 kb surrounding the centers of all HAS (n = 241). Red-shaded areas behind the solid red line depict the interquartile range of enrichment. (Horizontal dashed lines) Median enrichment of the corresponding feature in its genome-wide top 200 peaks including HAS and non-HAS peaks. (E) Average DNA fragment sizes of input and precipitated samples in the center of all HAS as precisely determined by paired-end sequencing. Top and bottom edges of the box indicate the 95% confidence interval.
The architecture of high-affinity binding sites
All MSL complex components including the noncoding RNA roX2 show a marked enrichment on HAS by our new definition, the extent of which, however, varies strongly (Fig. 3D, also consider the representative example in Fig. 3B). In keeping with our current discussion we assume that the degree of enrichment correlates with the “directness” of the interaction or proximity of the complex subunit to chromatin. The global analysis of the ChIP-seq data shows that the primary chromatin contacts for MSL-2 and MLE are at the 241 HAS (by our new definition). roX2 (Chu et al. 2011; Simon et al. 2011) has its strongest peaks at these sites as well, however, with a considerable spreading into neighboring regions. The broad enrichment of MSL-3 in the neighborhood of HAS is explained by the fact that the majority of the HAS are within transcribed genes on the X. The additional, local concentration and cross-linking efficiency of MSL-3 at these sites is very modest. MSL-1 and MOF show robust binding at HAS. However, in contrast to MSL-2 and MLE, there are many other sites in the genome, which are bound equally well or even stronger by both MSL-1 and MOF.
The different intensities of the MSL ChIP-seq signals suggested that MSL-2 and MLE were considerably closer to DNA than MOF or MSL-1. Recently, Henikoff and colleagues reported that a systematic paired-end sequencing analysis of chromatin fragments released by micrococcal nuclease (MNase) digestion can reveal structural detail of the local chromatin organization (Henikoff et al. 2011). We applied this strategy, using instead cross-linked and sonicated chromatin, to determine the average length of HAS-derived DNA fragments associated with each MSL protein aiming to gain insight into the topology of the MSL-DCC interactions at HAS (Fig. 3E). Even though all ChIP reactions were performed on the same input chromatin, the DNA fragments purified with the different proteins varied strongly in size. The shortest DNA fragments were recovered in an immunoprecipitation of MSL-2 (156 bp) and increasing lengths were found associated with MLE (162 bp), MSL-1 (168 bp), and MOF (170 bp). Intuitively, fragment sizes vary because of differences in chromatin interaction. Sonication breaks may directly occur adjacent to a DNA-bound protein. If proteins associate with chromatin indirectly as part of a larger assembly that leaves a broader “footprint” on DNA, the immunoprecipitated fragment will be longer. Following the assumption that the fragment length obtained in ChIP is inversely correlated with the proximity of a factor to DNA, we hypothesize that MSL-2 and MLE contact HAS DNA most directly followed by indirect association of MSL-1 and MOF (see model in Fig. 7C, below).
Figure 7.

Different modes of MSL protein binding in different chromatin contexts. (A) Nucleosome reads along MSL-1 peaks on promoters, MSL-3 peaks on gene bodies, and HAS as derived from MNase-seq data. Shaded areas above and below the solid lines describe the interquartile range of enrichment. (B) DNAse hypersensitivity along MSL-1 peaks on promoters, MSL-3 peaks on gene bodies, and HAS as derived from DNase-seq data (modEncode). Shaded areas on top and bottom of the solid lines describe the interquartile range of enrichment. (C) MSL complex architecture on three classes of binding sites as defined by high-resolution NGS mapping.
The roX gene loci are very peculiar high-affinity binding sites of the MSL-DCC that have been postulated as primary assembly locations of the complex (Oh et al. 2003). The binding of the MSL proteins on the roX2 HAS differs (Supplemental Fig. S3H) in that we observe an enrichment of all components about four times stronger when compared with the other HAS. The binding pattern could be the result of the superimposition of two phenomena: (1) the interaction of MSL proteins with the gene-internal enhancer element to activate transcription (Lee et al. 2004; Rattner and Meller 2004), and (2) ongoing assembly of MSL complexes on nascent roX RNA (Oh et al. 2003). Both scenarios are likely to differ substantially from the interactions of MSL-DCC at HAS.
Primary contacts of MSL-1 and MOF mainly occur at promoters but are not enriched on the X
As expected, the ChIP-seq signals of MSL-2, MSL-3, MLE, and H4K16ac exhibit a clear X-chromosomal enrichment (Supplemental Fig. S4A). To our surprise we found that the ones of MSL-1 (Fig. 4A) and MOF (Supplemental Fig. S4A) show no enhancement on the X. This is in contrast to immunofluorescence studies and a multitude of ChIP-chip results (Fig. 4B; Supplemental Figs. S4B, S5A), which provide ample proof for the fact that MSL-1 and MOF are enriched on the X chromosome as part of the MSL-DCC. This discrepancy can only be resolved by considering that our ChIP-seq analysis emphasizes the primary, most direct contacts with target chromatin and that the enrichment of MOF and MSL-1 on the X is due to indirect chromatin binding via other targeting components, namely MSL-2 and MLE at HAS and MSL-3 on transcribed target gene bodies.
Figure 4.

MSL-1 and MOF ChIP-seq profiles do not show an X-chromosomal enrichment. Chromosomal distribution of ChIP-seq (A) and ChIP-chip (B) enrichment signals for MOF. Each vertical line within a chromosome represents the maximum enrichment signal of a 20-kb window with a color scaling linearly from white (30% quantile) to dark red (99.9% quantile). For the sake of clarity, signals from heterochromatic arms have been omitted. (C) Chromosomal distribution of MSL-2, MLE, MSL-1, MOF, and MSL-3 peaks. Omitted are peaks mapping to the heterochromatic arms. (D) Fractional distribution of features bound on autosomes (A) versus the X chromosome (X) for the indicated dosage compensation proteins.
Consequently, the locations where MSL-1 and MOF signals peak must be sites where these two proteins come closest to target DNA. What are these sites? For a more precise characterization of MSL-1 and MOF binding sites we performed systematic peak calling on all MSL proteins including all ChIP-seq data (Fig. 4C). The chromosomal distributions of peaks confirm that, contrary to the cases for MSL-2, MLE, and MSL-3, there is no X-chromosomal enrichment of MSL-1 and MOF peaks. A detailed assessment of the genomic distribution of each MSL protein (Fig. 4D) reveals that a large fraction—more than 1000—of MSL-1 and MOF peaks map to promoters on all chromosomes with no preference for the X. On the X chromosome, additional HAS binding is prominent and MOF is also attracted to gene bodies.
Average enrichment profiles along active genes reveal a systematic increase of MOF and MSL-1 binding on the promoters and a slight enrichment on the bodies of X-chromosomal genes (Fig. 5A). The strong enrichment of MSL-1 and MOF on the transcribed sequences that was previously highlighted—the “spreading” fraction—is only obvious in cumulative ChIP-chip profiles, which are based on IP of mildly sheared chromatin (Fig. 5B).
Figure 5.

MSL-1 and MOF bind promoters of some active genes genome-wide. (A) Average ChIP-seq enrichment of MOF and MSL-1 along active genes on the X (red line, n = 1113) and the autosomes (dark blue line, n = 5341). Shaded areas on top and bottom of the solid lines describe the interquartile range of enrichment. Signals on HAS have been masked for this analysis. (B) Corresponding ChIP-chip enrichments. (C) Venn diagram depicting the overlap of MSL-1 and MOF peaks at promoters on both X and autosomes. Signals on HAS have been masked for this analysis. (D) Fractions of active genes that have an MSL-1 peak within the promoter. (E) Average DNA fragment sizes of input and precipitated samples in the center of MSL-1 promoter peaks as precisely determined by paired-end sequencing. Top and bottom edges of the box indicate the 95% confidence interval.
On a genome-wide scale the co-localization of MSL-1 and MOF at promoters is substantial: Almost all MSL-1-bound promoters are also targets for MOF (Fig. 5C). For MOF, such promoter binding has been described and related to its presence in the “NSL” complex (Prestel et al. 2010; Raja et al. 2010). However, so far no function for MSL-1 outside of the DCC and no MSL-2-independent chromatin association have been described. Since this latter finding appears provocative in light of the known MSL-1 biology (Copps et al. 1998; Li et al. 2005) we wished to substantiate the finding of MSL-2-independent MSL-1 binding to autosomal sites in an independent way. To this end we performed quantitative immuno-FISH experiments using high-resolution confocal microscopy (Supplemental Fig. S5A–C). In brief, we measured the immunofluorescence signals on autosomal loci that had been selected for binding of MSL-1/MOF in the absence of all other MSL proteins in the ChIP-seq profiles (e.g., Supplemental Fig. S5B). In the example shown, the mtRNApol locus was visualized by FISH and the co-localization of MSL proteins by immunostaining (Supplemental Fig. S5A). We indeed found MOF and MSL-1, but not MSL-3 or MLE, co-localizing with the FISH signal (Supplemental Fig. S5C). No signal for MSL-2 outside the X-chromosomal domain could be detected. We conclude that MOF and MSL-1 co-localize at many promoters genome-wide and independent of the MSL-DCC.
Intriguingly, only ∼20% of the active genes have such MSL-1/MOF promoter peaks (Fig. 5D). Fragment size analyses of the paired-end ChIP-seq data (Fig. 5E) show that the DNA fragments immuno-purified along with MSL-1 and MOF on promoters are larger than on HAS, supporting the notion of an alternative recruitment complex. Furthermore, MOF-associated fragments are much larger than those retrieved via MSL-1, suggesting a configuration in which MSL-1 is closer to chromatin. MOF may, therefore, be recruited via its interaction with the C terminus of MSL-1 (Morales et al. 2004; Kadlec et al. 2011). However, MOF can also be targeted to promoters via the alternative NSL complex. Comparing the available NSL1 ChIP-seq profiles in larval salivary glands and MSL-1 profiles in S2 cells we find a strong overlap of the two proteins on promoters (Supplemental Fig. S5D).
Dosage compensation correlates with MSL enrichment on gene bodies
The ChIP-seq analysis was able to differentiate the primary contacts of the MSL proteins. Which of these interactions are best correlated with the actual dosage compensation function, the activation of X-linked genes? We first tested if the presence of MSL-1 at promoters influenced the distribution of other MSL proteins on the gene bodies (Fig. 6A). Even though a slight increase of MSL-3 on active X-chromosomal genes that have MSL-1 bound at their promoters can be observed in comparison to those that lack MSL-1 binding, the overall impact on MSL complex distribution appears to be minor. Intriguingly, the enrichment of RNA polymerase II (pol II) is greater on the promoters with MSL-1 peaks (Supplemental Fig. S6C), but this is also true for autosomal genes. In general, there appears to be more pol II recruited to autosomal than to X-chromosomal genes. To correlate feature enrichment and transcription regulation more systematically, MSL binding on promoters and gene bodies were related with the reduction of gene expression after MSL-2 knockdown and with the distance to one of the 241 HAS. The correlation matrix derived (Fig. 6B) shows that dosage-compensated transcription correlates very well with body enrichments of MSL-DCC components, preferably roX2 and MSL-3 (Fig. 6B; Supplemental Fig. S6B). In contrast, promoter enrichments only correlate weakly with MSL-1, promoter enrichment being the poorest predictor for dosage compensation. The closer a gene is to one of the HAS loci, the more MSL proteins will be enriched on gene bodies (Fig. 6B; Supplemental Fig. S6C), confirming a previous interpretation of ChIP-chip data (Straub et al. 2008).
Figure 6.

MSL feature enrichment on the transcribed regions correlate well with compensated gene expression. (A) Distribution of MSL-1, MOF, MSL-3, and H4K16ac on X-chromosomal active genes with (red, n = 361) or without (dark blue, n = 752) MSL-1 promoter peaks. Shaded areas above and below the solid lines describe the interquartile range of enrichment. Signals on HAS have been masked for this analysis. (B) Correlation matrix of MSL feature enrichments on promoter or body regions of active genes and functional compensation and distance of the genes from HAS. Pearson correlation coefficients are color-coded as indicated by the scale bar on the right. Signals deriving from HAS have been masked for this analysis.
Chromatin organization at different MSL contact sites
We determined the nucleosome configuration at the major peak areas of MSL proteins—promoters, gene bodies, and HAS—using MNase-seq data obtained in the same cell line (Fig. 7A; Gilchrist et al. 2010). The MSL-3 peaks in coding sequences show a strong nucleosome position signal precisely underneath the MSL-3 signal. This result supports the earlier conclusion that the interaction of the MSL-DCC at coding regions is determined by MSL-3 interaction to H3K36me3-modified nucleosomes (Fig. 2). The earlier finding that HAS are characterized by a reduced nucleosome density (Alekseyenko et al. 2008; Straub et al. 2008) is also confirmed by our current analysis that shows a general reduction in nucleosomes with no evidence for regular positioning aligning around the binding sites (Fig. 7A). At promoters, strong nucleosome depletion at MSL-1 binding sites with a symmetric nucleosome phasing to both sides is noted. The data are nicely complemented by DNase sensitivity profiles, which reveal strong accessibility at MSL-1 promoter peaks and clear accessibility at HAS, whereas peaks on gene bodies do not display an increased accessibility (Fig. 7B).
Discussion
To the best of our knowledge, our data represent the first complete mapping of all known subunits of a chromatin-bound multiprotein complex. Taking advantage of the shear sensitivity of the MSL-DCC, we obtained first hints about the architecture of MSL complexes bound to different chromosomal targets. We were able to evaluate three different binding modes with respect to their relevance to the dosage compensation process and observed interactions of MSL subunits at promoters independent of an MSL-DCC. These data contrast the prevailing concepts that do not distinguish different MSL-DCC configurations during the initial association with and spreading along the X chromosome (Straub and Becker 2007; Gelbart and Kuroda 2009; Conrad and Akhtar 2011).
Three classes of MSL protein binding
Our analyses distinguish three modes of chromosomal interaction for MSL proteins based on the subunit that makes primary contact, on the local chromatin configuration, and with respect to the functional relevance for dosage compensation (Fig. 7C). The first, most common mode finds the MSL components on gene bodies, and correlates profoundly with the MSL-2-dependent transcriptional enhancement. The primary interface with transcribed chromatin appears to comprise MSL-3 interacting with nucleosomes. roX2 RNA is also prominent along the gene bodies (Supplemental Fig. S2). Depending on the shear forces applied to chromatin more or less MOF and MSL-1 can be detected associated with MSL-3.
A second MSL interaction mode is seen at HAS that frequently reside within the X-chromosomal transcription units. Here, MSL-2 and MLE are seen to establish primary contact, whereas MSL-1 and MOF appear to associate more indirectly. roX2 binding is prominent at these sites and nucleosome density is low. The presence of a HAS enhances the association of the MSL-DCC complex with active genes in the vicinity.
Finally, MSL-1 and MOF bind to a fraction of active promoters with no chromosomal preference. These promoters may also be bound by the MOF-containing NSL complex (Raja et al. 2010; Feller et al. 2012). Their chromatin structure resembles that of typical nucleosome-depleted promoters with regular nucleosome phasing on either side (Iyer 2012). This promoter association has no major influence on compensated gene expression.
Methodological considerations
Formaldehyde cross-linking as applied in ChIP protocols generates multiple covalent linkages between proteins and/or nucleic acids, i.e., DNA and RNA (Orlando et al. 1997). In a complex setting such as a chromatin-bound MSL-DCC, all types of cross-links are expected to occur, such that in addition to trapping direct protein–DNA interactions, indirect tethering of proteins to chromatin via cross-links to other proteins and RNA is expected. Explicit rules for the relative contributions of either type of cross-link are not available. Conditions are usually empirically determined for each target, since cross-linking efficiencies are very dependent on the individual molecular context. Application of ultrasound sonication to break the DNA backbone for chromatin fragmentation will also break other bonds and thus will lead to fragmentation of polypeptides, as we have shown for MSL proteins (Supplemental Fig. S1G).
The ChIP-seq procedure commonly favors chromatin fragments of a relatively small size (generally <250 bp). The fraction of total chromatin in fragments of this size range depends on the extent of chromatin shearing. We assume that the common ChIP-seq protocols are biased toward analyzing the most highly fragmented DNA and that a higher shear regime allows inclusion of the majority of chromatin fragments in the sequence analysis. All available data are consistent with the hypothesis that the discordance of MSL interactions we observe are due to a disruption of the large chromatin-bound MSL complexes, so that the analysis highlights the primary, direct chromatin contacts of the individual MSL proteins. Fragmenting chromatin <500 bp massively reduced the level of MSL-1 and MOF at transcribed regions (Fig. 2). At the same time, the X chromosome enrichment of the two proteins that is easily visible using immunofluorescence microscopy or ChIP-chip (>500-bp fragments) is lost, suggesting that the recruitment to the X chromosome is indeed indirect. The sharp peaks for MOF and MSL-1 obtained by ChIP-seq, although aesthetically pleasing, should not be interpreted as mapping with improved resolution or better signal-to-noise ratio. Only the reference to earlier ChIP-chip data, which reveal X enrichment, allowed derivation of novel information on the anatomy of the MSL complexes. In the absence of the dominant X-chromosomal aspect, minor interactions are appreciated that had gone unnoticed in the past. The promoter interactions of MSL-1 and MOF are retained, since they might show a fundamentally different quality.
Differences in ChIP-chip/ChIP-seq protocols due to differential shearing will probably neither affect the mapping of histone modifications, nor of proteins that directly contact DNA, such as transcription factors. Rather, they are likely to affect the analysis of proteins that are recruited to chromatin more indirectly, for example because they are peripheral subunits of larger assemblies. However, it has to be kept in mind that the removal of indirectly bound macromolecules by increased shearing might also increase the accessibility of epitopes of the remaining, directly bound proteins. In turn, some chromatin bound features might now be detected with increased sensitivity. This could provide an explanation for the rather unexpected appearance of promoter-bound MSL-1 on autosomes that remained largely undetected in ChIP under low-shear conditions as well as in immunofluorescence experiments where shearing is not applied.
Some discrepancies between data sets in the literature may represent similar cases and may yield insight into the anatomy of complex regulatory assemblies. For example, the profiles of Drosophila polycomb proteins look strikingly different if ChIP-chip (modEncode) and ChiP-seq (Enderle et al. 2011) profiles are compared, with loss of broad distributions in ChIP-seq data. It is possible that cases like the one we describe are confined to ribonucleoprotein assemblies and that the RNA component is particularly sensitive to shear forces. In this context it is interesting that PRC2 complexes may also contain noncoding RNA (Zhao et al. 2010). Another example relates to the global interactions of the histone kinase JIL-1, which covers all active transcription units in ChIP-chip profiles (Regnard et al. 2011) and appears reduced to promoter and enhancer peaks in a recent ChIP-seq study (Kellner et al. 2012). We hypothesize that a large fraction of JIL-1 does not directly bind to chromatin, but is targeted more indirectly though shear-sensitive interactions.
The anatomy of high-affinity sites for the MSL-DCC
Contrary to expectations we found that the direct interactions of MSL-2 with chromatin did not coincide with MSL-1 peaks, but with MLE peaks. The interactions of MLE with the X chromosome are largely confined to MSL-2 sites (Fig. 3). Since there is a very high degree of joint MSL-2/MLE peaks with HAS defined in previous studies, we tentatively extend our catalog of HAS (Straub et al. 2008) by defining them as joint MLE and MSL-2 binding sites in ChIP-seq studies. The 241 HAS share the same features described previously and most of them contain one or more GA-rich motifs that contribute to MSL-2 recruitment (Alekseyenko et al. 2008; Straub et al. 2008). The selective interactions of MLE with the base of the HAS-bound DCC suggests an important role of the helicase at these selected sites for complex assembly, perhaps in the context of roX RNA. Direct interaction of MLE and MSL-2 has been suggested earlier (Li et al. 2008; Morra et al. 2011). This is consistent with the notion that MLE activity is mainly required for complex dissemination from the HAS (Gu et al. 2000; Morra et al. 2008).
Interaction of the MSL-DCC with target gene bodies
All current models assume that all subunits of the MSL-DCC are present on the bodies of target genes (Gelbart and Kuroda 2009; Conrad and Akhtar 2011; Straub and Becker 2011). Our data suggest that the complex is particularly sensitive to shear forces on transcribed chromatin so that only the direct interactions of MSL-3 with nucleosomes are retained. Collectively, our results lend strong support to a model in which the MSL-DCC is tethered to target genes via interaction of MSL-3 with H3K36-methylated nucleosomes (Larschan et al. 2007). The interface between the remainder of the MSL proteins and MSL-3 is shear-sensitive and conceivably involves roX RNAs. It might also reflect a more flexible interaction of an effector module including MOF, which may reach out to acetylate chromatin in a larger chromosomal domain (Gelbart et al. 2009; Conrad et al. 2012). Comparing HAS and gene body binding we speculate about “inverted” binding modes, where the MSL-DCC uses one surface (MSL-2/MLE) to associate with HAS and is then stripped off these sites when the opposite interaction surface encounters suitably modified chromatin by looping (Fig. 7C). Our model is compatible with the simultaneous interaction of a single MSL complex with a HAS and a target gene. The realization of two distinct interaction surfaces may provide a mechanistic model for the distribution of the DCC from HAS to target genes. The dependency of gene body binding on HAS assembly is supported by the strong correlation between body signals and HAS proximity (Fig. 6B).
An alternative interpretation of the observed binding differences between HAS and gene bodies would be that MSL proteins are able to form more than just one canonical “DCC” but give rise to alternative assemblies with different subunit stoichiometry, depending on the site of chromatin interaction. The observed MSL-1–MOF co-localization at promoters in the absence of MSL-2 (see below) supports this idea. The lack of a perfect complex stoichiometry in biochemical preparations of solubilized MSL complexes may reflect such complex heterogeneity, although disruption of complexes during the fractionation procedure cannot be excluded (Smith et al. 2000).
The earlier description of MSL binding along transcribed genes was interpreted as evidence for a gradual and continuous 3′ enrichment of MSL-1, MOF, and MSL-3 along the transcription unit (Alekseyenko et al. 2006; Gilfillan et al. 2006; Kind et al. 2008). Such enrichment nicely fits the idea that dosage compensation functions at the level of transcription elongation (Larschan et al. 2011). We now show that the 3′ enrichment must be considered a numerical artifact due to the presence of long introns in the 5′ ends of a class of genes. These long introns contain a specific chromatin signature that could reflect replication origins or enhancers (Kharchenko et al. 2010; Kellner et al. 2012). Importantly, these regions are devoid of H3K36me3 and therefore cannot be targeted by the complex. In summary, MSL complex components cover intron-free parts of the active genes rather uniformly. This distribution is compatible with a wide variety of mechanistic models for gene activation, including effects on transcription elongation (Larschan et al. 2011). Indeed, there is a good correlation between the association of the MSL-DCC with genes bodies and MSL-2-dependent transcription. Surprisingly, MSL-3 and roX2 interactions are the features that correlate best with activation, and not H4K16 acetylation. This could be due to technical shortcomings such as a very low resolution of the roX2 mapping data and/or the stripping of body features by shear forces. However, it may also indicate that H4K16 acetylation is not the only functional consequence of the MSL-DCC association with target genes.
Roles for a novel MSL-1–MOF containing assembly independent of the DCC?
Promoter binding of MOF and MSL-1 were already described by Kind et al. (2008). However, the extent of the widespread association of MSL-1 with autosomal promoters we found was rather unexpected. We had shown earlier that ectopically expressed MSL-1 in females could be recruited to MOF tethered to a reporter locus (Prestel et al. 2010), indicating that such an association is possible and might just be difficult to detect in an unperturbed system. In females, MOF is mostly found in the context of the NSL complex, which binds to many housekeeping promoters (Feller et al. 2012). Previous biochemical studies suggested that the interactions of MOF with MSL-1 or NSL1, which share a common interaction domain, are mutually exclusive (Raja et al. 2010) and that MOF–promoter interactions were independent of MSL-1 (Kind et al. 2008). We now find a considerable overlap between MSL-1 and NSL1 at promoters. It is currently unclear whether the MSL-1 association has any functional implication. In the context of our study we asked whether the promoter associations of MSL-1 and MOF have any effect on dosage compensation. Our correlation studies clearly suggest that this is not the case. Even though an increased RNA polymerase II recruitment can be observed on MSL-1-bound promoters, this phenomenon also occurs on autosomes. In addition, only a minority of ∼20% of all active X-chromosomal genes show promoter binding of MSL-1. We conclude that the presence of MSL-1 at promoters is unrelated to dosage compensation. We substantiated the surprising lack of MSL-2 at these sites by an independent immunofluorescence approach. The precise nature of an assembly that contains MSL proteins but lacks MSL-2 remains to be explored. In light of the potential existence of several “MSL complexes,” we now systematically use the term “MSL-DCC” for an MSL complex that contains all five MSL subunits, at least one roX RNA, and functions in dosage compensation. We are aware of the fact that the existence of such a complex is currently only suggested by circumstantial evidence.
Methods
Chromatin IP
Male Drosophila S2 cells were cultured and processed for chromatin IP as previously described (Straub et al. 2008) with the following modifications: Chromatin was sheared using different instruments and energy settings (see Supplemental Table 1 for sample-technology relationship); Bioruptor (Diagenode) shearing was performed at setting “high” for 30 sec in 25 and 55 cycles to yield chromatin of an average size of 500 bp and 200 bp, respectively. Using a Covaris S220 (Covaris) we generated 180-bp chromatin with a peak incident power of 100 W, duty factor 20%, 200 cycles/burst for 30 min; 800-bp chromatin required a parameter adjustment to 60 W and a shearing time of 90 min. Chromatin fragment size distribution was evaluated on a Bioanalyzer (Agilent) (Supplemental Fig. S1). ChIP antibodies used are listed in Supplemental Table 1.
ChIP-chip sample and data processing
Whole genome amplification, microarray hybridization at ImaGenes, and data processing were performed exactly as described before (Straub et al. 2008).
ChIP-seq read mapping, normalization, and calculation of genomic coverage
Reads were mapped to the Drosophila genome (version dm3) using Bowtie version 0.12.7 (Langmead et al. 2009). Parameter adjustments in the case of single read data was “–m 1” and in the case of paired-end reads “–trim3 65 –X 1500”. On single read data we performed a read extension based on a fragment size determination by cross-correlation analysis of the forward and reverse strand reads (lengths are specified in Supplemental Table 1). We next calculated for each sample a per-base genomic coverage vector by cumulating the total spans of all sequenced fragments.
For obtaining an average coverage vector corrected for background read distribution and incorporating all replicate samples, we first performed an arcsine transformation of the raw coverage vectors adjusting for library size differences using
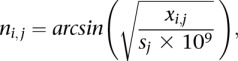 |
where i = genomic position, x = number of fragments covering i, j = sample, s = size factor of raw coverage vectors calculated according to Anders and Huber (2010).
The transformed vectors of replicate samples were then averaged. The average background from each average IP vector was subtracted and the resulting values transformed to Z-scores. These values served as average enrichment over input measurement in all of our analyses on continuous ChIP-seq data. We performed our analysis in parallel on data processed using either spp (Kharchenko et al. 2008) or CisGenome (Ji et al. 2008) and obtained similar results.
Calculation of the coverage of roX2 RNA based on published data (Chu et al. 2011; Simon et al. 2011) was performed exactly as described for the protein targets.
Peak calling
Local peaks of enrichment were identified using the Cisgenome2 tool seqpeak (Ji et al. 2008) on the raw bowtie mappings. Peaks were called including all replicates and input controls using the read extension parameter “–e 150” in addition to default parameters. In the case of paired-end reads we used a one-sided read subset for compatibility reasons. We applied a FDR cutoff of 0.5% to the peak result list.
Gene structure classification
Each gene was divided into 20 nonoverlapping, consecutive, and equally sized bins. For each bin we calculated the average intron density as number of bases in introns divided by the length of the bin in base pairs. The resulting vectors were aligned from 5′ to 3′ and a euclidean distance matrix was computed. We then performed hierarchical clustering using the “ward” agglomeration as provided by the function “hclust” in R.
Data analysis
For all analyses the Drosophila genome annotation version gadfly 537 served as a reference. All downstream data visualizations were performed in R (R-project.org). Data processing details are provided in the Supplemental Methods.
Additional data sets used: GSE22618 (H4K16ac/RNA Polymerase S2ph ChIP-chip), GSE12292 (MSL-1/MSL-2 ChIP-chip), GSE31332 & GSE28180 (roX2), GSE20472 (MNase-seq), GSE8557 (H3K36me3 ChIP-chip), GSE13217 (H3.3 ChIP-chip), modEncode_3324 (DNAse I), modEncode_296 (H3K27ac ChIP-chip), modEncode_292 (H3K18ac ChIP-chip).
Data access
ChIP-chip and ChIP-seq data have been submitted to the NCBI Gene Expression Omnibus (GEO) (http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE37865.
Acknowledgments
We thank Dirk Eick for providing antibodies against RNA polymerases; and Christian Beisel, Henk Stunnenberg, and the associated sequencing facilities for performing the initial sequencing of the MSL-2 ChIPs. The research leading to these results was funded by the Deutsche Forschungsgemeinschaft (Be 1140/6-1) and the European Research Council under the European Union's Seventh Framework Programme (FP7/2007-2013)/ERC grant agreement no. 293948.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.146407.112.
References
- Alekseyenko AA, Larschan E, Lai WR, Park PJ, Kuroda MI 2006. High-resolution ChIP-chip analysis reveals that the Drosophila MSL complex selectively identifies active genes on the male X chromosome. Genes Dev 20: 848–857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alekseyenko AA, Peng S, Larschan E, Gorchakov AA, Lee O-K, Kharchenko P, McGrath SD, Wang CI, Mardis ER, Park PJ, et al. 2008. A sequence motif within chromatin entry sites directs MSL establishment on the Drosophila X chromosome. Cell 134: 599–609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Huber W 2010. Differential expression analysis for sequence count data. Genome Biol 11: R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu C, Qu K, Zhong FL, Artandi SE, Chang HY 2011. Genomic maps of long noncoding RNA occupancy reveal principles of RNA-chromatin interactions. Mol Cell 44: 667–678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad T, Akhtar A 2011. Dosage compensation in Drosophila melanogaster: Epigenetic fine-tuning of chromosome-wide transcription. Nat Rev Genet 13: 123–134 [DOI] [PubMed] [Google Scholar]
- Conrad T, Cavalli FMG, Holz H, Hallacli E, Kind J, Ilik I, Vaquerizas JM, Luscombe NM, Akhtar A 2012. The MOF chromobarrel domain controls genome-wide H4K16 acetylation and spreading of the MSL complex. Dev Cell 22: 610–624 [DOI] [PubMed] [Google Scholar]
- Copps K, Richman R, Lyman LM, Chang KA, Rampersad-Ammons J, Kuroda MI 1998. Complex formation by the Drosophila MSL proteins: Role of the MSL2 RING finger in protein complex assembly. EMBO J 17: 5409–5417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enderle D, Beisel C, Stadler MB, Gerstung M, Athri P, Paro R 2011. Polycomb preferentially targets stalled promoters of coding and noncoding transcripts. Genome Res 21: 216–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan X, Lamarre-Vincent N, Wang Q, Struhl K 2008. Extensive chromatin fragmentation improves enrichment of protein binding sites in chromatin immunoprecipitation experiments. Nucleic Acids Res 36: e125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fauth T, Müller-Planitz F, König C, Straub T, Becker PB 2010. The DNA binding CXC domain of MSL2 is required for faithful targeting the Dosage Compensation Complex to the X chromosome. Nucleic Acids Res 38: 3209–3221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feller C, Prestel M, Hartmann H, Straub T, Soeding J, Becker PB 2012. The MOF-containing NSL complex associates globally with housekeeping genes, but activates only a defined subset. Nucleic Acids Res 40: 1509–1522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelbart ME, Kuroda MI 2009. Drosophila dosage compensation: A complex voyage to the X chromosome. Development 136: 1399–1410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelbart ME, Larschan E, Peng S, Park PJ, Kuroda MI 2009. Drosophila MSL complex globally acetylates H4K16 on the male X chromosome for dosage compensation. Nat Struct Mol Biol 16: 825–832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilchrist DA, Santos Dos G, Fargo DC, Xie B, Gao Y, Li L, Adelman K 2010. Pausing of RNA polymerase II disrupts DNA-specified nucleosome organization to enable precise gene regulation. Cell 143: 540–551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilfillan GD, Straub T, de Wit E, Greil F, Lamm R, van Steensel B, Becker PB 2006. Chromosome-wide gene-specific targeting of the Drosophila dosage compensation complex. Genes Dev 20: 858–870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu W, Wei X, Pannuti A, Lucchesi JC 2000. Targeting the chromatin-remodeling MSL complex of Drosophila to its sites of action on the X chromosome requires both acetyl transferase and ATPase activities. EMBO J 19: 5202–5211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henikoff JG, Belsky JA, Krassovsky K, MacAlpine DM, Henikoff S 2011. Epigenome characterization at single base-pair resolution. Proc Natl Acad Sci 108: 18318–18323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer VR 2012. Nucleosome positioning: Bringing order to the eukaryotic genome. Trends Cell Biol 22: 250–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji H, Jiang H, Ma W, Johnson DS, Myers RM, Wong WH 2008. An integrated software system for analyzing ChIP-chip and ChIP-seq data. Nat Biotechnol 26: 1293–1300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadlec J, Hallacli E, Lipp M, Holz H, Sanchez-Weatherby J, Cusack S, Akhtar A 2011. Structural basis for MOF and MSL3 recruitment into the dosage compensation complex by MSL1. Nat Struct Mol Biol 18: 142–149 [DOI] [PubMed] [Google Scholar]
- Kelley RL, Meller VH, Gordadze PR, Roman G, Davis RL, Kuroda MI 1999. Epigenetic spreading of the Drosophila dosage compensation complex from roX RNA genes into flanking chromatin. Cell 98: 513–522 [DOI] [PubMed] [Google Scholar]
- Kellner WA, Ramos E, Van Bortle K, Takenaka N, Corces VG 2012. Genome-wide phosphoacetylation of histone H3 at Drosophila enhancers and promoters. Genome Res 22: 1081–1088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharchenko PV, Tolstorukov MY, Park PJ 2008. Design and analysis of ChIP-seq experiments for DNA-binding proteins. Nat Biotechnol 26: 1351–1359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kharchenko PV, Alekseyenko AA, Schwartz YB, Minoda A, Riddle NC, Ernst J, Sabo PJ, Larschan E, Gorchakov AA, Gu T, et al. 2010. Comprehensive analysis of the chromatin landscape in Drosophila melanogaster. Nature 471: 480–485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kind J, Vaquerizas JM, Gebhardt P, Gentzel M, Luscombe NM, Bertone P, Akhtar A 2008. Genome-wide analysis reveals MOF as a key regulator of dosage compensation and gene expression in Drosophila. Cell 133: 813–828 [DOI] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL 2009. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10: R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larschan E, Alekseyenko AA, Gortchakov AA, Peng S, Li B, Yang P, Workman JL, Park PJ, Kuroda MI 2007. MSL complex is attracted to genes marked by H3K36 trimethylation using a sequence-independent mechanism. Mol Cell 28: 121–133 [DOI] [PubMed] [Google Scholar]
- Larschan E, Bishop EP, Kharchenko PV, Core LJ, Lis JT, Park PJ, Kuroda MI 2011. X chromosome dosage compensation via enhanced transcriptional elongation in Drosophila. Nature 471: 115–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsson J, Meller VH 2006. Dosage compensation, the origin and the afterlife of sex chromosomes. Chromosome Res 14: 417–431 [DOI] [PubMed] [Google Scholar]
- Lee C, Reichman T, Baik T, Mathews M 2004. MLE functions as a transcriptional regulator of the roX2 gene. J Biol Chem 279: 47740–47745 [DOI] [PubMed] [Google Scholar]
- Li F, Parry DAD, Scott MJ 2005. The amino-terminal region of Drosophila MSL1 contains basic, glycine-rich, and leucine zipper-like motifs that promote X chromosome binding, self-association, and MSL2 binding, respectively. Mol Cell Biol 25: 8913–8924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F, Schiemann AH, Scott MJ 2008. Incorporation of the noncoding roX RNAs alters the chromatin-binding specificity of the Drosophila MSL1/MSL2 complex. Mol Cell Biol 28: 1252–1264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucchesi JC 2009. The structure-function link of compensated chromatin in Drosophila. Curr Opin Genet Dev 19: 550–556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyman LM, Copps K, Rastelli L, Kelley RL, Kuroda MI 1997. Drosophila male-specific lethal-2 protein: Structure/function analysis and dependence on MSL-1 for chromosome association. Genetics 147: 1743–1753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mank JE 2009. The W, X, Y and Z of sex-chromosome dosage compensation. Trends Genet 25: 226–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meller VH, Rattner BP 2002. The roX genes encode redundant male-specific lethal transcripts required for targeting of the MSL complex. EMBO J 21: 1084–1091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales V, Straub T, Neumann MF, Mengus G, Akhtar A, Becker PB 2004. Functional integration of the histone acetyltransferase MOF into the dosage compensation complex. EMBO J 23: 2258–2268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morra R, Smith ER, Yokoyama R, Lucchesi JC 2008. The MLE subunit of the Drosophila MSL complex uses its ATPase activity for dosage compensation and its helicase activity for targeting. Mol Cell Biol 28: 958–966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morra R, Yokoyama R, Ling H, Lucchesi JC 2011. Role of the ATPase/helicase maleless (MLE) in the assembly, targeting, spreading and function of the male-specific lethal (MSL) complex of Drosophila. Epigenet Chromatin 4: 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh H, Park Y, Kuroda MI 2003. Local spreading of MSL complexes from roX genes on the Drosophila X chromosome. Genes Dev 17: 1334–1339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orlando V, Strutt H, Paro R 1997. Analysis of chromatin structure by in vivo formaldehyde cross-linking. Methods 11: 205–214 [DOI] [PubMed] [Google Scholar]
- Prestel M, Feller C, Straub T, Mitlöhner H, Becker PB 2010. The activation potential of MOF is constrained for dosage compensation. Mol Cell 38: 815–826 [DOI] [PubMed] [Google Scholar]
- Raja SJ, Charapitsa I, Conrad T, Vaquerizas JM, Gebhardt P, Holz H, Kadlec J, Fraterman S, Luscombe NM, Akhtar A 2010. The nonspecific lethal complex is a transcriptional regulator in Drosophila. Mol Cell 38: 827–841 [DOI] [PubMed] [Google Scholar]
- Rattner BP, Meller VH 2004. Drosophila male-specific lethal 2 protein controls sex-specific expression of the roX genes. Genetics 166: 1825–1832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reenan RA, Hanrahan CJ, Ganetzky B 2000. The mle(napts) RNA helicase mutation in Drosophila results in a splicing catastrophe of the para Na+ channel transcript in a region of RNA editing. Neuron 25: 139–149 [DOI] [PubMed] [Google Scholar]
- Regnard C, Straub T, Mitterweger A, Dahlsveen IK, Fabian V, Becker PB 2011. Global analysis of the relationship between JIL-1 kinase and transcription. PLoS Genet 7: e1001327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shogren-Knaak M, Ishii H, Sun J-M, Pazin MJ, Davie JR, Peterson CL 2006. Histone H4-K16 acetylation controls chromatin structure and protein interactions. Science 311: 844–847 [DOI] [PubMed] [Google Scholar]
- Simon MD, Wang CI, Kharchenko PV, West JA, Chapman BA, Alekseyenko AA, Borowsky ML, Kuroda MI, Kingston RE 2011. The genomic binding sites of a noncoding RNA. Proc Natl Acad Sci 108: 20497–20502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith ER, Pannuti A, Gu W, Steurnagel A, Cook RG, Allis CD, Lucchesi JC 2000. The Drosophila MSL complex acetylates histone H4 at lysine 16, a chromatin modification linked to dosage compensation. Mol Cell Biol 20: 312–318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straub T, Becker PB 2007. Dosage compensation: The beginning and end of generalization. Nat Rev Genet 8: 47–57 [DOI] [PubMed] [Google Scholar]
- Straub T, Becker PB 2011. Transcription modulation chromosome-wide: Universal features and principles of dosage compensation in worms and flies. Curr Opin Genet Dev 21: 147–153 [DOI] [PubMed] [Google Scholar]
- Straub T, Grimaud C, Gilfillan GD, Mitterweger A, Becker PB 2008. The chromosomal high-affinity binding sites for the Drosophila dosage compensation complex. PLoS Genet 4: e1000302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Ohsumi TK, Kung JT, Ogawa Y, Grau DJ, Sarma K, Song JJ, Kingston RE, Borowsky M, Lee JT 2010. Genome-wide identification of polycomb-associated RNAs by RIP-seq. Mol Cell 40: 939–953 [DOI] [PMC free article] [PubMed] [Google Scholar]