

Abstract
Zinc-finger nucleases (ZFNs) are important tools for genome engineering. Despite intense interest by many academic groups, the lack of robust noncommercial methods has hindered their widespread use. The modular assembly (MA) of ZFNs from publicly available one-finger archives provides a rapid method to create proteins that can recognize a very broad spectrum of DNA sequences. However, three- and four-finger arrays often fail to produce active nucleases. Efforts to improve the specificity of the one-finger archives have not increased the success rate above 25%, suggesting that the MA method might be inherently inefficient due to its insensitivity to context-dependent effects. Here we present the first systematic study on the effect of array length on ZFN activity. ZFNs composed of six-finger MA arrays produced mutations at 15 of 21 (71%) targeted loci in human and mouse cells. A novel drop-out linker scheme was used to rapidly assess three- to six-finger combinations, demonstrating that shorter arrays could improve activity in some cases. Analysis of 268 array variants revealed that half of MA ZFNs of any array composition that exceed an ab initio B-score cutoff of 15 were active. These results suggest that, when used appropriately, MA ZFNs are able to target more DNA sequences with higher success rates than other current methods.
Zinc-finger nucleases (ZFNs) have shown great potential as tools for genome engineering and gene therapy (Mackay and Segal 2010; Urnov et al. 2010). Two arrays of engineered zinc fingers must bind their DNA targets at a precise spacing (typically five, six, or seven base pairs) to allow their C-terminal FokI cleavage domains to dimerize and form an active nuclease. It has been challenging to engineer zinc-finger arrays with properties appropriate to produce highly active ZFNs. The proprietary methods of Sangamo Biosciences and Sigma-Aldrich appear to produce arrays that recognize a broad spectrum of DNA sequences, sufficient to create ZFNs that can disrupt any desired human gene or be used in clinical trials (Urnov et al. 2010). However, the high cost of these commercial reagents has severely limited their use. In contrast, noncommercial engineering methods have been more restricted in their capabilities. Rapid and simple modular assembly (MA) methods have been developed based on fingers or modules that had been engineered (Barbas and coworkers [Gonzalez et al. 2010]) or identified from nature (Kim and coworkers/ToolGen [Bae et al. 2003]) and bind a broad diversity of 3-bp DNA sites. However, Ramirez et al. (2008) reported that MA had unexpectedly high failure rates with only 6% of three-finger MA array pairs predicted to produce an active ZFN. More recently, success rates ∼25% were achieved using specialized sets of modules (S Kim et al. 2011; Zhu et al. 2011). Oligomerized pool engineering (OPEN) was introduced as a method to account for context-dependent effects by optimizing all three modules together in the context of the target sequence (Maeder et al. 2008). However, OPEN was complicated, laborious, and limited to arrays that recognize all 16 GNN (e.g., GAG, GCT…) and a few TNN triplets. Three-finger arrays that recognize 5′-GNNGNNGNN-3′ are limited to <4% of all possible 9-bp sites. Since two arrays are required for a ZFN, <0.16% of all 18-bp sequences could be targeted. As one example of this limitation, it would not be possible to design OPEN arrays for 90 (87%) of the 104 target sites used in the Ramirez et al. (2008) study; thus, the expected failure rate of OPEN would be greater than the unexpected failure rate of MA (76%) on these targets. Context-dependent assembly (CoDA) enabled the rapid assembly of parts of previously successful OPEN arrays (Sander et al. 2011). CoDA ZFNs were shown to successfully cleave 50% of their chromosomal targets. However, the range of targetable sequences was a subset of OPEN and insufficient to target, for example, all protein-coding regions in the genomes of zebrafish and Arabidopsis thaliana (Sander et al. 2011). Thus, public capabilities are far from the aspirations to use ZFNs to precisely edit single-nucleotide polymorphisms (SNPs) or genetic mutations causing disease.
A common interpretation for the failure rates of three-finger MA arrays was their insensitivity to context-dependent effects; modules engineered in one context may not perform well when placed in a different sequence context (Cathomen and Joung 2008). However, an alternative interpretation was suggested by Sander et al. (2009), who observed that three-finger arrays composed of high-affinity Barbas modules tended to have high affinity, whereas arrays of low affinity modules bound poorly. Approximately 60% of the GNN-modules were considered to have high affinity, corresponding well with the 60% of GNN arrays reported to bind well by Ramirez et al. (2008). These results led us to hypothesize that the primary inefficiency of three-finger MA arrays might not be a fundamental disregard for context dependencies, but rather that several modules lacked sufficient affinity. This hypothesis was reinforced by the recent demonstration that the specificity of MA arrays was similar to that of naturally occurring zinc-finger proteins and therefore was unlikely to be the primary cause of their poor performance in ZFNs (Lam et al. 2011). In principle, modules could be re-engineered to have higher affinity. As a more pragmatic solution, longer arrays of modules could be used to increase affinity. Anecdotal evidence supported this concept (Gordley et al. 2009; Guo et al. 2010; Perez-Pinera et al. 2012). However, we showed recently that a caveat to the latter approach is that the addition of modules can, in some cases, reduce the activity of a ZFN, perhaps by allowing subgroups of fingers to bind additional unexpected locations (Shimizu et al. 2011). These considerations led us to perform a systematic investigation of number and composition of modules on ZFN activity.
Results
Longer MA arrays generally increase ZFN activity, but the relationship is not linear
To systematically examine the relationship between array length and ZFN activity, we designed extended-MA ZFNs to eight loci in the human genome near SNPs associated with coronary artery disease. Target sites were chosen that contained spacers of five, six, or seven base pairs between the two zinc-finger binding sites, requiring ZFNs to be constructed with zinc finger–FokI linkers of TGGS, TGAAAR, and TGPGAAAR, respectively (Handel et al. 2009). Unlike CoDA target sites, all sites here contained mixtures of ANN, CNN, GNN, and TNN type triplets (Supplemental Table S1; Supplemental Appendix SI). Arrays composed of three to six fingers were constructed using the Barbas set of zinc-finger modules, which are the most expansive in terms of sequence recognition (Bhakta and Segal 2010). Since the sequential addition of modules is laborious, we developed a “drop-out linker” strategy by which the three-, four-, and five-module arrays could be created from a six-module array using silent restriction sites in coding regions (Fig. 1; Supplemental Fig. S1). In this way, only two six-finger arrays would need to be synthesized or assembled for each heterodimeric ZFN. The two sets of four arrays could then be generated in parallel in one day to enable the rapid testing of all 16 combinations of arrays.
Figure 1.

The drop-out linker scheme. SphI, BsmI, and HindIII sites were introduced into the zinc-finger coding region as silent mutations. Digestion with one restriction enzyme followed by ligation allows the full set of arrays to be created in parallel in one day.
Using a single-strand annealing (SSA) recombination reporter assay in HEK293T cells, we examined the activity of the various combinations of arrays (Fig. 2; Table 1). Generally, ZFNs composed of two three-finger arrays were least active, whereas ZFNs composed of two six-finger arrays were more active (defined as a percentage of the activity of a control ZFN [GZF3-L3 + GZF1-R3]) (Szczepek et al. 2007). However, activity did not correlate linearly with the number of modules. The optimal lengths for the left and right arrays were often different. For example, increasing the left array of the CS2-1 ZFN from three to six fingers increased ZFN activity, whereas increasing the right array decreased activity. The optimum was L5 + R3. For CS7-3, L6 + R5 was more active than L6 + R6.
Figure 2.
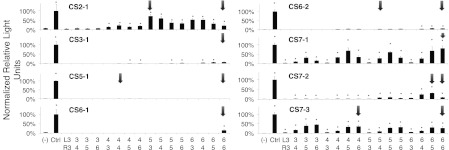
Activity of the CS series of ZFNs determined by a SSA assay. The number of fingers in the left and right arrays for each ZFN are indicated. Note that the CS3-1, 5-1, 6-1, and 6-2 series started with four- or six-finger arrays instead of three-finger arrays to reduce the assembly effort required prior to the invention of the drop-out linker strategy. Based on the data, the missing ZFNs seemed likely to be inactive and were not tested subsequently. (-) SSA reporter only as a negative control. (Ctrl) GZF3-L3 + GZF1-R3 control nuclease to which all activities are normalized. (Error bars) The standard deviation of normalized duplicates from at least two experiments. (*) P < 0.01 compared to the negative control based on a one-tailed homoscedastic t-test. (Black arrows) ZFNs used in genomic cleavage assays.
Table 1.
Summary of ZFN activity based on the SSA and genomic assays
MA ZFNs are active at chromosomal target sites
To streamline the methodology even further, we demonstrated that we could also omit the SSA assay. Cells were treated directly with the 16 combinations of ZFNs targeting two additional endogenous loci and analyzed using the Surveyor assay, which detects the appearance of mutations due to nonhomologous end joining at the cleavage site. Active MA ZFNs were obtained for both loci (Fig. 3; Supplemental Fig. S2). Consistent with the differential array behaviors observed in the SSA assay, all left arrays of the T2-X6 ZFN supported activity, but only R4 produced an active nuclease.
Figure 3.

Functional selection of ZFNs using the Surveyor assay in HEK293T cells. ZFN CS2-1-L5 + R3, as a positive control, and all 16 combinations of ZFN T2-X6 were assayed in duplicate. The number of fingers in the left and right arrays for each ZFN are indicated above each pair of lanes. Numbers below each lane indicate the percentage of modified alleles averaged over the duplicates. (Arrows) Appearance and position of the Surveyor cleavage band. (Black box) ZFN pair T2-X6 L5 + R4 that was used for Illumina sequencing analysis.
High-throughput sequencing demonstrated that seven of the 10 L6 + R6 ZFNs (70%) produced indels (Table 1, Exploratory study; Supplemental Fig. S3; Supplemental Appendix SIII) at 0.5% or greater (a value considered “active” in other ZFN studies) (Gupta et al. 2012). In addition, seven array-length variants that had been found to be more active by SSA or the Surveyor assay were generally found to be more active at their endogenous sites. Some ZFNs remained inactive, perhaps due to an unfavorable chromatin environment at the endogenous locus. Of the 13 ZFNs for which both SSA and indel data were available, the correlation of percent indel activity to percent SSA activity was modest (R2 = 0.25). However, there was perfect correlation between an indel threshold of ≥0.5% and an SSA threshold of ≥8%.
Development of a scoring system to help predict outcomes
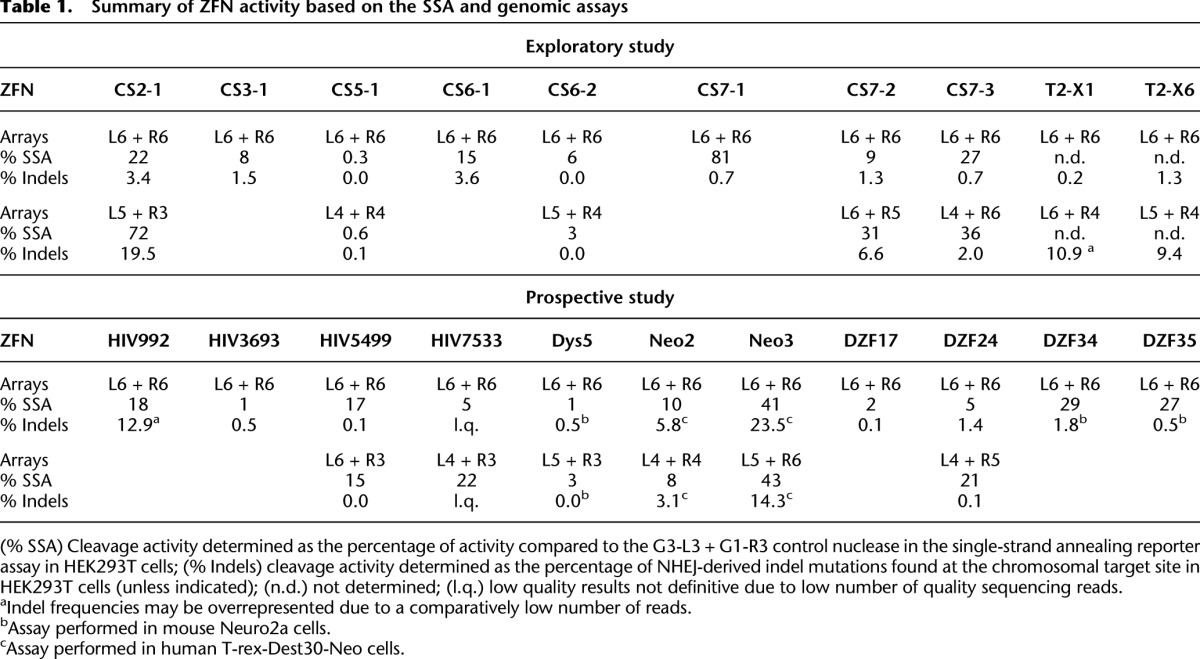
To apply this “extended-MA” approach as a useful method, we developed a scoring system to rank potential binding sites based on predicted zinc-finger binding affinity. Using the measured affinities of Barbas GNN modules (Segal et al. 1999), previous studies showed that the relative affinity of new 3 × GNN arrays (recognizing sites of the composition 5′-GNNGNNGNN-3′) could be accurately predicted by calculating the value of ΔΔG (Sander et al. 2009). Unfortunately, this method is limited to the prediction of 3 × GNN arrays since similar affinity data do not currently exist for ANN, CNN, or TNN modules. We therefore created an ab initio “B-score” based on the theory that binding energy should be proportional to the number of bivalent hydrogen bonds, such as between G and Arg or between A and Gln or Asn (Fig. 4A; Supplemental Table S2; Supplemental Discussion S1). The B-score correlated well (R2 = 0.73–0.86) (Fig. 4B,C) with the measured affinities of 3 × GNN arrays (Segal et al. 1999; Sander et al. 2009) and moderately well (R2 = 0.52) (Fig. 4D) with the affinities of six-finger arrays of mixed ANN, CNN, or TNN composition (MS Kim et al. 2011; Shimizu et al. 2011). The weak correlation with the latter data set was somewhat expected since it is known that the affinities of arrays longer than three fingers often do not scale linearly. However, the trend predicted by the B-score was more accurate for this set than any currently available method, such as summing the number of GNN modules (R2 = 0.12). Therefore, though not a perfect predictor, the B-score was used as a predictor of relative binding affinity. The SSA scores for 92 ZFN array-length variants were evaluated by receiver operating characteristic (ROC) (Fig. 4E; Supplemental Appendix SI). The Comb.B score was found to be the best classifier for an SSA threshold of ≥8% (area under the curve [AUC] = 0.77). Importantly, the Comb.B score was more accurate than the number of GNN modules (AUC = 0.66), which was recently suggested to have the best correlation with the activity of 3 × GNN MA ZFNs (Zhu et al. 2011). A Comb.B score of 15 was found to be the best compromise between true and false positives.
Figure 4.
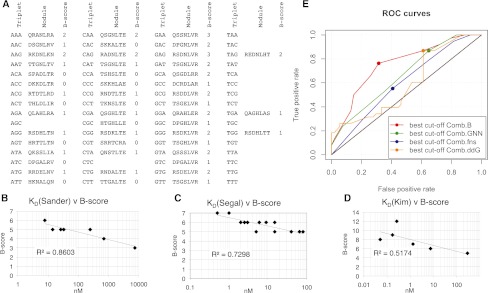
The ab initio B-score as a predictor of relative affinity and activity of MA ZFNs. (A) The B-scores are shown for each Barbas zinc-finger module (Bhakta and Segal 2010). B-scores were summed and compared to the measured affinities of (B) seven three-finger arrays (Sander et al. 2009); (C) 16 three-finger arrays (Segal et al. 1999); and (D) six six-finger arrays of mixed composition (MS Kim et al. 2011; Shimizu et al. 2011). (E) Receiver operating characteristic (ROC) analysis of four predictors of ZFN activity (≥8% SSA activity) was performed for the 92 array variants of the CS ZFN series using the Daim package of the R statistics program. (Comb.B) The combined B-score of the modules in the left and right arrays. (Comb.GNN) The combined number of GNN modules. (Comb.fns) The combined number of fingers. (Comb.ddG) The combined ΔΔG of the GNN modules (Sander et al. 2009).
A prospective study based on B-scores
The most efficient method for most investigators would be to (1) use the B-score to predict sites to which active ZFNs could be prepared; (2) screen just one ZFN combination on such sites for activity; and (3) optionally test all 16 combinations of arrays on active sites to determine the optimal nuclease configuration. ZFNs composed of six-finger drop-out linker arrays would be an ideal initial configuration because the 16 combinations of subarrays could be quickly constructed and tested, if desired. We therefore performed a prospective study on 11 additional ZFN targets that each had a B-score for the L6 + R6 arrays of ≥15 (Table 1, Prospective study; Supplemental Appendix SIII). Eight of the 11 L6 + R6 ZFNs were successful in the indel assay (73%). In contrast to the earlier ZFN set, analysis of all 16 array-length combinations by SSA revealed few examples of shorter arrays providing improved activity (Supplemental Fig. S4). Six array variants were examined and found to have similar or reduced activity at their endogenous targets (Table 1).
Long interfinger linkers do not improve ZFN activity
The ZFNs made in this study exclusively use canonical TGEKP interfinger linkers, which have been shown to be the optimal linker length for six-finger arrays (Peisach and Pabo 2003; Neuteboom et al. 2006). However, proteins with four or more fingers have the potential to bind to off-target sites using subsets of three fingers (Shimizu et al. 2011). Other studies have suggested that the use of noncanonical TGSQKP linkers between pairs of fingers can improve the specificity and consequently, ZFN activity, presumably by disrupting the binding of three-finger subsets (Moore et al. 2001; Gupta et al. 2012). This strategy has been adapted widely by Sangamo Biosciences and Sigma-Aldrich (Doyon et al. 2008; Perez et al. 2008; Hockemeyer et al. 2009). In contrast to these studies, we observed that the use of disruptive linkers dramatically decreased the SSA activity of all configurations of extended-MA ZFNs and did not rescue the activity of any inactive ZFNs (Fig. 5). These data suggest that the use of disruptive linkers is not recommended for extended-MA ZFNs.
Figure 5.

Influence of “disruptive” interfinger linkers on extended-MA ZFN activity. ZFN activity was determined by the SSA assay for ZFNs with long linkers (TGSQKP) inserted between fingers 2 and 3 and fingers 4 and 5. (Std Link) All canonical linkers (TGEKP). The array configuration for the Std Link ZFN was GZF3-L3 + GZF1-R3, CS2-1 L5 + R3, CS5-1 L4 + R4, CS6-2 L5 + R4, CS7-3 L4 + R6, and T2-X6 L5 + R4.
Longer MA arrays are not associated with increased cytotoxicity
To better understand the mechanism by which additional fingers affect performance (Supplemental Discussion S2), the affinity and specificity of the three- and five-finger arrays of ZFN CS2-1 were investigated by quantitative electromobility shift assays (Fig. 6A; Supplemental Fig. S4). Consistent with the activity data, CS2-1 L3 did not display detectable binding (KD > 532 nM), whereas L5 bound tightly (KD = 0.08 nM). In contrast, both R3 and R5 bound their target with similar affinity (KD = 0.56 nM). Therefore, the reduction in ZFN activity in comparing CS2-1-R3 to CS2-1-R5 is likely not due to loss of affinity, which is similar to observations we reported previously (Shimizu et al. 2011). The reduction in activity may be due to the ability of subsets of the multiple fingers to bind additional specific or nonspecific targets. As one measure of specificity, the proteins were tested for binding to a nontarget DNA (proteins L3 and L5 on the R6 target and vice versa). All proteins failed to shift the nontarget DNA even at the highest protein concentrations used in this assay, suggesting a >1000-fold discrimination between target and nontarget. Specificity was further examined using the Bind-n-Seq (Zykovich et al. 2009) target site selection assay (Fig. 6B). Compared to L3, L5 recognized a site that was not only longer but also a closer match with the intended target site. In contrast, the binding site length and composition of R5 improved minimally compared to R3. These results demonstrate that, for the case of CS2-1, ZFN failure was primarily the fault of just one array in the pair. Optimizing the array length improved both the affinity and specificity of the underperforming array.
Figure 6.

An analysis of affinity, specificity, and cytotoxicity. (A) EMSA was used to determine the affinity of CS2-1 three- and five-finger arrays for their specific (cognate) target as well as a nontarget (e.g., the L3 array on the R3 binding site). A large ratio of nontarget:specific binding is an indicator of good specificity. (B) In vitro binding specificity was also determined using the Bind-n-Seq target site selection assay (Zykovich et al. 2009). The binding preference of CS2-1-L3 appears to differ from the intended target site at three positions (red boxes). (C) Cytotoxicity was assessed as a decrease in the percentage of ZFN-expressing cells on day 5 compared to day 1. To follow only those cells expressing nuclease, a GFP expression vector was cotransfected with the indicated ZFN expression vectors. (GFP) Cells cotransfected with GFP and empty ZFN vector as a positive control. (GZF3-wt) A nuclease known to be cytotoxic (Szczepek et al. 2007) as a negative control. (Error bars) The standard deviation of normalized duplicates from at least two experiments. (*) P < 0.00001 compared to the GFP-only positive control based on a one-tailed homoscedastic t-test. (D) Cleavage activity is a summary of the SSA data from other figures and is shown here for reference.
As another assay of the specificity of short and long arrays, we examined the cytotoxicity of three inactive and three active ZFNs. ZFNs with poor specificity have more off-target cleavage events, leading to cytotoxicity. CS7-1-L3 + R3 was inactive with three-finger arrays, HIV7533-L6 + R6 was inactive with six-finger arrays (even though the L4 + R3 version was active), and CS2-1-L3 + R5 was inactive with arrays of mixed length (Fig. 6D). Cytotoxicity was assessed as a decrease in the percentage of ZFN-expressing cells on day 5 compared to day 1 (Fig. 6C). None of the inactive ZFNs were observed to be cytotoxic, arguing against the possibility that the long array-ZFNs only appear inactive because they kill the cells expressing them. HIV7533-L4 + R3 was the shortest active combination of arrays in our study, CS7-1-L6 + R6 was active with the longest arrays, and CS2-1-L5 + R3 was active with mixed array lengths. Importantly, the active ZFNs with the shortest and longest arrays were not observed to be cytotoxic, demonstrating that long arrays per se are not problematic. The third ZFN of mixed array length showed a significant level of cytotoxicity. Considering all of the data, it seems likely that such cytotoxicity is due to the specifics of that particular ZFN and not a general deficiency of the methodology. These observations generally support the use of extended MA to create active and nontoxic nucleases.
Discussion
Combining the data from the exploratory and prospective studies, L6 + R6 extended-MA arrays produced active ZFNs at 15 of 21 (71%) of endogenous target sites, greater than the success rates recently reported for other MA methods (25%) (S Kim et al. 2011; Zhu et al. 2011) or CoDA (50%) (Sander et al. 2011) and similar to the rate of an engineered two-finger archive (82%) (Gupta et al. 2012). From a practical perspective, this means it might only be necessary to test one or two L6 + R6 ZFNs to different sites to find one that is active. Activity can be improved by using shorter arrays in some cases, and the drop-out linker can facilitate this rapid empirical testing. Shorter arrays can also be useful to maximize the spectrum of sequences that can be targeted, since in some cases the lack of appropriate modules may prevent the construction of two six-finger arrays. Toward that end, the SSA scores of all 268 ZFN array variants examined in this study suggest that any combination of MA arrays would be successful in 38% of cases; but with a combined B-score of ≥15, they could produce an active ZFN in 52% of cases (Supplemental Table S4), which again would require testing of only one or two ZFNs. Thus, when choosing sites that are not L6 + R6 arrays, the combined B-score is particularly helpful. The accuracy (69%) of using the B-score was found to derive primarily from avoiding true negative unsuccessful short-array target sites (Fig. 7).
Figure 7.

The utility of the B-score for extended-MA. Distribution of combined B-scores for successful (red bars) and unsuccessful (blue bars) ZFNs based on the 268 array combinations in this study. The optimal cutoff of 15 is indicated (green line).
Another potential parameter for optimization is the use of long disruptive linkers between sets of two fingers. However, in contrast to previous studies, we observed only deleterious effects from the use of long linkers. It could be that even more empirical testing would be required to realize a benefit, such as systematically evaluating all possible linker substitutions and combinations. It should also be noted that most other studies have used long linkers in the context of two-finger modules rather than one-finger modules. Until better guidance becomes available, long linkers should be avoided for extended-MA ZFNs.
The very comprehensive lexicon of existing one-finger modules enables an extended-MA ZFN site approximately once every 52 bp (Supplemental Discussion S3). To illustrate the impact of the broader sequence recognition, a 60-kb region of DNA (hg18, chr9:22060000-22119999) was searched for potential ZFN sites using websites corresponding to three publicly available engineering methods.
Our website (http://www.genomecenter.ucdavis.edu/segallab/segallabsoftware) using extended MA found 3474 ZFN sites with a B-score ≥15 (spacers = 5, 6, or 7). The website outputs in BED format, which can be easily uploaded as a custom track in the UCSC Genome Browser (http://genome.ucsc.edu/) (Fig. 8A). Of the 3474 ZFNs, 50% (1737) would be expected to be active.
The ToolGen website (http://toolgen.co.kr/ZFNfinder/sws.cgi) using MA with a “recommended” set of modules (spacers = 5, 6, or 7) found 5401 3 + 3 ZFNs and 1791 4 + 4 ZFNs. 26% of potential cleavage sites could be targeted successfully by 4 + 4 ZFNs but only 9.1% by 3 + 3 ZFNs (Kim et al. 2009). Of the 3610 unique 3 + 3 and 1791 4 + 4 ZFNs, 15% (794) would be expected to be active (not shown).
The Zinc Finger Consortium ZiFiT website (http://zifit.partners.org/ZiFiT/ChoiceMenu.aspx) using CoDA found 50 sites (spacers = 5, 6, or 7) (Fig. 8A). Of the 50 ZFNs, 50% (25) would be expected to be active (Sander et al. 2011).
Figure 8.
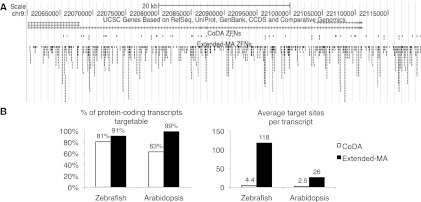
Spectrum of sequences targetable using extended MA. (A) An image from the UCSC Genome Browser shows potential extended-MA and CoDA ZFNs sites found within a 60-kb region at the human 9p21 locus (hg18, chr9:22060000-22119999). Each vertical black bar represents a target site. (B) A comparison of the transcript targeting capabilities reported for CoDA and calculated from the data in this study for extended MA.
These enhanced capabilities should also allow extended MA to target 91% of 28,527 unique protein-coding transcripts in the zebrafish genome (Ensemble Zv9 database), with an average of 118 sites per transcript, and 99% of 27,251 unique protein-coding transcripts in Arabidopsis (The Arabidopsis Information Resource 10 release) with an average of 26 sites per transcript (Fig. 8B). In comparison, CoDA was reported to target only 81% and 63% of these coding sequences, respectively, with an average of 4.4 and 2.5 sites per transcript (Sander et al. 2011).
The extended-MA method therefore provides dramatically improved genome engineering capabilities compared to other publicly available ZFN assembly methods. These results demonstrate that, when used appropriately, modular assembly can outperform methods that were developed to address its deficiencies (Cathomen and Joung 2008; Maeder et al. 2008; Sander et al. 2011). We believe that these results clarify a common misconception in the field that modular assembly is fundamentally inefficient. We note, however, that transcription activator-like effector nucleases (TALENs) appear to have an even greater success rate and target an even broader spectrum of DNA sequences than is currently possible with extended MA (Bogdanove and Voytas 2011; Reyon et al. 2012). Robust assembly methods have already enabled widespread adaptation of TALENs for genome engineering applications in contrast to the limited ZFN methods that inspired this study.
Methods
Construction of zinc-finger arrays
In most cases, coding regions were synthesized by BioBasic, Inc. Arrays of three, four, five, and six fingers were created using the drop-out linker strategy described in Figure 1 and Supplemental Figure S1, sequential cloning of additional fingers by PCR, or using the SuperZiF system (Gonzalez et al. 2010). Additional fingers were digested with XhoI/AgeI and cloned between the XhoI/XmaI sites of these vectors so as to preserve canonical TGEKP linkers between all fingers. The sequences of all zinc-finger coding regions are provided in Supplemental Appendices SI and SII. Most heterodimeric ZFNs were expressed in vector pPGK-FokI and contained the obligate heterodimer modifications DD + RR (Szczepek et al. 2007). CS6-2, T2-X1, and T2-X6 were expressed in pCMV-FokI (DA + RV) (Szczepek et al. 2007). Dys5, Neo2, Neo3, DZF17, DZF24, DZF34, and DZF35 were expressed in pCMV-FokI (wt) and did not contain obligate heterodimer modifications. ZFNs used on target sites with 5-, 6-, or 7-bp spacers used zinc-finger–FokI linkers TGGS, TGAAAR, and TGPGAAAR, respectively (Handel et al. 2009).
Single-strand annealing (SSA) recombination reporter assay
The SSA assay is a plasmid-based reporter assay for detecting ZFN-induced repair of a split luciferase gene. A ZFN-induced double-strand break between homologous regions of the split luciferase will allow the SSA repair pathway to reconstruct an active luciferase gene. Construction of the SSA luciferase reporter plasmid, pSSA Rep 3-1, has been described previously (Szczepek et al. 2007; Bhakta and Segal 2010). The pSSA Rep3-1 and control GZF3-L3 + GZF1-R3 ZFN plasmids are available from Addgene (ID 5091–5094). Briefly, ZFN binding sites were introduced into the left and/or right arms of a split firefly luciferase gene by PCR and cloned into the BglII/EcoRI sites of the vector. All primers used for SSA construction are listed in Supplemental Table S5. In poly-lysine treated, 24-well plates, HEK293T cells at 80% confluency in DMEM supplemented with 10% fetal calf serum and 1 unit/mL of penicillin and 1 μg/mL of streptomycin were cotransfected with 100 ng of each ZFN monomer expression plasmid, 25 ng of pRL-TK-Renilla Luciferase (as a transfection control), and 25 ng of SSA reporter plasmid using Lipofectamine 2000 (Invitrogen). Cells were harvested 48 h post-transfection by removing media, washing with 500 μL of 1× DPBS followed by lysis in 100 μL of 1× Passive Lysis Buffer (Promega) with 1× Complete protease inhibitors (454 Life Sciences [Roche]). Clarified cell lysis supernatants (20 μL) were used to determine the luciferase activity using DualGlo or BrightGlo reagents (Promega) in a Veritas microplate luminometer (Turner Biosystems). All experiments were performed in duplicates and repeated on at least two different days.
Electromobility shift assay (EMSA)
The DNA binding domains of CS2-1-L3, -L5, -R3, and -R5 were subcloned into the prokaryotic expression vector pMal-c2× (New England Biolabs) that had been modified to contain a C-terminal His6 tag. Proteins were expressed in BL21 (DE3) E. coli (Invitrogen) for 6 h at 37°C, purified over amylose resin in Zinc Buffer A (ZBA: 100 mM Tris, pH 7.5, 90 mM KCl, 1 mM MgCl2, and 100 μm ZnCl2), and eluted in ZBA, 10 mM maltose, and 5 mM DTT. Complementary pairs of 5′-biotin labeled forward and 5′-poly-T reverse oligonucleotides were annealed to obtain double-stranded target DNAs. The sequences of all oligonucleotide targets are listed in Supplemental Table S6. Protein-DNA binding reactions were performed for 1 h at room temperature in ZBA, 150 mM KCl, 5 mM DTT, 10% glycerol, 0.1 mg/mL BSA, 0.05% NP-40, 35 pM target DNA, and purified ZFPs at the indicated concentrations. Gel electrophoresis was performed on 10% TBE-polyacrylamide gel in 0.5× TBE buffer at 4°C. The LightShift Chemiluminescent EMSA Kit (Pierce) was used according to the manufacturer’s protocol. After blotting on a Biodyne B nylon membrane (Pierce) for 1 h at 100 V at 4°C, the DNA was cross-linked by a UV cross-linker (Stratagene) for 4 min. Equilibrium binding constants (KD) were calculated from protein titration experiments imaged on X-ray film. All experiments were performed in at least duplicates and repeated on two different days. The reported values represent the results of at least two experiments, with a standard deviation of ±50%.
Binding site specificity assay using massively parallel sequencing (Bind-n-Seq)
Bind-n-Seq was performed essentially as described (Zykovich et al. 2009). Briefly, the coding regions CS2-1-L3, -L5, -R3, and -R5 were subcloned into pDest-GST-MBP, expressed in BL21 (DE3) E. coli (Invitrogen), and purified over amylose resin in ZBA, 10 mM maltose, 5 mM DTT. The maltose was removed by overnight dialysis in 2 L of ZBA, 5 mM DTT. Bar-coded 93-mer double-stranded oligonucleotide targets containing Illumina primer binding sites and a 21-nt random region were incubated with various concentrations of proteins and salt (50 nM protein, 1 mM KCl; 50 nM protein, 50 mM KCl; 50 nM protein, 100 mM KCl; 5 nM protein, 100 mM KCl; and 410 nM [CS2-1-L3, L5] or 200 nM [CS2-1-R3, R5] protein, 100 mM KCl). Bound complexes were precipitated using amylose resin and enriched by six wash steps in the corresponding salt buffer. Eluted DNA was sequenced on an Illumina GAII sequencer. Motifs were determined using a custom motif finder that will be described elsewhere.
Surveyor assay
HEK293T cells (5 × 105) were seeded in six-well plates. The next day, cells were transfected at 90% confluency with 1 μg of each ZFN using Lipofectamine 2000 (Invitrogen). Cells were harvested 72 h post-transfection and genomic DNA was extracted using a Macherey-Nagel tissue prep kit or Qiagen Puregene Core kit A according to manufacturer’s instructions. The ZFN target site region was amplified from 100 ng of genomic DNA (2 min at 95°C; 15 sec at 95°C, 30 sec at 58°C, 1 min at 72°C, 35 cycles; 5 min at 72°C). The sequences of all primers are listed in Supplemental Table S7. PCR products (3 μL) were diluted in 1× Taq Pro Buffer (Denville), 50 mM KCl. The amplicon mixture was heat denatured to allow for wild-type and mutant alleles to reanneal and form heteroduplexes (5 min at 95°C; 95°C to 85°C at -2°C/sec; 85°C to 25°C at -0.1°C/sec). The heteroduplex product was digested at the mismatch locus with Surveyor nuclease (Transgenomic) in 1× Enhancer Solution at for 20 min at 42°C. Digestion was completed by adding 1 μL of stop solution and the product was resolved by running on 10% TBE-polyacrylamide gel. Gels were stained with ethidium bromide and visualized using a UV imager. Band intensities were quantified using ImageJ as described previously (Guschin et al. 2010).
Mutation analysis at endogenous loci using massively parallel sequencing
Genomic DNA from ZFN-treated cells was isolated as in the Surveyor assay. Genomic regions flanking the ZFN sites were amplified with primers containing Illumina sequencing ends and adapter sequence, followed by a 5-bp barcode and genomic priming site. The sequences of all primers are listed in Supplemental Table S8. Specific fragments containing the binding sites were amplified from genomic DNA (200 ng) 72 h post-transfection using Phusion polymerase (New England Biolabs) in a 100 μL reaction (2 min at 95°C; 15 sec at 95°C, 30 sec at 57 or 60°C, 30 sec at 72°C, 31 cycles; 5 min at 72°C). The amplicons were purified and separated on 1% agarose gel. The band of interest was extracted and eluted in Elution Buffer (Qiagen). Samples were checked for quality on a BioAnalyzer and sequenced using 100-bp or 150-bp paired-end reads on an Illumina HiSeq sequencer, 160-bp paired-end on Illumina MiSeq sequencer, or 85-bp paired-end reads on an Illumina GAII sequencer. Analysis was performed using a custom bioinformatics pipeline. In short, reads were sorted according to their respective barcodes and trimmed for sequence quality, presence of adaptor sequence, and minimum length. Pairs of quality-filtered reads were surveyed for the presence of their specific genomic priming sites. Read pairs that did not contain perfect matches to both primer sequences were discarded. Pairs of sequences were compared to each other to search for overlap between the ends of the reads. When overlap was found, both sequences were combined into a single DNA sequence. Each of these resulting sequences was compared to the sequence and length of the wild-type (WT) target site sequence and the number of instances of each particular sequence was recorded.
Cytotoxicity assay
Toxicity was assessed by measuring the ratio of transfected cells on day 5 compared to day 1, as described by (Cornu and Cathomen 2010). HEK293T cells (5 × 105) were seeded in six-well plates in 2 mL of DMEM/BCS/PenStrep. Cells were transfected using Lipofectamine-2000 and 1 μg of pPNLK-GFP and 1 μg of each ZFN or PGK-empty (3 μg of total DNA). On day 1 (24 h post-transfection), cells were harvested for flow cytometry. Cells were washed with 1 mL of DPBS. Cells were released with 300 μL of trypsin then harvested in an additional 900 μL of DMEM complete medium. A portion of this (350 μL) was applied to a new six-well plate + 2 mL DMEM complete as the Day 5 sample. The remaining cells were washed with DPBS, resuspended in 300 μL of DPBS, and transferred to tubes for flow cytometry on ice. Cells (50,000) were scanned for GFP using a BD Biosciences LSRII. On day 5, cells were similarly harvested and scanned. All experiments were performed in at least duplicates and repeated on two different days.
Data access
Illumina massively parallel sequencing data are available at the NCBI BioProject archive (www.ncbi.nlm.nih.gov/bioproject) under accession number PRJNA179355. A website implementing the B-score and drop-out linker strategies is available at http://www.genomecenter.ucdavis.edu/segallab/segallabsoftware.
Acknowledgments
We thank Zhen Jia, Alice Adriaenssens, Sara Venturini, Lauren E. Cosby, and Matthew T. Brown for their technical contributions to this work. Funding was provided by the W.M. Keck Foundation and the UC Davis Clinical and Translational Sciences Center through NIH grant RR024146. I.M.H. was supported by NSF grant DBI 0733857. D.G.O. and C.A.G. were supported by an NIH Director’s New Innovator Award (DP2OD008586), the Hartwell Foundation, and a Basil O’Connor Starter Scholar Award from the March of Dimes.
Author contributions: D.J.S. and C.A.G. conceived of the method; M.S.B., L.X., C.A.G., and D.J.S. designed research; M.S.B., D.G.O, K.T.D., S.H.L., J.F.M, M.C.W., Y.Y., and H.L. performed experiments; I.M.H., A.Z., and D.J.S. performed bioinformatics analyses; M.S.B., L.X., C.A.G., and D.J.S. wrote the manuscript.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.143693.112.
Freely available online through the Genome Research Open Access option.
References
- Bae KH, Kwon YD, Shin HC, Hwang MS, Ryu EH, Park KS, Yang HY, Lee DK, Lee Y, Park J, et al. 2003. Human zinc fingers as building blocks in the construction of artificial transcription factors. Nat Biotechnol 21: 275–280 [DOI] [PubMed] [Google Scholar]
- Bhakta MS, Segal DJ 2010. The generation of zinc finger proteins by modular assembly. Methods Mol Biol 649: 3–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogdanove AJ, Voytas DF 2011. TAL effectors: Customizable proteins for DNA targeting. Science 333: 1843–1846 [DOI] [PubMed] [Google Scholar]
- Cathomen T, Joung JK 2008. Zinc-finger nucleases: The next generation emerges. Mol Ther 16: 1200–1207 [DOI] [PubMed] [Google Scholar]
- Cornu TI, Cathomen T 2010. Quantification of zinc finger nuclease-associated toxicity. Methods Mol Biol 649: 237–245 [DOI] [PubMed] [Google Scholar]
- Doyon Y, McCammon JM, Miller JC, Faraji F, Ngo C, Katibah GE, Amora R, Hocking TD, Zhang L, Rebar EJ, et al. 2008. Heritable targeted gene disruption in zebrafish using designed zinc-finger nucleases. Nat Biotechnol 26: 702–708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez B, Schwimmer LJ, Fuller RP, Ye Y, Asawapornmongkol L, Barbas CF III 2010. Modular system for the construction of zinc-finger libraries and proteins. Nat Protoc 5: 791–810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordley RM, Gersbach CA, Barbas CF III 2009. Synthesis of programmable integrases. Proc Natl Acad Sci 106: 5053–5058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo J, Gaj T, Barbas CF III 2010. Directed evolution of an enhanced and highly efficient FokI cleavage domain for zinc finger nucleases. J Mol Biol 400: 96–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta A, Christensen RG, Rayla AL, Lakshmanan A, Stormo GD, Wolfe SA 2012. An optimized two-finger archive for ZFN-mediated gene targeting. Nat Methods 9: 588–590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guschin DY, Waite AJ, Katibah GE, Miller JC, Holmes MC, Rebar EJ 2010. A rapid and general assay for monitoring endogenous gene modification. Methods Mol Biol 649: 247–256 [DOI] [PubMed] [Google Scholar]
- Handel EM, Alwin S, Cathomen T 2009. Expanding or restricting the target site repertoire of zinc-finger nucleases: The inter-domain linker as a major determinant of target site selectivity. Mol Ther 17: 104–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hockemeyer D, Soldner F, Beard C, Gao Q, Mitalipova M, DeKelver RC, Katibah GE, Amora R, Boydston EA, Zeitler B, et al. 2009. Efficient targeting of expressed and silent genes in human ESCs and iPSCs using zinc-finger nucleases. Nat Biotechnol 27: 851–857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HJ, Lee HJ, Kim H, Cho SW, Kim JS 2009. Targeted genome editing in human cells with zinc finger nucleases constructed via modular assembly. Genome Res 19: 1279–1288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Lee MJ, Kim H, Kang M, Kim JS 2011. Preassembled zinc finger arrays for rapid construction of zinc finger nucleases. Nat Methods 8: 7. [DOI] [PubMed] [Google Scholar]
- Kim MS, Stybayeva G, Lee JY, Revzin A, Segal DJ 2011. A zinc finger protein array for the visual detection of specific DNA sequences for diagnostic applications. Nucleic Acids Res 39: e29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam KN, van Bakel H, Cote AG, van der Ven A, Hughes TR 2011. Sequence specificity is obtained from the majority of modular C2H2 zinc-finger arrays. Nucleic Acids Res 39: 4680–4690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackay JP, Segal DJ, ed. 2010. Engineered zinc finger proteins: Methods and protocols. Springer Science and Business Media, New York [Google Scholar]
- Maeder ML, Thibodeau-Beganny S, Osiak A, Wright DA, Anthony RM, Eichtinger M, Jiang T, Foley JE, Winfrey RJ, Townsend JA, et al. 2008. Rapid “open-source” engineering of customized zinc-finger nucleases for highly efficient gene modification. Mol Cell 31: 294–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore M, Klug A, Choo Y 2001. Improved DNA binding specificity from polyzinc finger peptides by using strings of two-finger units. Proc Natl Acad Sci 98: 1437–1441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuteboom LW, Lindhout BI, Saman IL, Hooykaas PJ, van der Zaal BJ 2006. Effects of different zinc finger transcription factors on genomic targets. Biochem Biophys Res Commun 339: 263–270 [DOI] [PubMed] [Google Scholar]
- Peisach E, Pabo CO 2003. Constraints for zinc finger linker design as inferred from X-ray crystal structure of tandem Zif268-DNA complexes. J Mol Biol 330: 1–7 [DOI] [PubMed] [Google Scholar]
- Perez EE, Wang J, Miller JC, Jouvenot Y, Kim KA, Liu O, Wang N, Lee G, Bartsevich VV, Lee YL, et al. 2008. Establishment of HIV-1 resistance in CD4+ T cells by genome editing using zinc-finger nucleases. Nat Biotechnol 26: 808–816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Pinera P, Ousterout DG, Brown MT, Gersbach CA 2012. Gene targeting to the ROSA26 locus directed by engineered zinc finger nucleases. Nucleic Acids Res 40: 3741–3752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez CL, Foley JE, Wright DA, Muller-Lerch F, Rahman SH, Cornu TI, Winfrey RJ, Sander JD, Fu F, Townsend JA, et al. 2008. Unexpected failure rates for modular assembly of engineered zinc fingers. Nat Methods 5: 374–375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyon D, Tsai SQ, Khayter C, Foden JA, Sander JD, Joung JK 2012. FLASH assembly of TALENs for high-throughput genome editing. Nat Biotechnol 30: 460–465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sander JD, Zaback P, Joung JK, Voytas DF, Dobbs D 2009. An affinity-based scoring scheme for predicting DNA-binding activities of modularly assembled zinc-finger proteins. Nucleic Acids Res 37: 506–515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sander JD, Dahlborg EJ, Goodwin MJ, Cade L, Zhang F, Cifuentes D, Curtin SJ, Blackburn JS, Thibodeau-Beganny S, Qi Y, et al. 2011. Selection-free zinc-finger-nuclease engineering by context-dependent assembly (CoDA). Nat Methods 8: 67–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal DJ, Dreier B, Beerli RR, Barbas CF III 1999. Toward controlling gene expression at will: Selection and design of zinc finger domains recognizing each of the 5′-GNN-3′ DNA target sequences. Proc Natl Acad Sci 96: 2758–2763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu Y, Söllü C, Meckler JF, Adriaenssens A, Zykovich A, Cathomen T, Segal DJ 2011. Adding fingers to an engineered zinc finger nuclease can reduce activity. Biochemistry 50: 5033–5041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szczepek M, Brondani V, Buchel J, Serrano L, Segal DJ, Cathomen T 2007. Structure-based redesign of the dimerization interface reduces the toxicity of zinc-finger nucleases. Nat Biotechnol 25: 786–793 [DOI] [PubMed] [Google Scholar]
- Urnov FD, Rebar EJ, Holmes MC, Zhang HS, Gregory PD 2010. Genome editing with engineered zinc finger nucleases. Nat Rev Genet 11: 636–646 [DOI] [PubMed] [Google Scholar]
- Zhu C, Smith T, McNulty J, Rayla AL, Lakshmanan A, Siekmann AF, Buffardi M, Meng X, Shin J, Padmanabhan A, et al. 2011. Evaluation and application of modularly assembled zinc-finger nucleases in zebrafish. Development 138: 4555–4564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zykovich A, Korf I, Segal DJ 2009. Bind-n-Seq: High-throughput analysis of in vitro protein–DNA interactions using massively parallel sequencing. Nucleic Acids Res 37: e151. [DOI] [PMC free article] [PubMed] [Google Scholar]